
Alkali and Alkaline Earth Metal Complexes Ligated by an Ethynyl Substituted Cyclopentadienyl Ligand



Abstract
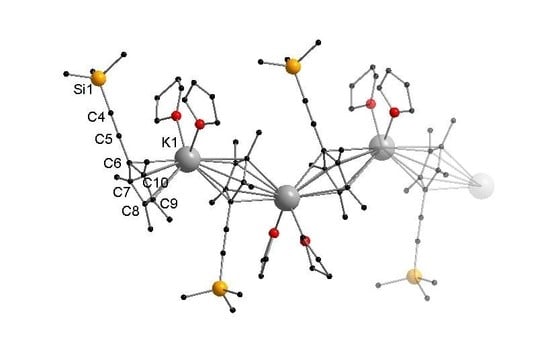
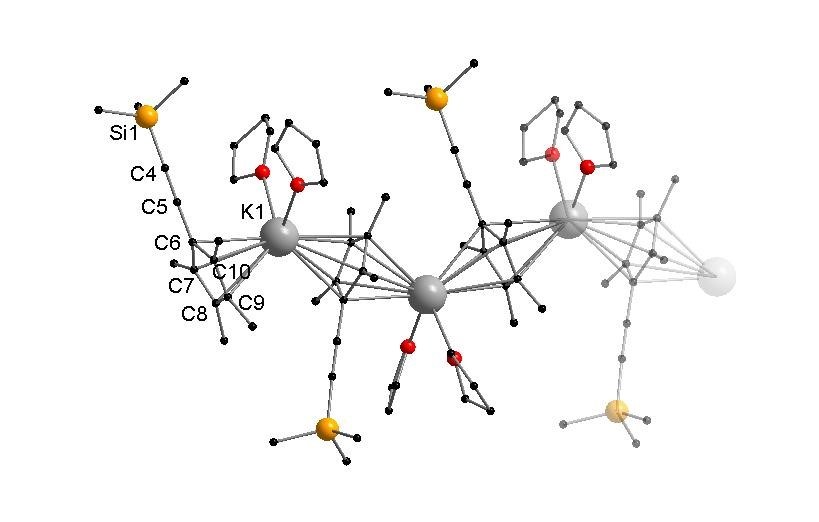
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
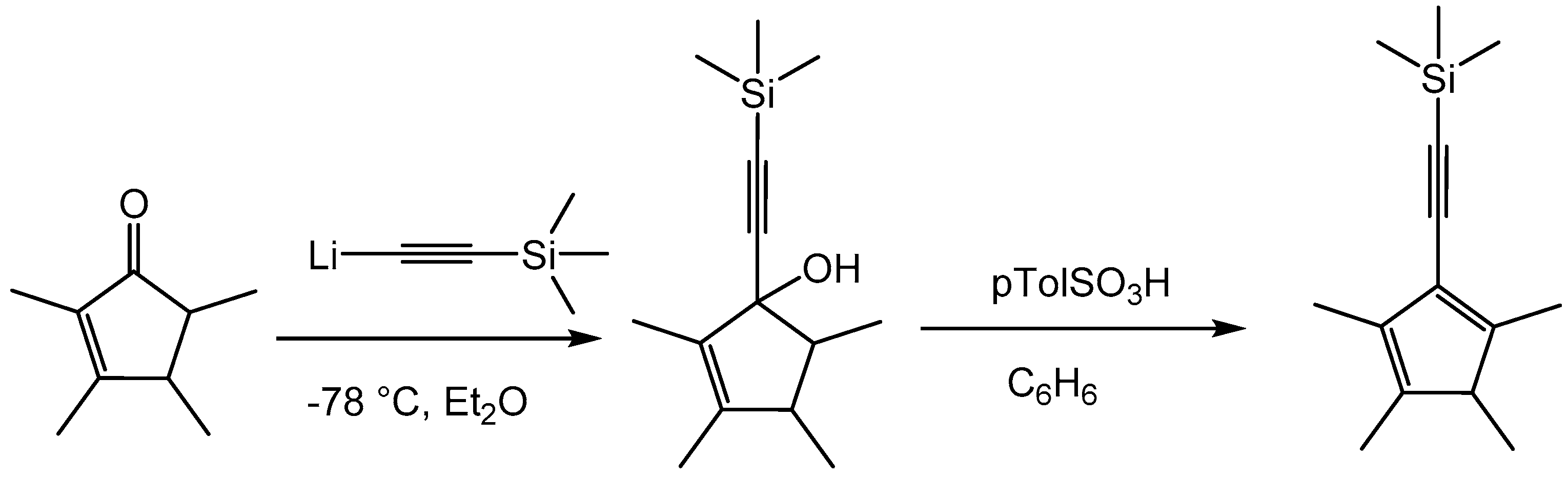
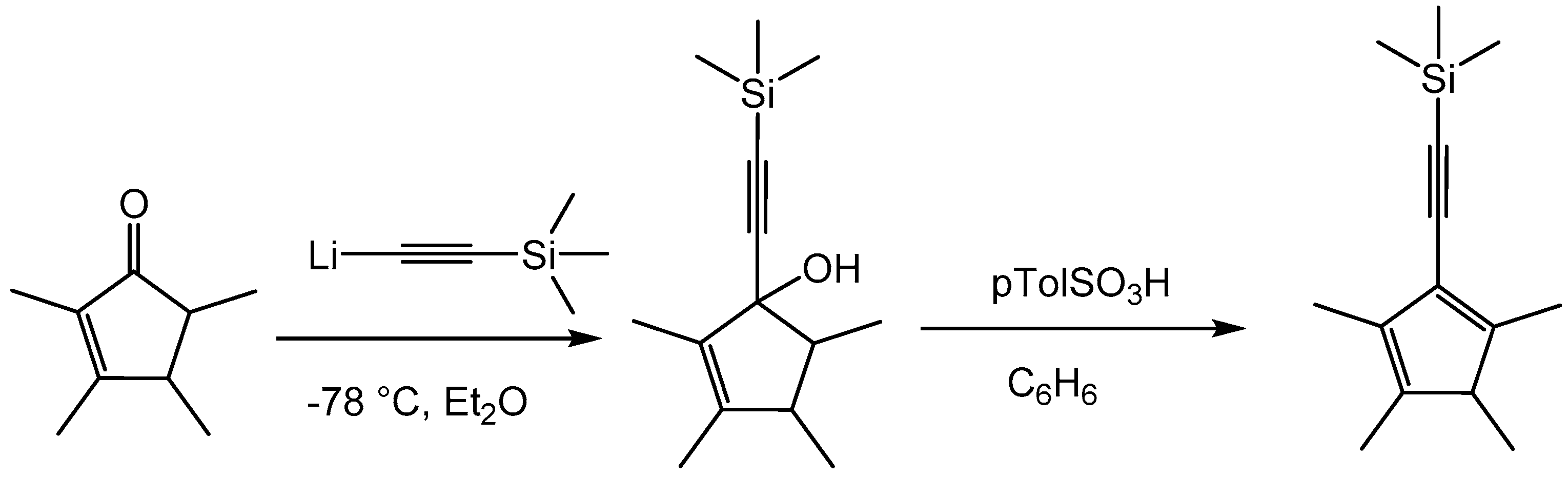
2. Results and Discussion
3. Experimental
3.1. General Procedures
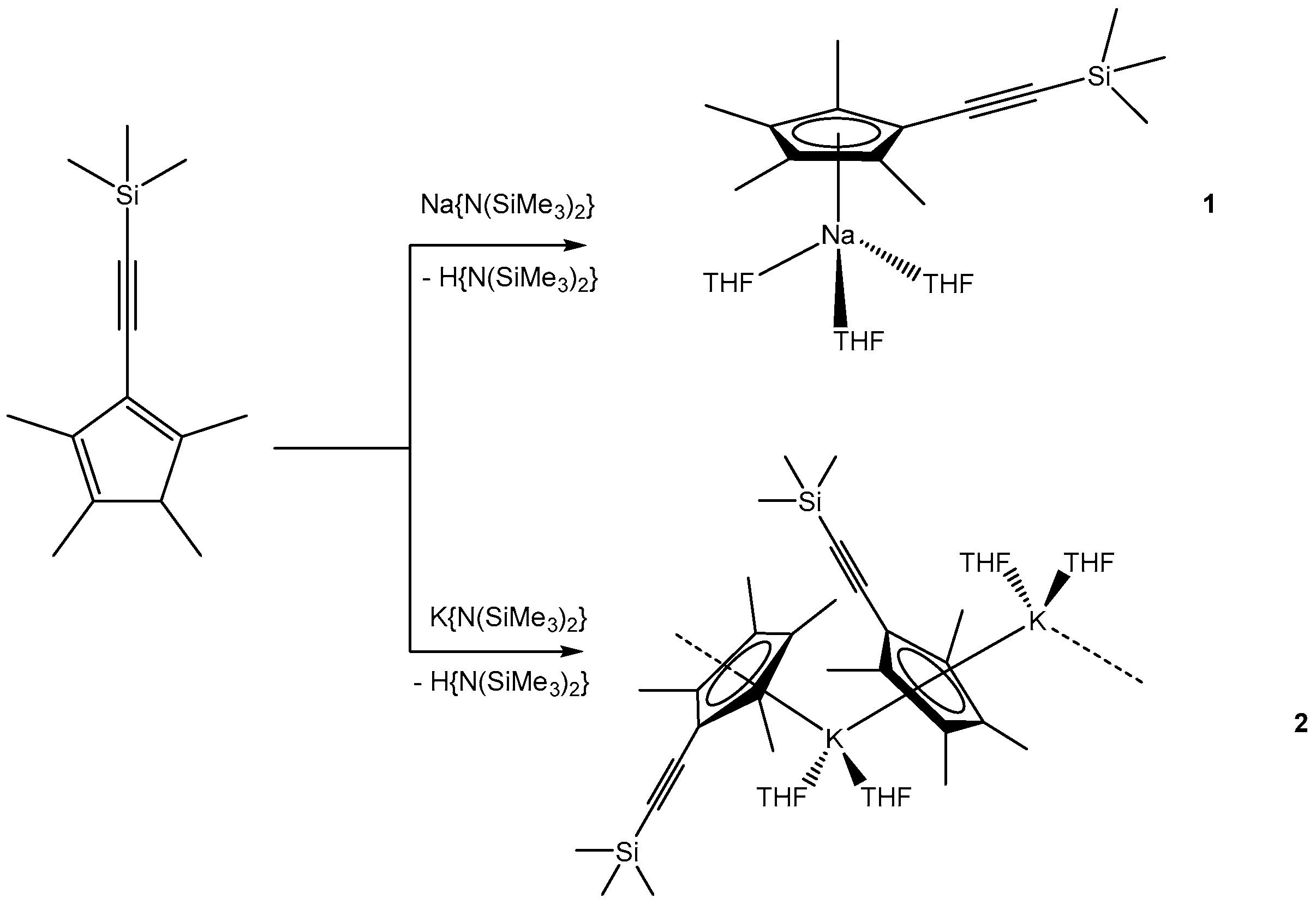
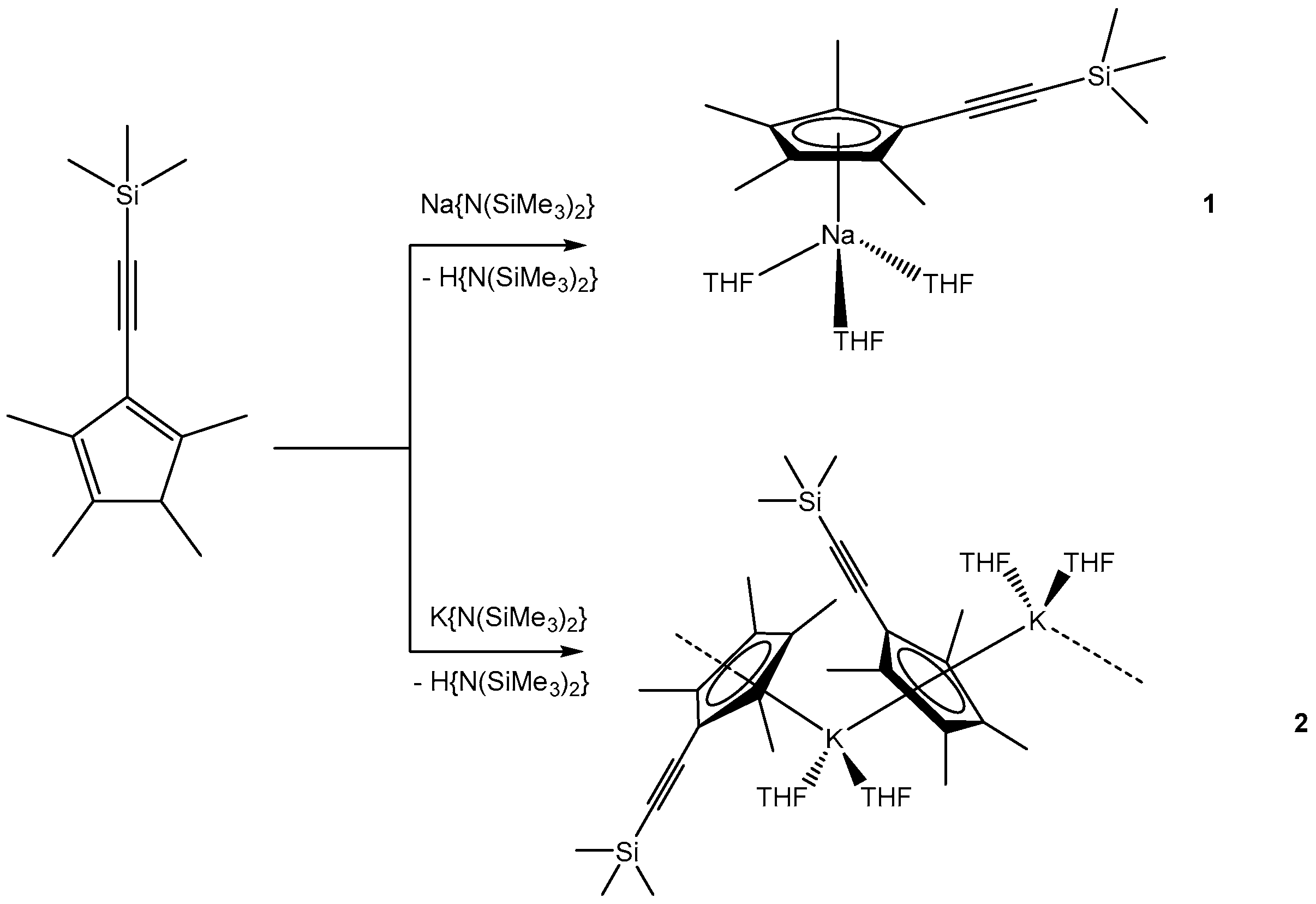
3.1.1. [NaCpMe4(C≡CSiMe3)(THF)3] (1)
3.1.2. [KCpMe4(C≡CSiMe3)(THF)2]n (2)
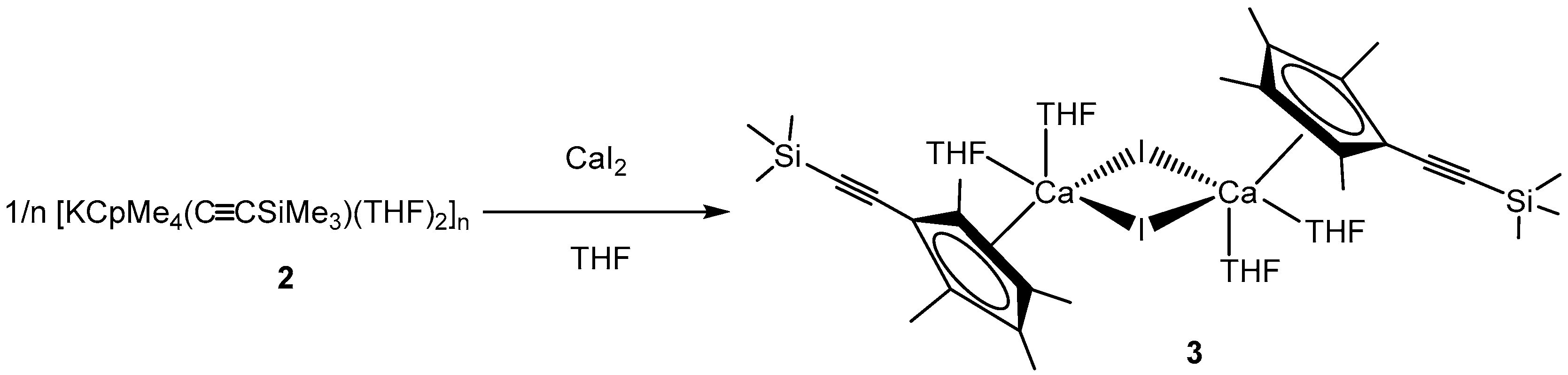
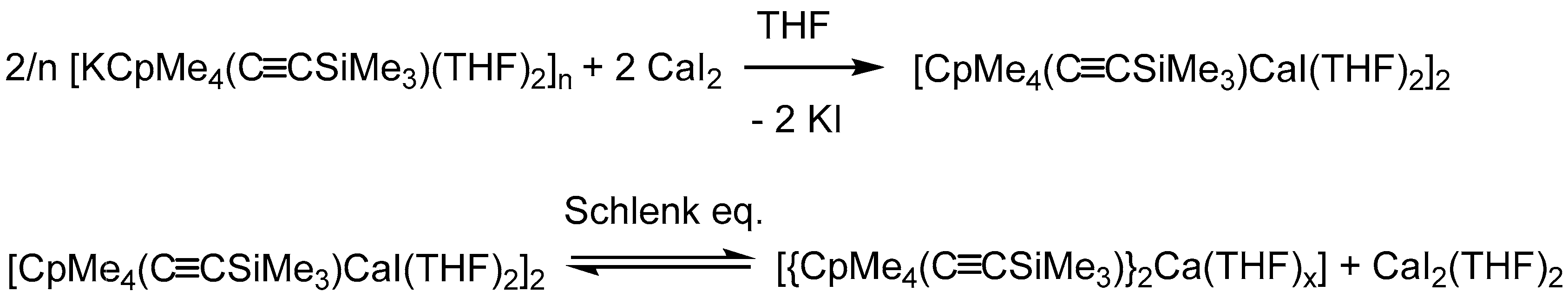
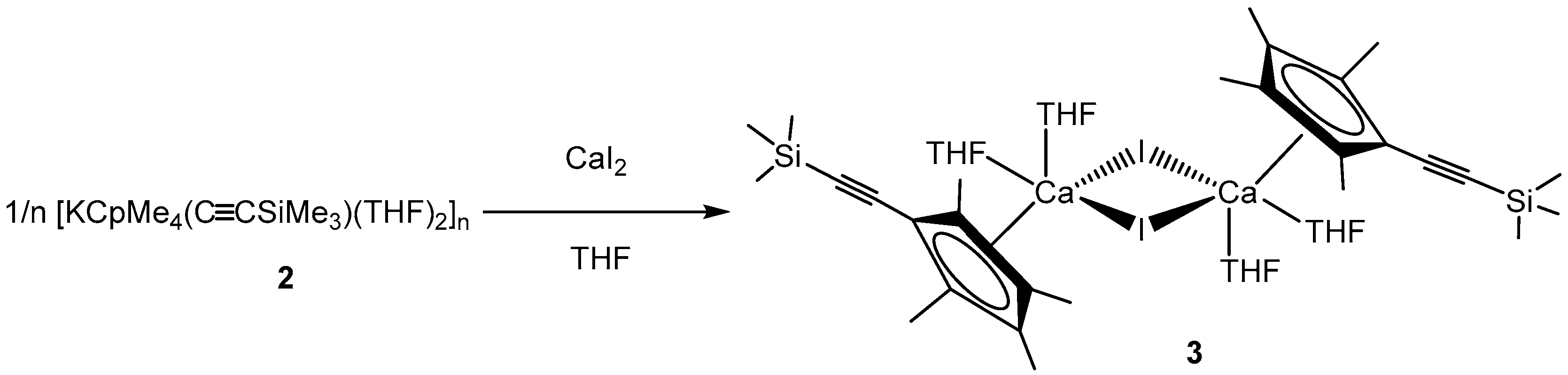
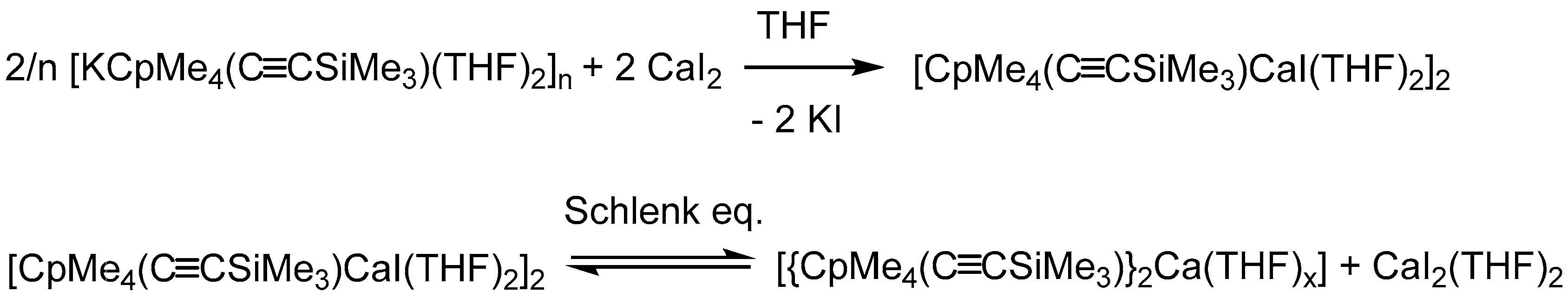
3.1.3. [CpMe4(C≡CSiMe3)CaI(THF)2]2 (3)
3.2. X-ray Crystallographic Studies of 1–3
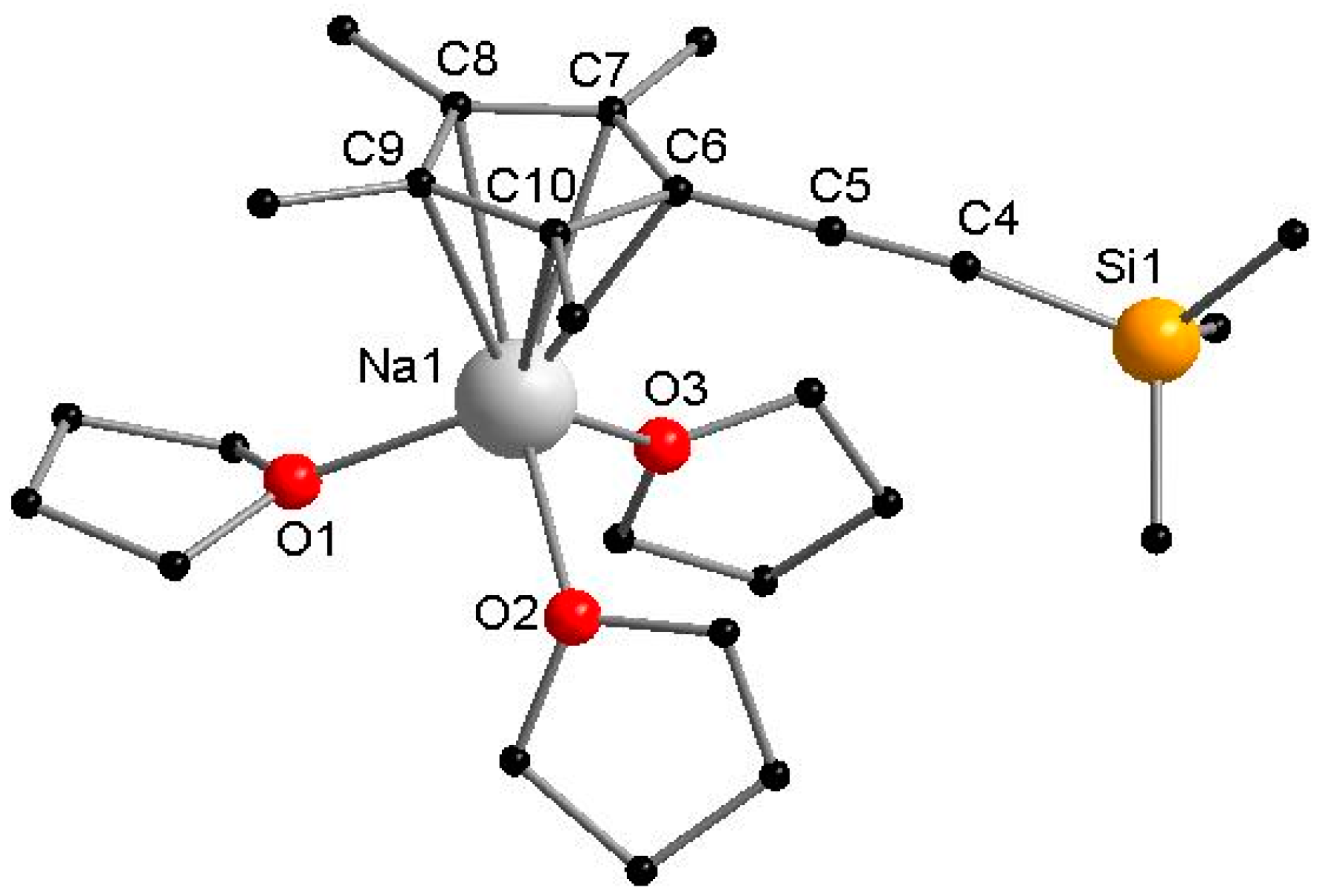
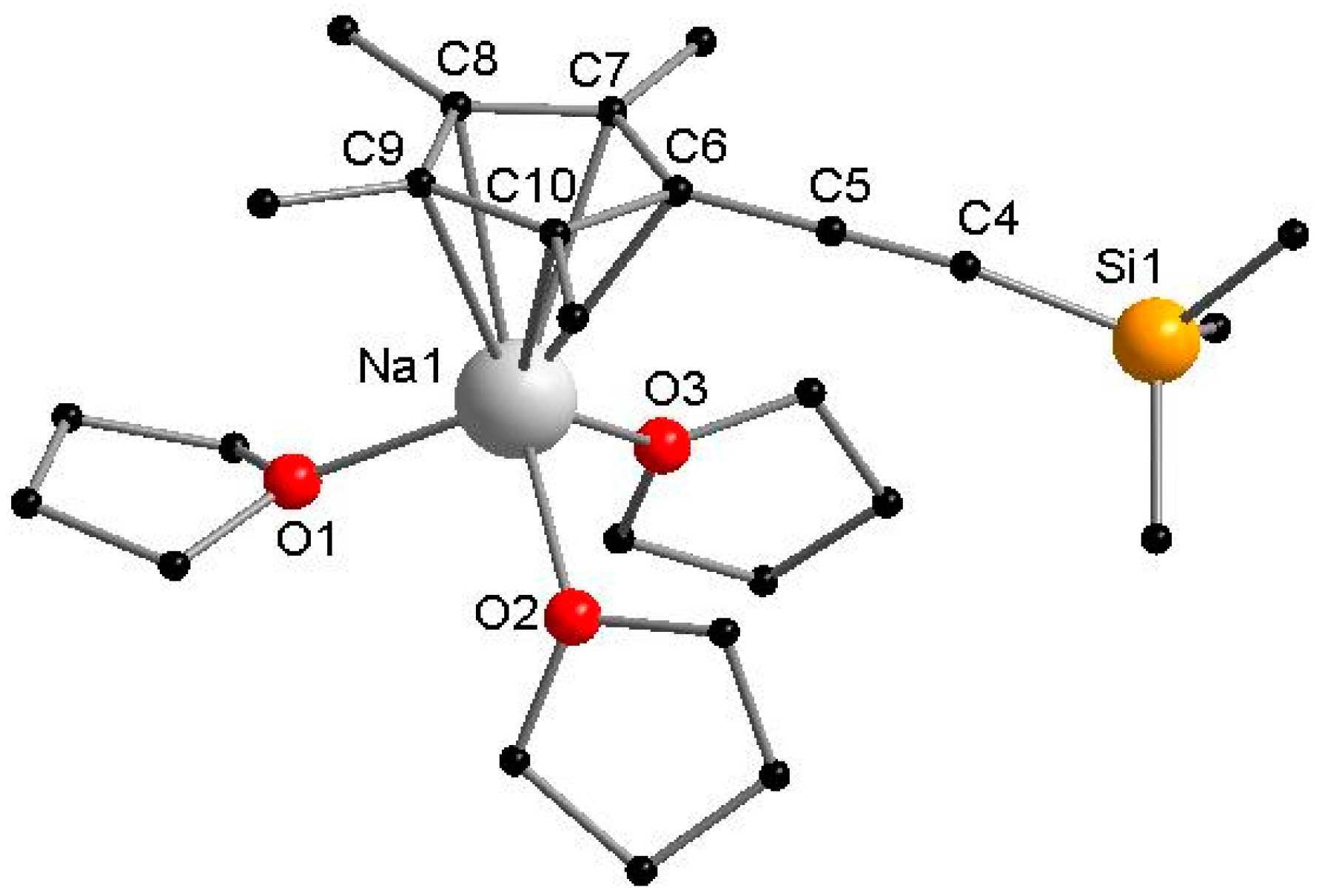
3.2.1. [NaCpMe4(C≡CSiMe3)(THF)3]
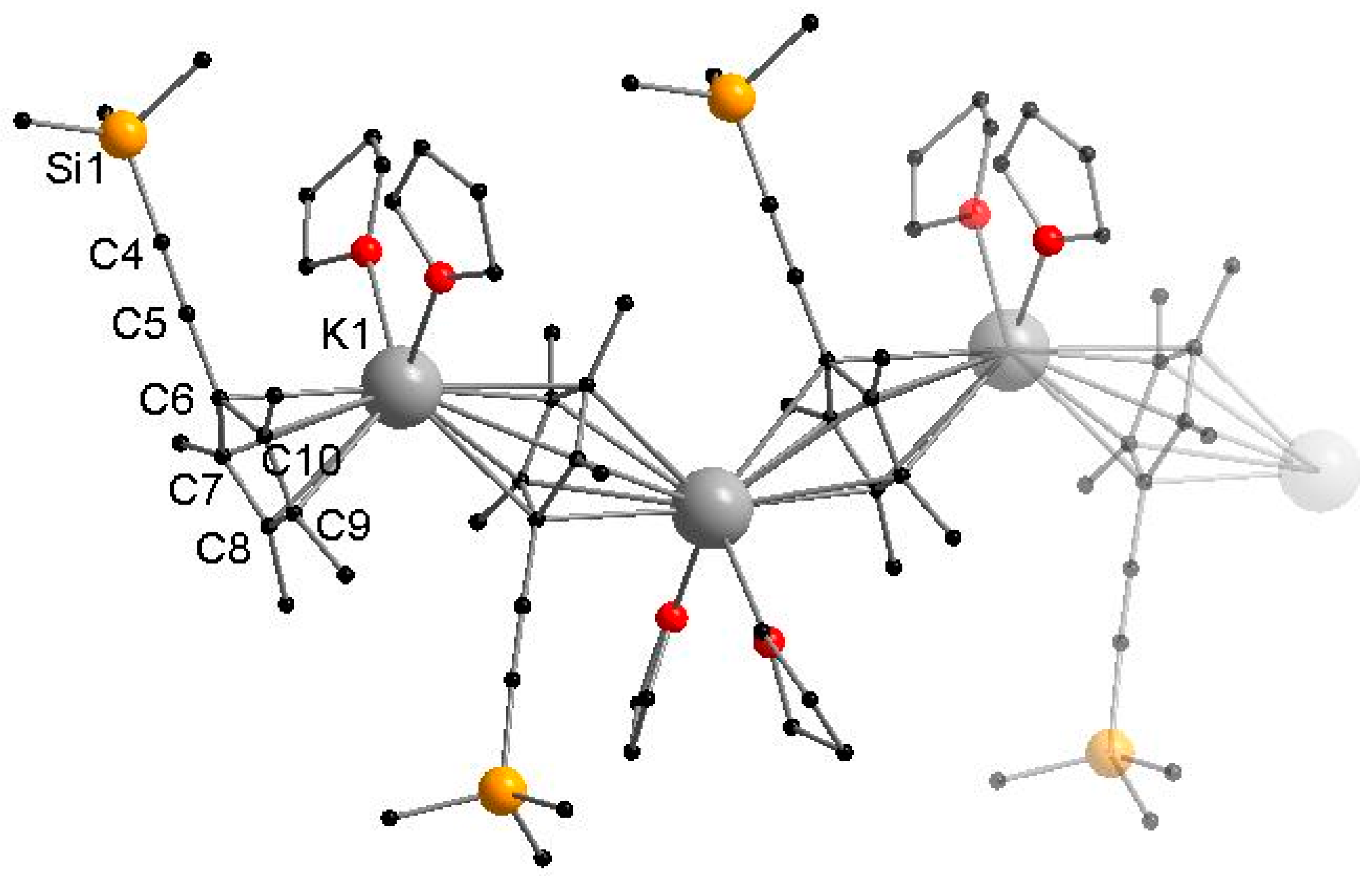
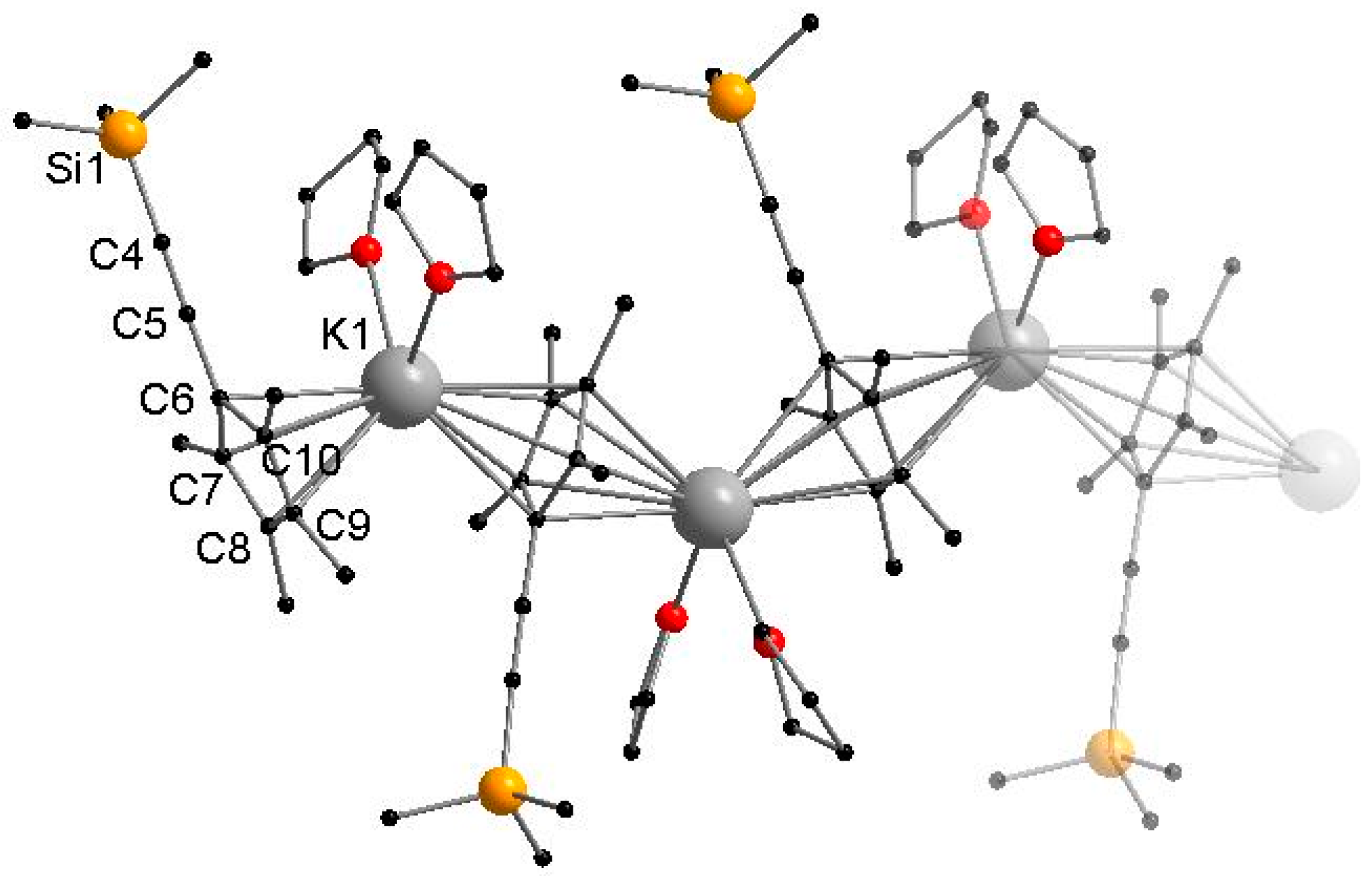
3.2.2. [KCpMe4(C≡CSiMe3)(THF)2]n (2)
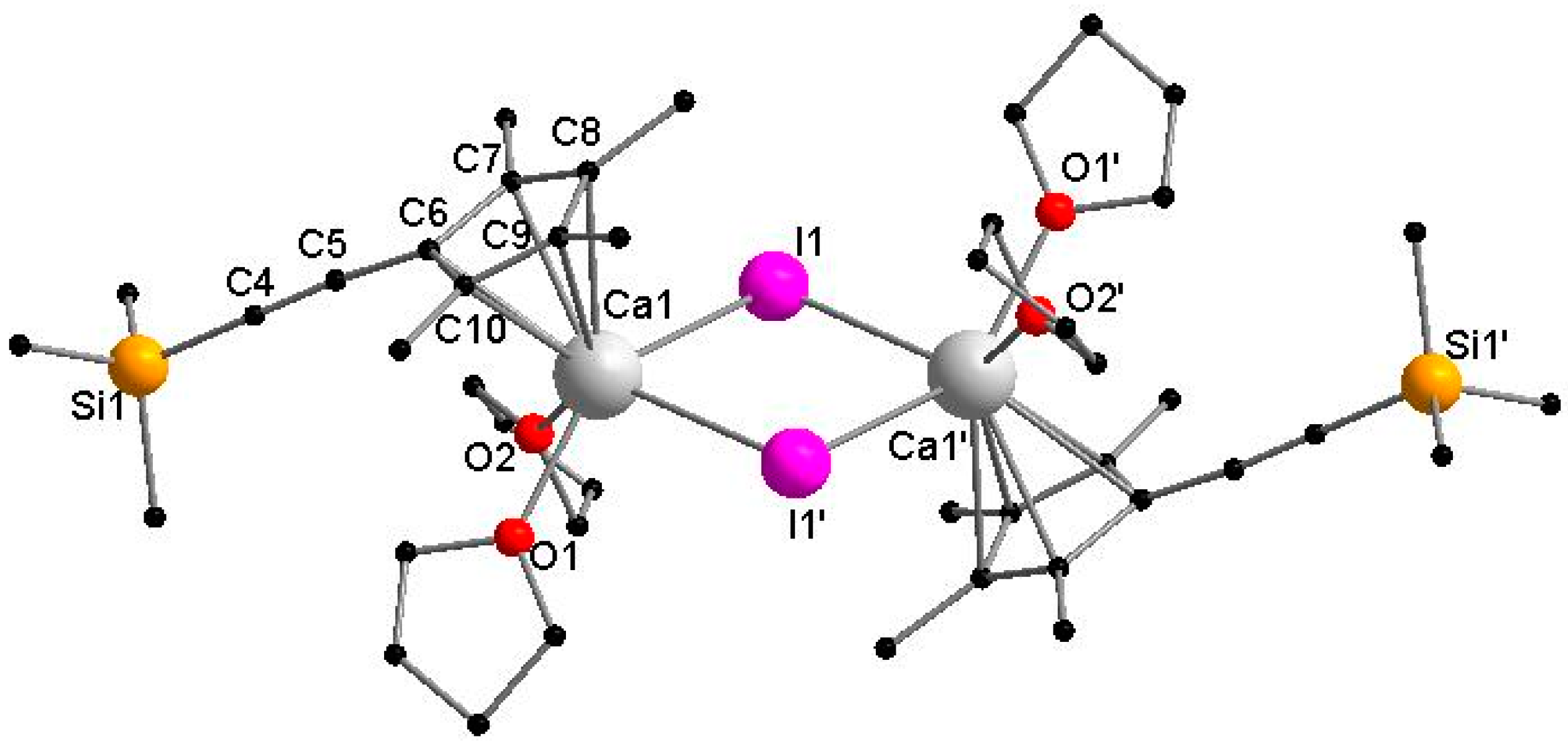
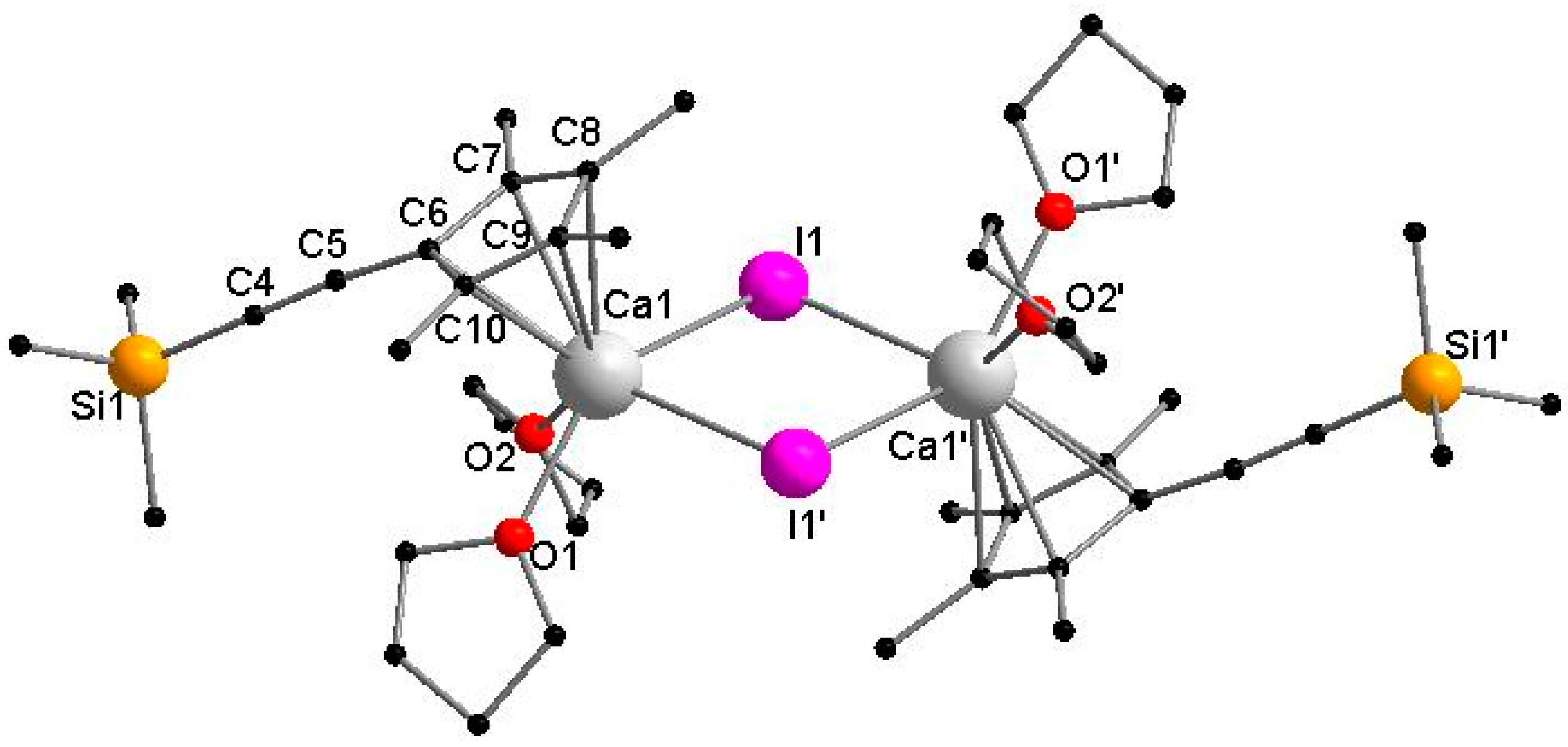
