1. Introduction
With the ability to adopt numerous coordination modes, flexible
N,
N′-bis(aryl)formamidinates (and by extension aryl-functionalised amidinates) have earned a special place in coordination chemistry [
1,
2,
3,
4,
5]. Not only does the anionic NCHN bite provide a variety of different nitrogen-based coordination modes (e.g., monodentate κ(
N), bidentate κ(
N,
N′), or various bridging modes e.g.,
μ-1κ(
N):2κ(
N′),
μ-1κ(
N,
N′):2κ(
N,
N′) to list a few) [
6,
7], the nitrogen-bound aromatic substituents can also provide additional coordination modes. The potential to form metal–arene interactions, such as η
6 coordination, has been largely observed in group one chemistry [
8,
9,
10,
11,
12,
13], though some examples are known in
f-block chemistry [
14]. In almost all examples of this aromatic coordination, the phenyl rings contained alkyl-substituents in either the 2,6 positions (e.g.,
iPr, Et, Me), or in the 2,4,6 positions (e.g., Me). This is likely due to a combination of steric pressure, which starves the metal centre from coordination of additional donors, and increased electron donation from the aromatic ring caused by the alkyl substituents. Another means to engage the aromatic component in coordination is through the addition of donor functionalities (e.g., OMe, F), especially in the
ortho-positions, thereby transforming the formamidinate ligand into a tri- [
15,
16,
17], or tetra-dentate (e.g.,
N,
N′,
X or
N,
N′,
X,
X′) [
17], chelate, with examples across a variety of different metal classes [
18]. For
s-block chemistry however, the use of such ligands has been restricted to very few examples, namely the use of
N,
N′-bis(2-fluorophenyl)formamidine (FForm) [
19].
Nearly 15 years ago, FForm was complexed to the group one metals Li, Na, and K [
19]. Akin to the transition metal complexes of Cotton and co-workers [
20], the presence of the fluorine atom on the
ortho-position of the aromatic rings permitted an additional coordinating site. This further led to partial, or complete, exclusion of bound donor molecules (e.g., Et
2O, thf), by the formation of either binuclear, or for potassium, polymeric constructs (e.g., [Na(FForm)(Et