3.2.3. [CaCpMe4(C≡CSiMe3)I(THF)2]2 (3)
4. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Thiele, J. Ueber Abkömmlinge des Cyclopentadiëns. Eur. J. Inorg. Chem. 1901, 34, 68–71. [Google Scholar] [CrossRef]
- Fischer, E.; Jira, R.Z. Di-cyclopentadienyl-kobalt (II). Naturforschung B 1953, 8, 327–328. [Google Scholar]
- Fischer, E.; Hafner, W.; Stahl, H.Z. Über Cyclopentadienyl-metall-carbonyl-wasserstoffe des Chroms, Molybdäns und Wolframs. Anorg. Allg. Chem. 1955, 282, 47–62. [Google Scholar] [CrossRef]
- Ziegler, K.; Froitzheim-Kühlhorn, H.; Hafner, K. Metallorganische Verbindungen XXI: Metallverbindungen des Cyclopentadiens. Chem. Ber. 1956, 89, 434–443. [Google Scholar] [CrossRef]
- Panda, T.K.; Gamer, M.T.; Roesky, P.W. An improved synthesis of sodium and potassium cyclopentadienide. Organometallics 2003, 22, 877–878. [Google Scholar] [CrossRef]
- Borisov, A.; Makhaev, V. The synthesis of potassium and sodium cyclopentadienides by the interaction of cyclopentadiene with alkalis. Organomet. Chem. USSR 1989, 2, 680. [Google Scholar]