2O)]
2 or [K(FForm)]
∞) [
19]. This contrasts the group one complexes of the non-fluorinated
N,
N′-di(aryl)formamidinate ligands [
21,
22,
23], which readily retain coordinating solvent. Since then, we have expanded the use of fluorinated formamidinate ligands to
f-block chemistry [
9,
16,
17,
24,
25,
26,
27,
28], in a variety of different contexts [
5]. One of the fluorinated formamidinate ligands used was
N,
N′-bis(2,6-difluorophenyl)formamidinate (DFForm) in both trivalent [
16,
17], and divalent [
16] rare-earth complexes. Despite the presence of the additional fluorine atoms, the observation of any M–F interaction was rare, typically only occurred in unsolvated species, and interactions were displaced on coordination of donor solvents [
16,
17]. It is likely that the smaller ionic radii of the trivalent rare-earths, compared with the larger potassium ion [
29], create a significant strain in the NCHN bite of the DFForm ligand when it coordinates the fluorine atoms, and therefore donor solvent coordination is preferred. Although DFForm has been used in some transition metal complexes, it has no precedent in
s-block chemistry. We hypothesised that the additional two fluorine atoms over FForm could engage in further coordination chemistry, generating different coordination modes from FForm, and quite spectacular results have been obtained by way of new formamidinate binding modes.
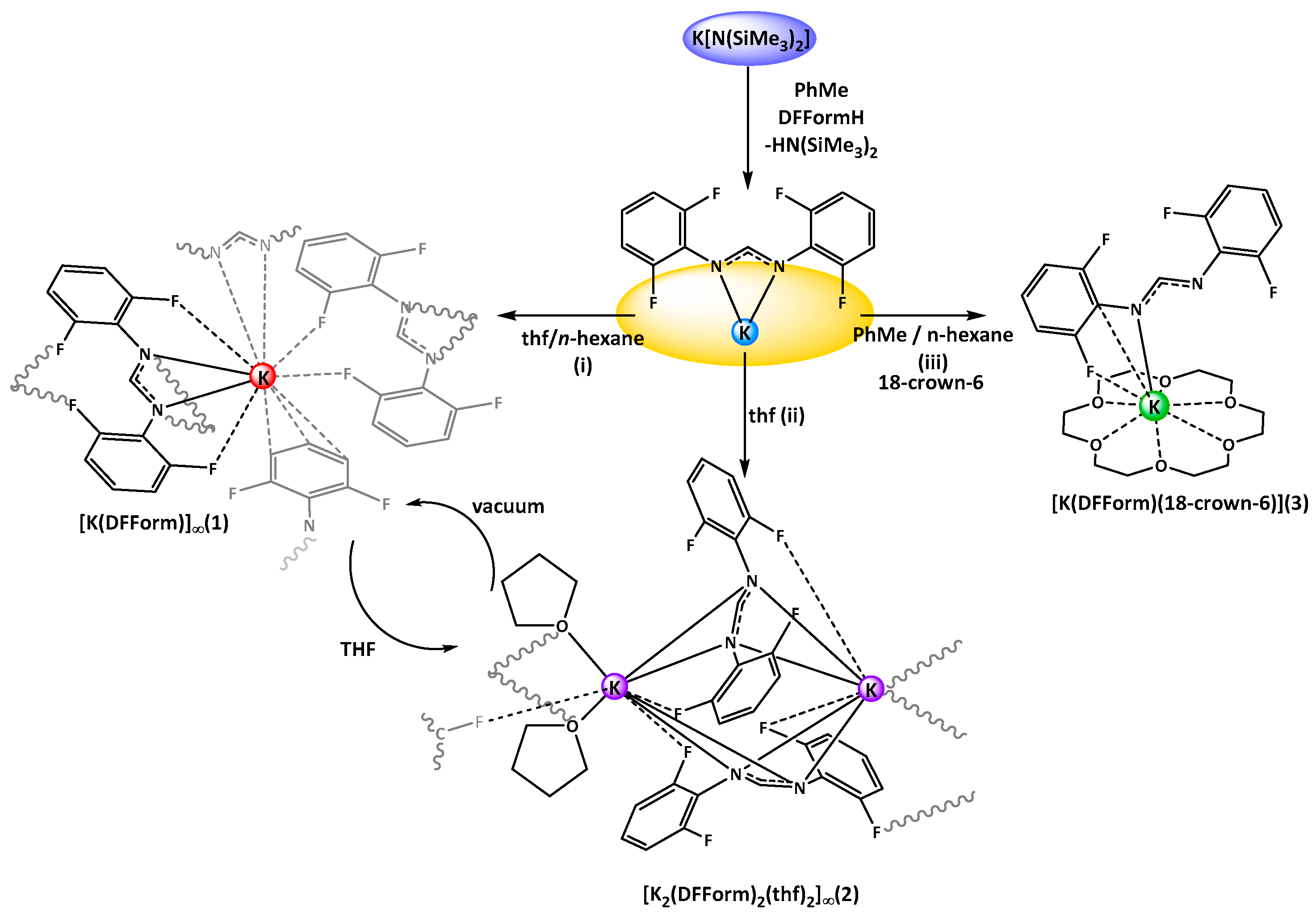
2. Results and Discussion
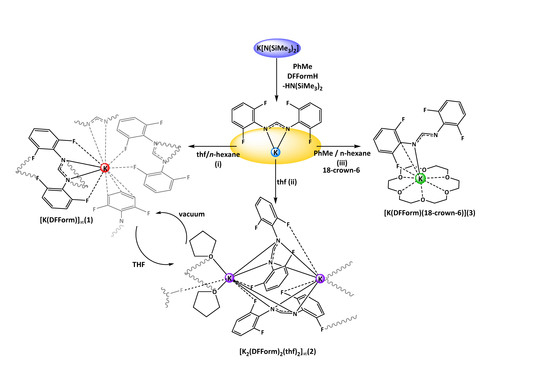
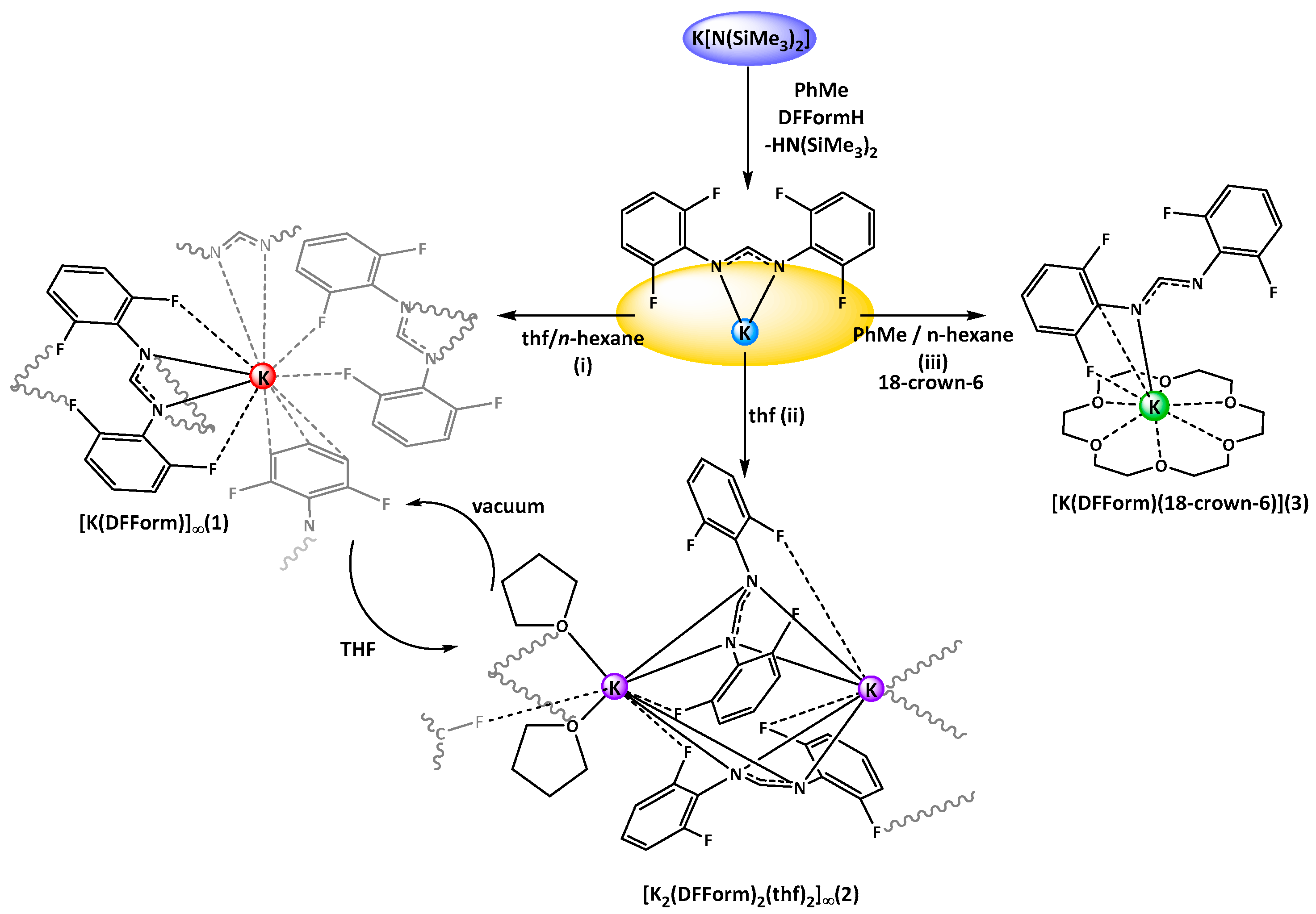
Treatment of K[N(SiMe
3)
2] with DFFormH in toluene resulted in the formation of a colourless, poorly soluble white powder. Upon dissolution in thf, concentration, and layering with
n-hexane, white crystals of targeted [K(DFForm)]
∞ (
1,
Scheme 1i) were obtained. The structure of
1 was determined by X-ray crystallography, revealing that
1 is a two-dimensional polymer. The binding of the DFForm ligand in
1 is complex, and is discussed starting from the asymmetric unit (ASU), and then extending in both dimensions of the polymeric network.