- Panda, T.K.; Gamer, M.T.; Roesky, P.W.; Yoo, H.; Berry, D.H. Sodium and Potassium Cyclopentadienide. In Inorganic Syntheses; John Wiley & Sons, Inc.: New Jersey, NJ, USA, 2014; Volume 36. [Google Scholar]
- Kohl, F.X.; Jutzi, P. An improved synthesis of 1,2,3,4,5-pentamethylcyclopentadiene. J. Organomet. Chem. 1983, 243, 119–121. [Google Scholar] [CrossRef]
- DeVries, L. Communications-Preparation of 1,2,3,4,5-Pentamethyl-cyclopentadiene, 1,2,3,4,5,5-Hexamethyl-cyclopentadiene, and 1,2,3,4,5-Pentamethyl-cyclopentadienylcarbinol. J. Org. Chem. 1960, 25, 1838. [Google Scholar] [CrossRef]
- Sawtelle, S.M.; Johnston, R.F.; Cook, C.C. An investigation of electron transfer properties of (Cp)Mn(CO)3 and substituted Cp derivatives. Inorg. Chim. Acta 1994, 221, 85–92. [Google Scholar] [CrossRef]
- Ye, B.; Cramer, N. Chiral cyclopentadienyl ligands as stereocontrolling element in asymmetric C–H functionalization. Science 2012, 338, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.P.; Norton, J.R.; Buccella, D.; Sites, L.A.; Kleinbach, S.S.; Jarem, D.A.; Bocage, K.M.; Nataro, C. Synthesis, Electrochemistry, and Reactivity of Half-Sandwich Ruthenium Complexes Bearing Metallocene-Based Bisphosphines. Organometallics 2009, 28, 3804–3814. [Google Scholar] [CrossRef]