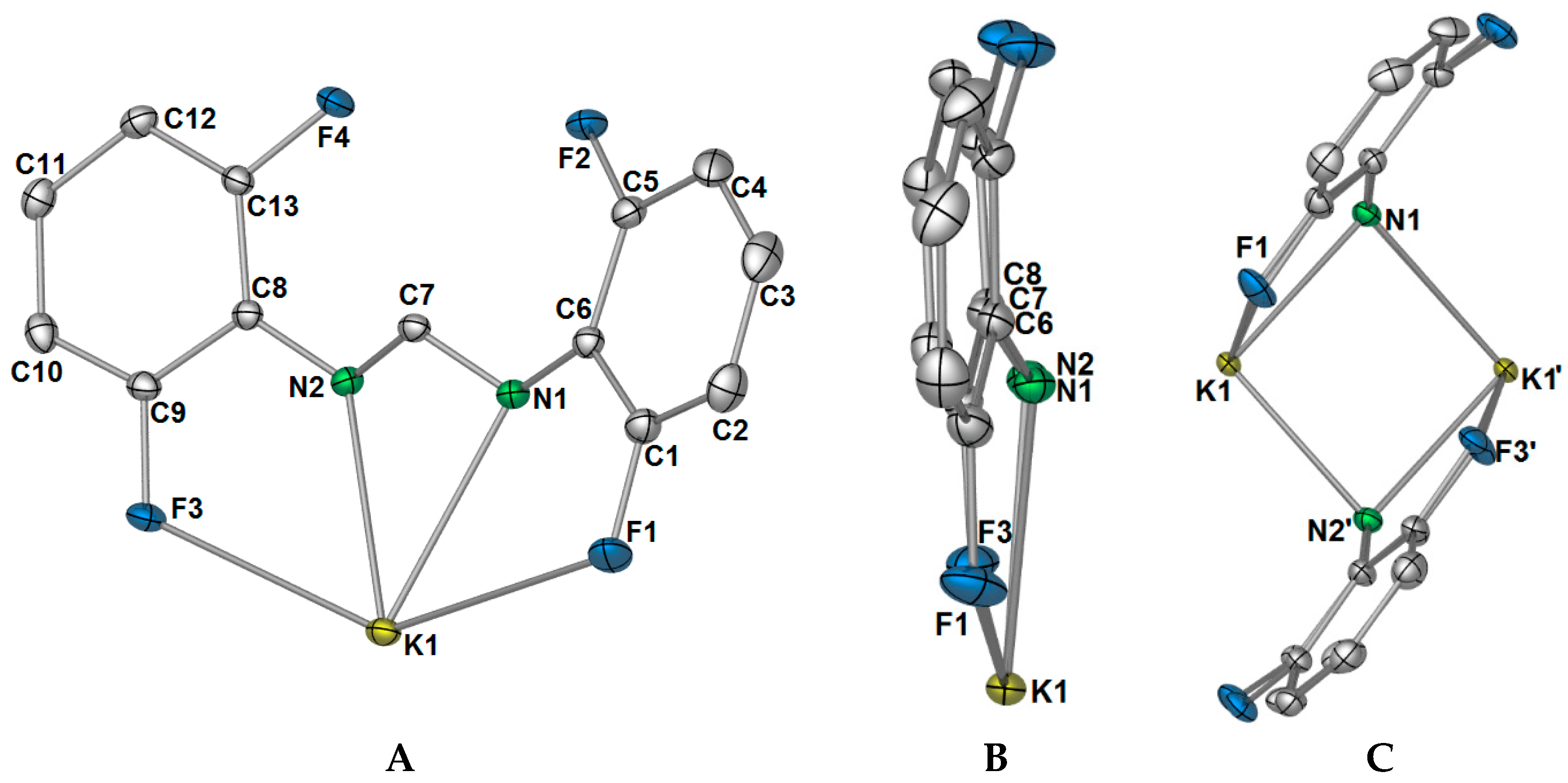
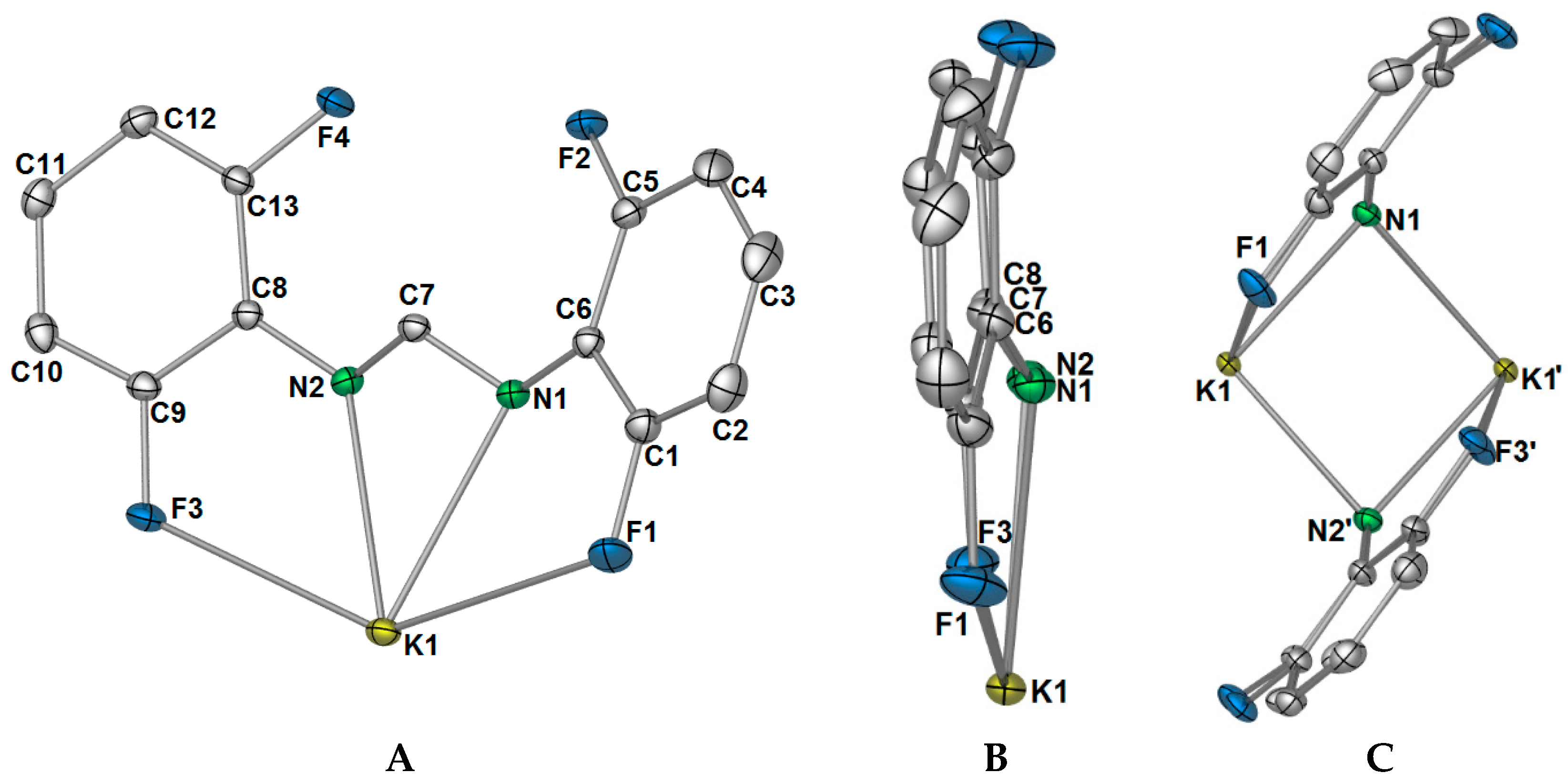
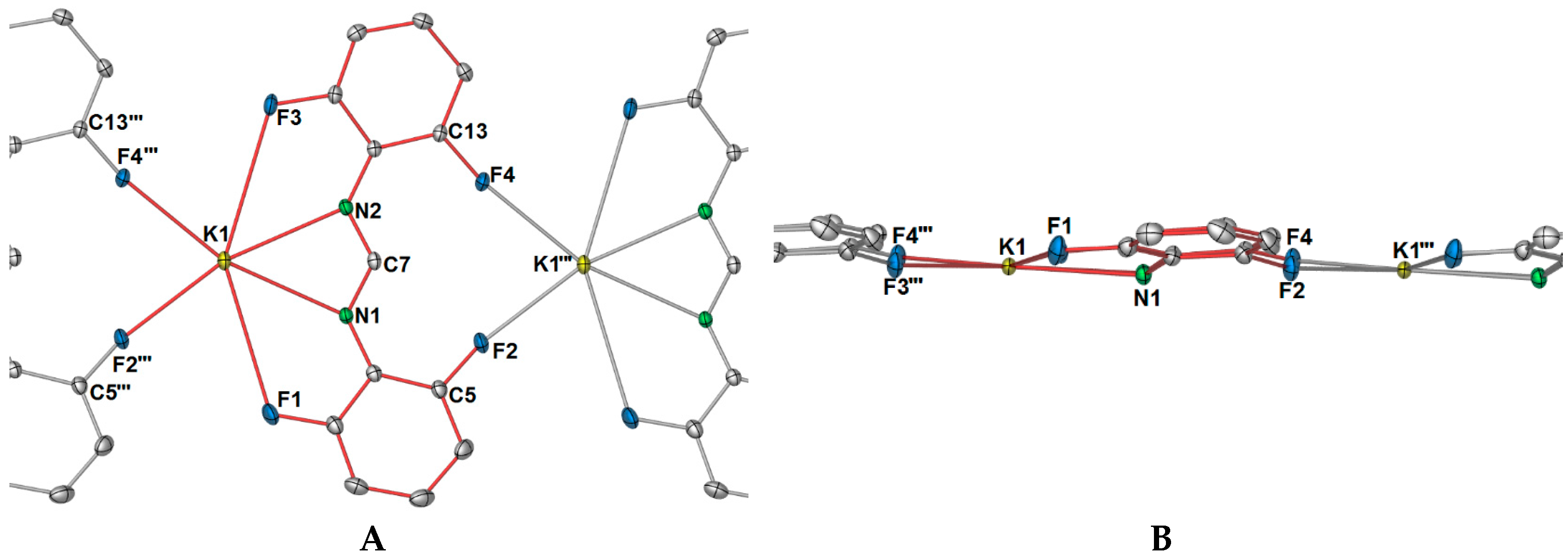
Complex
1 crystallised in the triclinic space group
P-1, with only one potassium ion and one DFForm ligand in the ASU (
Figure 1A). The DFForm ligand of the ASU is bound (
F,
N,
N′,
F′) to the ASU potassium ion. This tetradentate binding of the DFForm ligand contrasts that of the FForm ligand in [K(FForm)]
∞ [
19], where the ASU contains one FForm ligand bound η
4(
N,(Ar-
C6,5),
F) to potassium. As the K ion is bound by the DFForm NCHN bite in an almost symmetrical manner, and does not favour one nitrogen donor (as observed in [K(FForm)], the K···F–C bonding is weak, and thus the C–F bonds in
1 (of either C1/F1 or C9/F3,
Figure 1) are almost unchanged from those of DFFormH (C–F: 1.3596(17)–1.3625(18)) [
30]. By contrast, the asymmetrical NCHN binding of FForm to K in [K(FForm)]
∞ (along with the coordination across the aromatic component), brings the fluorine atom into a closer proximity to the potassium atom (K–F: 3.029(4)), and weakens the C–F bond (C–F: 1.377(6) Å) [
19]. Another example of such tetradentate (
F,
N,
N′,
F′) DFForm coordination was observed in the homoleptic cerium DFForm complex, [Ce(DFForm)
3] [
17], where one of three DFForm ligands is tetradentate, with the other two being tri-dentate (
F,
N,
N′). In this example, all Ce···F–C interactions were identical at 2.92 Å (range: 2.9187(13)–2.9213(13)), and consequentially each C–F bond was also strained to a similar degree (range: 1.374(1)–1.376(1) Å). However, considering that a ten-coordinate cerium(III) is smaller than a nine-coordinate potassium (difference in ionic radii: −0.3 Å) [
29], the tetradentate DFForm ligand for the cerium complex had to bend the aryl-rings towards the cerium ion to bring the fluorine atoms into proximity, causing a strain on the C
ipso–N–CH angle (range: 126.4(2)°–128.3(2)°). However, due to the larger ionic radii of potassium, this phenomenon is not observed in
1 (range: 120.39(9)°–120.75(9)°).
The differences in coordination between the DFForm and FForm ligands to potassium becomes considerably more apparent with expansion of the coordination mode of the ligands through bridging. Initial extension of the coordination of the DFForm ligand in
1 shows that the nitrogen atoms are further bridging to an adjacent potassium ion in a
μ-(
N,
N′):(
N,
N′) manner (
Figure 1C, also
Figure 2A). Such formamidinate bridging is known for other
s- and
f-block complexes [
16,
22,
31]. This bridging is mirrored by a symmetry equivalent DFForm ligand, generating a potassium nitrogen based cube of volume: 6.78 Å
3 (
Figure 1C). In stark contrast, the FForm system shows a twisted
μ-(
N,
CipsoCortho,
F):(
N,
N′,
F′) FForm binding, where the NCHN bite is shared asymmetrically across two anent potassium atoms. It should be further noted that the aromatic group, nitrogen atoms, and backbone/ipso carbon atoms of DFForm are not flat and that the DFForm ligand is tilted (
Figure 1B). The two nitrogen atoms coordinate to potassium in an almost symmetrical manner, but C7 is puckered away from the nitrogen atoms (K1–N1/2(cent)–C7: 141.10(8)°), so it is almost in line with the two
ipso carbon atoms of the phenyl rings (C6–C7–C8: 177.26(6)°, c.f. (K(FForm): 168.9(3)°). This puckered nature of the DFForm ligand is typical of other formamidinate complexes which bridge in a
μ-(
N,
N′):(
N,
N′) fashion (e.g., [K(
p-TolForm)(dme)]
∞ (K1–N1/2(cent)–C″7″: 144.8(3)°,
p-TolForm =
N,
N′-bis(4-methylphenyl)formamidinate) [
32].