- Powell, C.E.; Humphrey, M.G. Nonlinear optical properties of transition metal acetylides and their derivatives. Coord. Chem. Rev. 2004, 248, 725–756. [Google Scholar] [CrossRef]
- Yam, V.W.W.; Lo, K.K.; Wong, K.M.C. Luminescent polynuclear metal acetylides. J. Organomet. Chem. 1999, 578, 3–30. [Google Scholar] [CrossRef]
- Dias, H.R.; Flores, J.A.; Wu, J.; Kroll, P. Monomeric copper(I), silver(I), and gold(I) alkyne complexes and the coinage metal family group trends. J. Am. Chem. Soc. 2009, 131, 11249–11255. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Dash, C.; Celik, M.A.; Yousufuddin, M.; Frenking, G.; Dias, H.R. Tris(alkyne) and Bis(alkyne) Complexes of Coinage Metals: Synthesis and Characterization of (cyclooctyne)3M+ (M = Cu, Ag) and (cyclooctyne)2Au+ and Coinage Metal (M = Cu, Ag, Au) Family Group Trends. Organometallics 2013, 32, 3135–3144. [Google Scholar] [CrossRef]
- Kabalka, G.W.; Wang, L.; Pagni, R.M. Sonogashira coupling and cyclization reactions on alumina: A route to aryl alkynes, 2-substituted-benzo [b] furans and 2-substituted-indoles. Tetrahedron 2001, 57, 8017–8028. [Google Scholar] [CrossRef]
- Yi, C.; Hua, R. Efficient copper-free PdCl2(PCy3)2-catalyzed sonogashira coupling of aryl chlorides with terminal alkynes. J. Org. Chem. 2006, 71, 2535–2537. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Dai, M.; Chen, J.; Yang, Z. Copper-free Sonogashira coupling reaction with PdCl2 in water under aerobic conditions. J. Org. Chem. 2005, 70, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Astruc, D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC)“click” reaction and its applications. An overview. Coord. Chem. Rev. 2011, 255, 2933–2945. [Google Scholar] [CrossRef]
- Crowley, J.D.; Bandeen, P.H. A multicomponent CuAAC “click” approach to a library of hybrid polydentate 2-pyridyl-1,2,3-triazole ligands: New building blocks for the generation of metallosupramolecular architectures. Dalton Trans. 