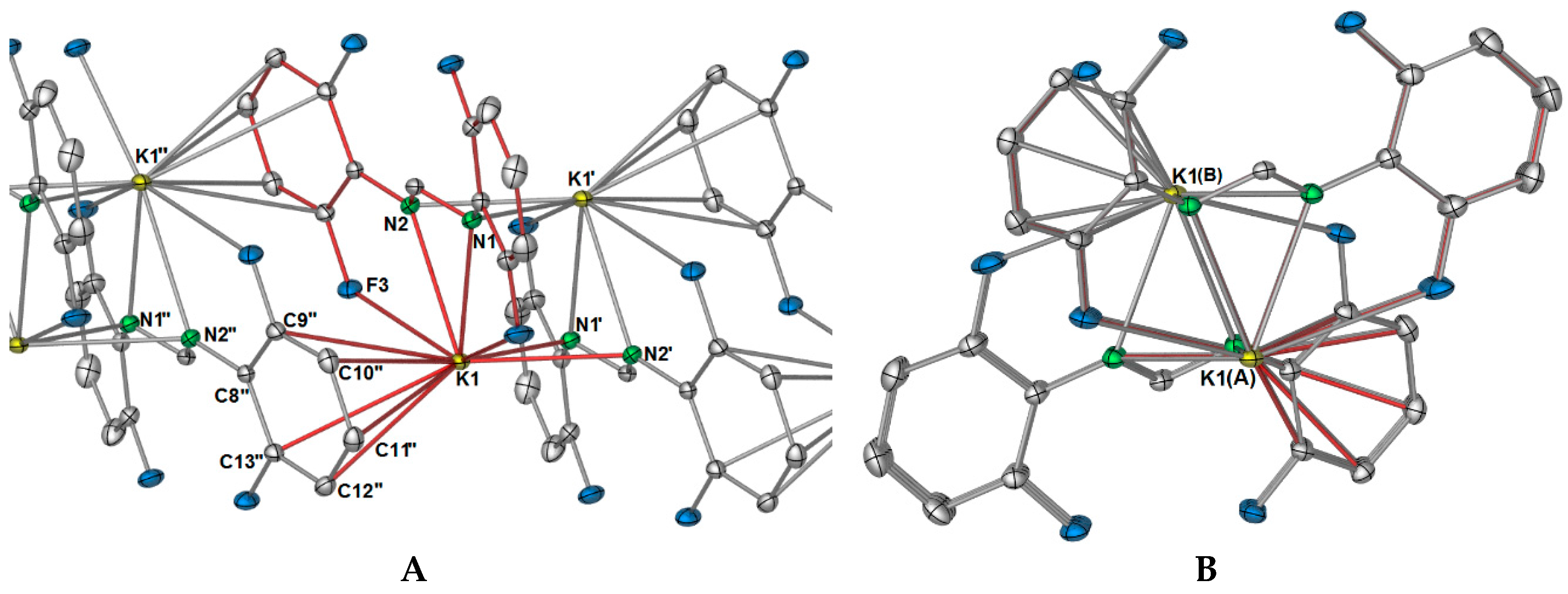
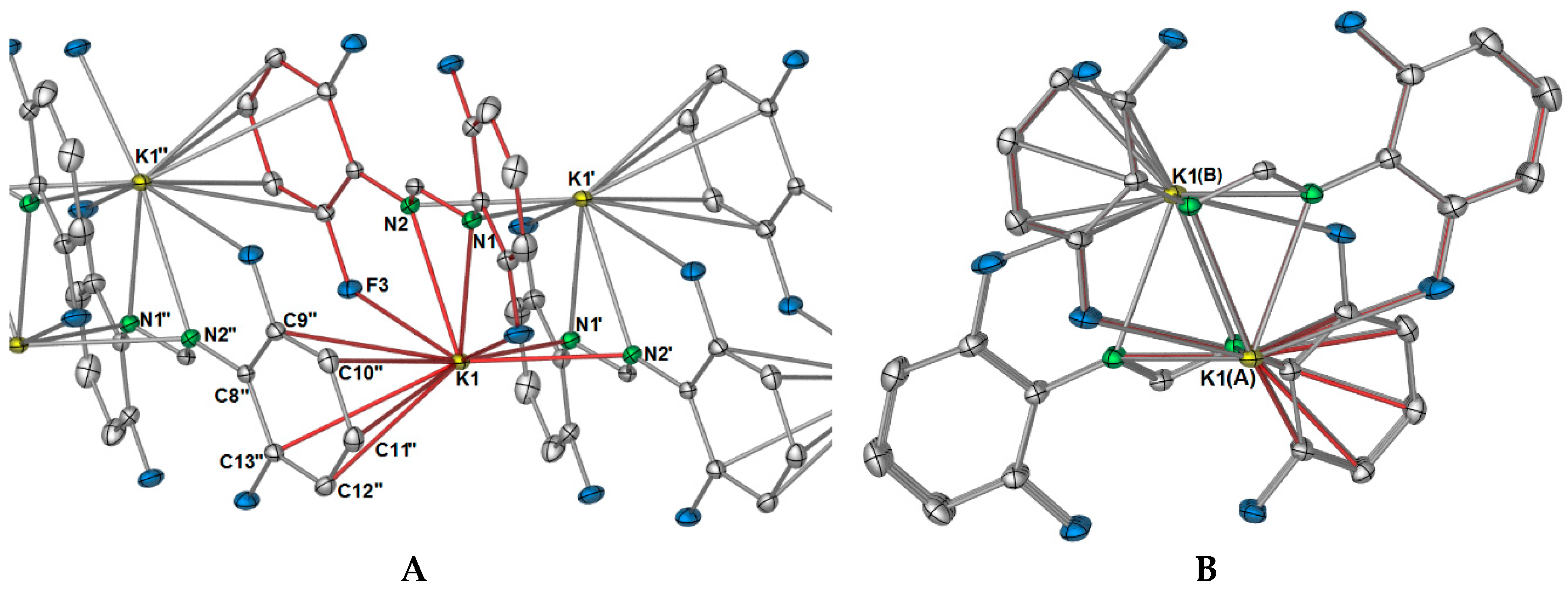
The polymeric network of
1 is complicated. One might expect that, as the DFForm ligand bridges in a
μ-(
N,
N′):(
N,
N′) manner between potassium ions, and that this is the repeating dinuclear unit (e.g., [K
2(
μ-(
N,
N′):(
N,
N′)-DFForm)
2]
∞), but this is not the case. Instead, one dimension of the polymer is generated through aromatic interactions of one 2,6-difluorophenyl group (
Figure 2A), where the aromatic ring of N2 binds to K1′, and the aromatic ring (but without the
ipso carbon) of N2′ coordinates to K1, both in a η
5(C2,3,4,5,6) manner. Thus, this direction of the polymeric network has an “A, B” alternating potassium ion arrangement where A = K and B = K′ and K″ (
Figure 2). For the FForm system, the one and only dimension of the polymeric network is generated by additional nitrogen based bonding to two other potassium ions, namely through one (
N,
F) interaction, and one (
N′,
Cispo′) interaction, making the overall coordination of each FForm ligand shared across four potassium ions as
μ4-(
N,
CipsoCortho,
F):(
N,
N′,
F′):(
N,
F):(
N′,
Cispo′). The DFForm ligand is also further bridging to a fourth symmetry equivalent potassium ion, and this binding is completely different from that in the FForm system.
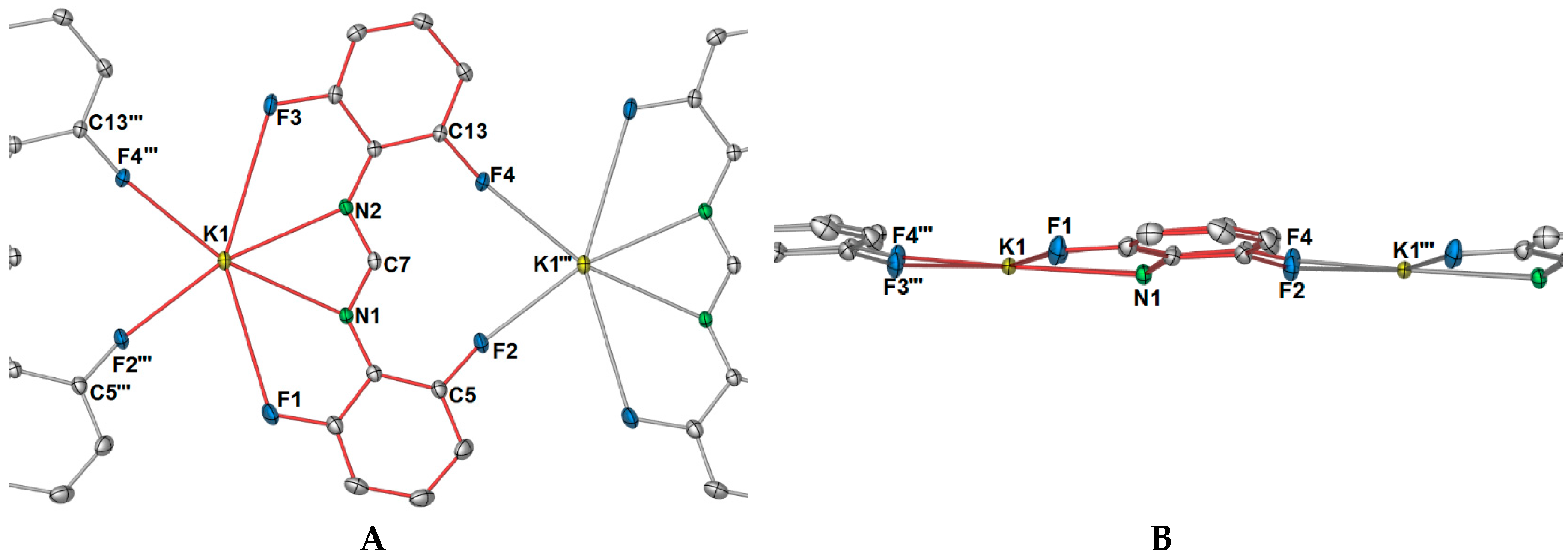
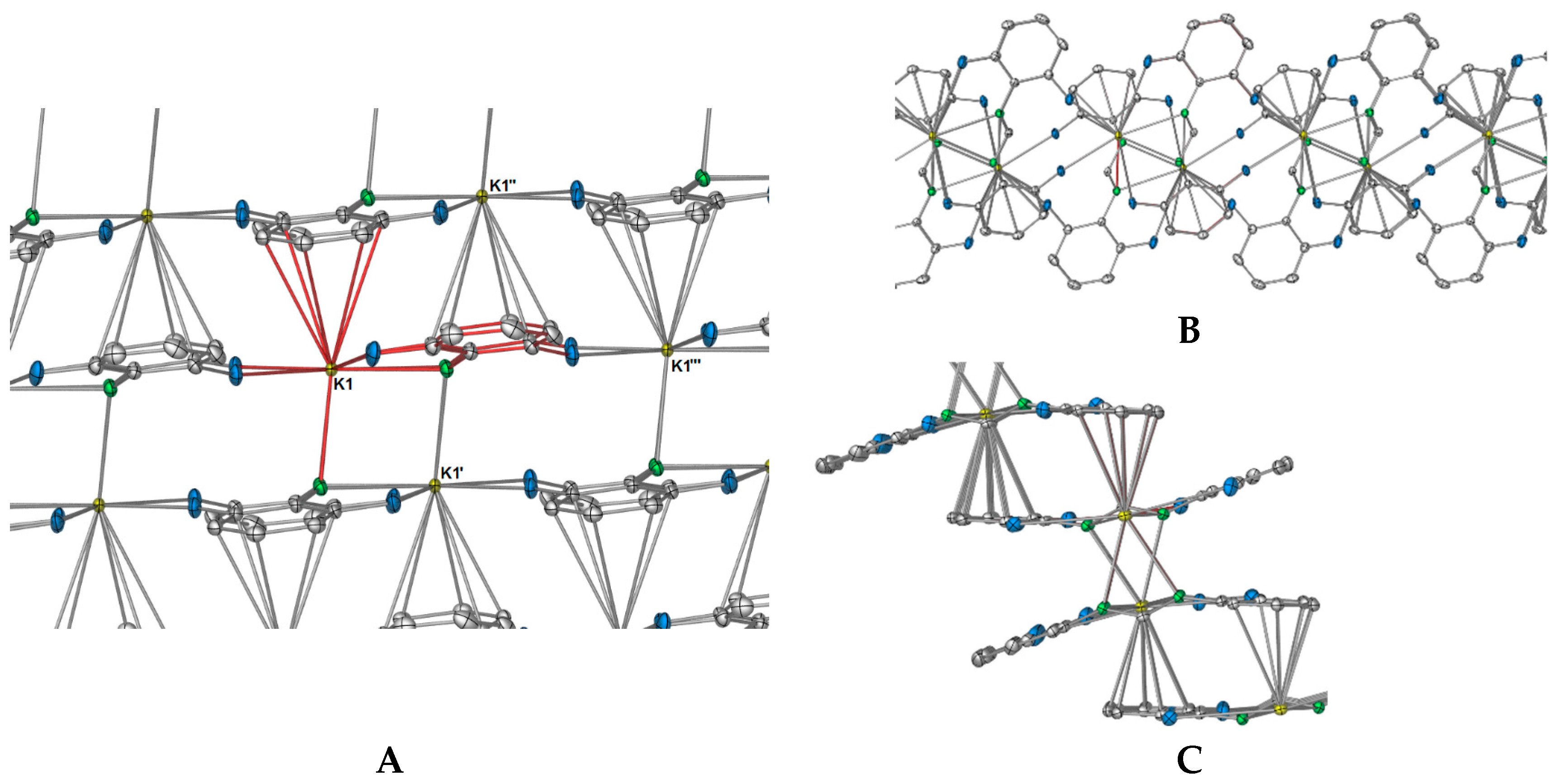
As shown in
Figure 2A, there is an apparent coordination gap in axial positions of the potassium ions, and it is in this position that the other two fluorine atoms (namely, F2 and F4) of the DFForm ligand become relevant, and expand the one-dimensional polymeric network into a two-dimensional polymer. The further fluorine atoms (F2 and F4) coordinate to an adjacent potassium ion in a
μ-(
F″,
F′′′) manner, generating a ten-membered ring (
Figure 3A). Because of this additional coordination, the DFForm ligand is nearly planar across the K and K′′′ atoms, with the bond angle of K–N1/N2
cent–K′′′ being 175.66(1)°. Although the auxiliary fluorine atoms are coordinated at a considerably shorter distance than the K–F1 and K–F3 analogues, there is still only a minor shortening of the C–F bonds from those of DFFormH (C–F: 1.3596(17)–1.3625(18)) [
30]).