2010, 39, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, H.; Lin, C.; Liu, Z.; Wang, C.; Zhang, Y. Cobalt-Catalyzed Cyclization of Aliphatic Amides and Terminal Alkynes with Silver-Cocatalyst. J. Am. Chem. Soc. 2015, 137, 12990–12996. [Google Scholar] [CrossRef] [PubMed]
- Pudelski, J.K.; Callstrom, M.R. Structure, reactivity, and electronic properties of [4]ferrocenophanes and [4]ruthenocenophanes prepared via a novel heteroannular cyclization reaction. Organometallics 1994, 13, 3095–3109. [Google Scholar] [CrossRef]
- Yao, X.; Li, C.J. Water-Triggered and Gold(I)-Catalyzed Cascade Addition/Cyclization of Terminal Alkynes with ortho-Alkynylaryl Aldehyde. Org. Lett. 2006, 8, 1953–1955. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, C.; Dinnebier, R.E.; Olbrich, F.; Smaalen, S.V. Disordered crystal structure of pentamethylcyclopentadienylsodium as seen by high-resolution X-ray powder diffraction. Acta Crystallogr. Sect. B Struct. Sci. 2001, 57, 673–679. [Google Scholar] [CrossRef]
- Pidcock, E. Achiral molecules in non-centrosymmetric space groups. Chem. Commun. 2005, 3457–3459. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Brady, J.C.; Fujimoto, C.H.; Giarikos, D.G.; Ziller, J.W. The bent metallocene geometries of potassium polyalkyl cyclopentadienyl THF solvates: Monosolvated [(THF)K(μ-C5Me5)]n, disolvated [(THF)2K(μ-C5Me5)]n and the tethered olefin complex [(THF)K(μ-C5Me4SiMe2CH2CH=CH2)]n. J. Organomet. Chem. 2002, 649, 252–257. [Google Scholar] [CrossRef]
- McCormick, M.J.; Sockwell, S.C.; Davies, C.E.; Hanusa, T.P.; Huffman, J.C. Synthesis of a monopentamethylcyclopentadienyl halide complex of calcium. The X-ray crystal structure of [(Me5C5)Ca(μ-I)(THF)2]2. Organometallics 1989, 8, 2044–2049. [Google Scholar] [CrossRef]
- McCormick, M.; Williams, R.; Levine, L.; Hanusa, T. Solution synthesis of calcium, strontium and barium metallocenes. Polyhedron 1988, 7, 725–730. [Google Scholar] [CrossRef]
- Westerhausen, M.Z. Recent Developments in the Organic Chemistry of Calcium–An Element with Unlimited Possibilities in Organometallic Chemistry? Anorg. Allg. Chem. 2009, 635, 13–32. [Google Scholar] [CrossRef]