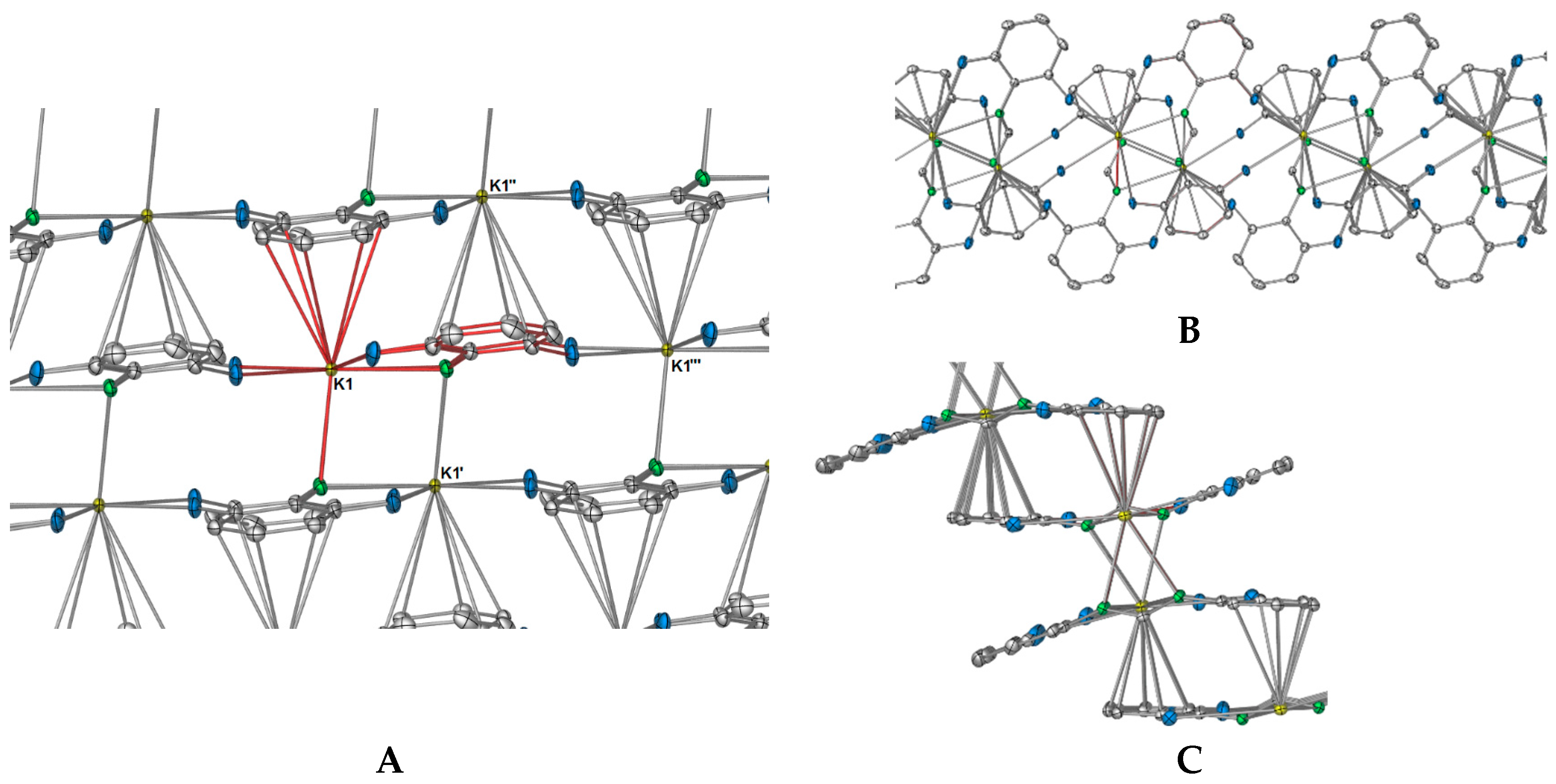
In summation, each DFForm ligand binds four symmetry equivalent potassium ions in a
μ4-1κ(
N,
N′,
F,
F′):2κ(
N,
N′):3η
5(
Ar-
C(2,3,4,5,6)):4κ(
F″,
F′′′) manner, giving the potassium ion a coordination number of 11. Such an interesting binding mode exemplifies how the simple addition of other donors to a ligand system can dramatically alter the coordination network. Furthermore, it appears the
1 is the first crystallographically characterised example across all metal classes, where one
N,
N′-bis(aryl)formamidinate ligand generates a two-dimensional polymer network, all other examples are restricted to one dimension [
18]. The complete polymeric network of
1 is displayed in
Figure 4, showing both how the DFForm bridges across four potassium ions (
Figure 4A) and the two dimensions of the polymer (
Figure 4B,C).
Crystals of
1 were air- and moisture-sensitive, but under an inert atmosphere the compound appeared stable. Complex
1 was repeatedly obtained by simple exposure of thf solutions of “[K(DFForm)(thf)
x]” to vacuum, giving
1 upon drying. The poor solubility in non-coordinating solvents made analysis by
1H NMR and
19F NMR spectroscopy difficult, giving only broad resonances in both spectra (NCHN at 8.88 ppm and F2,6 at −127.2 ppm).All fluorine atoms of the DFForm ligand are equivalent, but clear spectra were generated when
1 was dissolved in thf-d
8. In this solvent, the NC
HN resonance appeared as a pentet, owing to
5JH–F coupling with the
ortho-fluorine atoms, as the pentet collapsed to a singlet with
19F decoupling. A broadening of the F resonance (corresponding to F1–F4) was also observed in the
19F NMR spectrum when it was performed without
1H decoupling. It is likely that upon dissolution in thf-d
8, the polymeric network is dissociated, and a simpler DFForm coordination mode is adopted e.g., [K(DForm)(thf)
x] (2 <
x < 6). Attempts to crystallise a potential monomeric derivative were not successful, but upon concentration of a thf solution of
1, and storage at −35 °C, crystals of a thf-coordinated species were isolated, namely [K
2(DFForm)
2(thf)
2]
∞ (
2,
Scheme 1ii), identified as a one-dimensional polymer. Complex
2 is probably a transient intermediate between the putative monomeric [K(DFForm)(thf)
x] solution species and polymeric
1.