- Evans, W.J.; Meadows, J.H.; Hanusa, T.P. Organolanthanide and organoyttrium hydride chemistry. 6. Direct synthesis and proton NMR spectral analysis of the trimetallic yttrium and yttrium–zirconium tetrahydride complexes, {[(C5H5)2YH]3H}{Li(THF)4} and {[(CH3C5H4)2YH]2[(CH3C5H4)2ZrH]H}. J. Am. Chem. Soc. 1984, 106, 4454–4460. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seifert, T.; Roesky, P.W. Alkali and Alkaline Earth Metal Complexes Ligated by an Ethynyl Substituted Cyclopentadienyl Ligand. Inorganics 2017, 5, 28. https://doi.org/10.3390/inorganics5020028
Seifert T, Roesky PW. Alkali and Alkaline Earth Metal Complexes Ligated by an Ethynyl Substituted Cyclopentadienyl Ligand. Inorganics. 2017; 5(2):28. https://doi.org/10.3390/inorganics5020028
Chicago/Turabian StyleSeifert, Tim, and Peter W. Roesky. 2017. "Alkali and Alkaline Earth Metal Complexes Ligated by an Ethynyl Substituted Cyclopentadienyl Ligand" Inorganics 5, no. 2: 28. https://doi.org/10.3390/inorganics5020028
APA StyleSeifert, T., & Roesky, P. W. (2017). Alkali and Alkaline Earth Metal Complexes Ligated by an Ethynyl Substituted Cyclopentadienyl Ligand. Inorganics, 5(2), 28. https://doi.org/10.3390/inorganics5020028