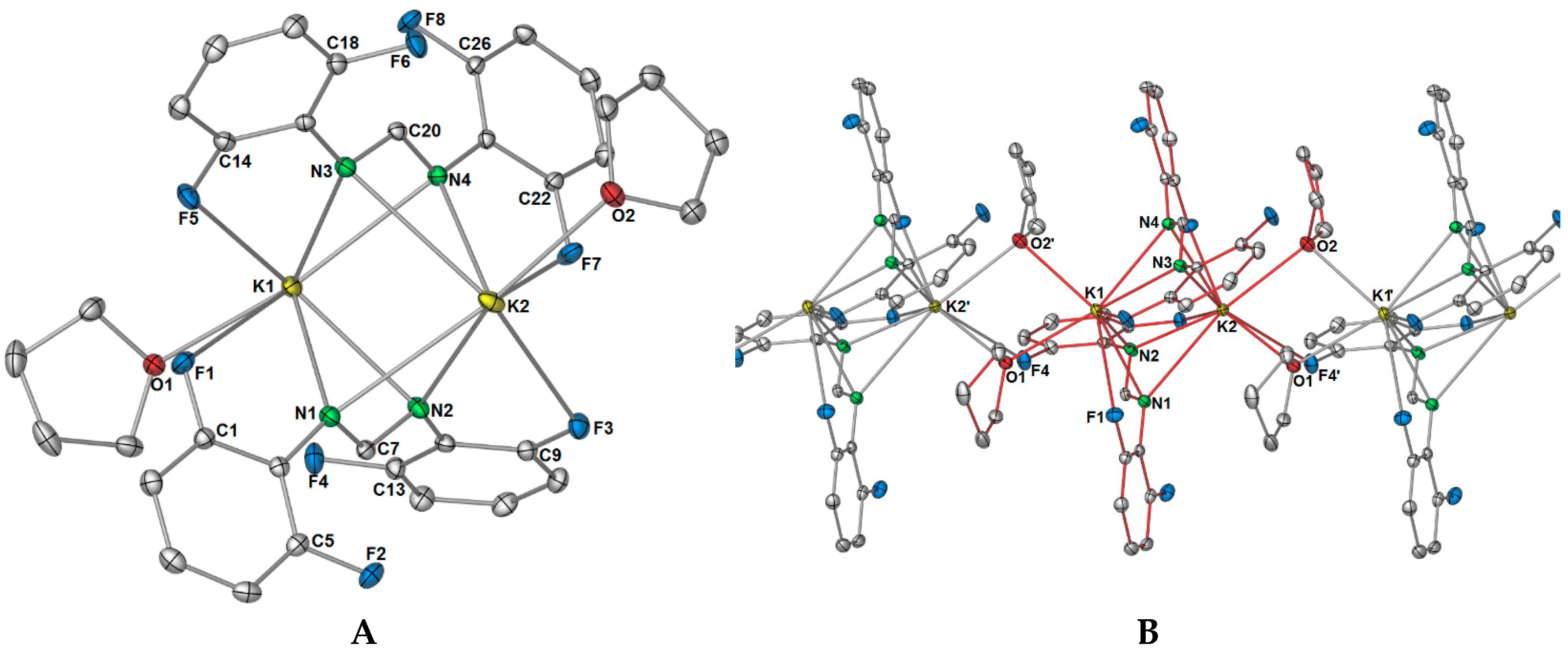
X-ray data for
2 were solved and refined in the monoclinic space group
P2
1, with two potassium ions, two DFForm ligands, and two coordinating thf molecules occupying the asymmetric unit (
Figure 5A). For the ASU component, the two DFForm ligands bridge between both potassium centres in a
μ-1κ(
N,
N′,
F):2κ(
N,
N′,
F′) manner, and N1 and N3 coordinate closer to K1, and N2 and N4 coordinate closer to K2. The K···F–C coordination in this arrangement is overall shorter than those observed for the tetradentate (
N,
N′,
F,
F′) DFForm coordination in
1, but longer than the auxiliary fluorine K···F–C coordination in
1. All the C–F bond lengths exhibit only a slight elongation, with the exception of the C13–F4 bond, which is notably longer than the others. An explanation behind the elongation of only C13–F4 is due to the involvement of this fluorine atom in additional bridging to an adjacent potassium atom. This, in conjunction with the two bridging thf ligands, leads to a one-dimensional polymer (
Figure 5B).
The thf ligands in
2 bridge in an almost symmetrical manner between the two potassium atoms, though O2′ coordinates closer to K2′ than K1. This type of
μ-1κ(
O):2κ(
O) bridging of two thf molecules is no stranger to group one chemistry [
18], for example in the polymeric sodium diphenyloxidomethanide (Ph
2CO)
2− polymer [Na
2(Ph
2CO)(thf)
2] [
33], or the potassium 2,4,6-tris(trifluoromethyl)phenolate (OAr
CF3) complex [K
2(OAr
CF3)
2(thf)
4(
μ-
O-thf)
2] [
34]. However, examples where the polymeric structure is generated by two thf ligands connecting the dinuclear units is restricted to only one other example in group one chemistry, namely [K
4(COT)
2(thf)
6]
∞ [
35]. One difference between the COT (cyclooctatetraenyl) system and
2 is that asymmetric bridging of the thf ligands is more apparent, as both thf ligands favour one metal centre over the other (e.g., K1–O1: 2.839(3), K1–O2: 2.846(5), K2–O1:2.781(3), K2–O2: 2.783(4)). It should also be noted that there are examples where three thf ligands, not two, bridge the monomeric units to create a polymeric network [
32,
36]. Exposure of crystalline
2 to vacuum immediately causes fracturing of the crystals, giving
1. Furthermore, when crystals of
2 are isolated and allowed to stand at room temperature, some degree of thf liberation is apparent as the elemental analysis performed on these crystals gave a lower than expected carbon value. The best fit was obtained when the composition was calculated with loss of 0.4 thf molecules from
2. By examining the structure of
2, it seems that upon the liberation of bound thf, the DFForm ligand changes from the asymmetric
μ-1κ(
N,
N′,
F):2κ(
N,
N′,
F′) coordination to a
μ-(
N,
N′,
F,
F′):(
N,
N′) binding mode, and the auxiliary fluorine and aromatic carbon atoms become free to engage with adjacent potassium ions, building the complex polymeric network of
1. Owing to the rapid loss of thf from
2, additional characterisation was difficult. Dissolution in C
6D
6 gave rapid formation of a powder, presumably
1, as a large excess of thf was observed in the
1H NMR spectrum.
Although no monomeric [K(DFForm)(thf)
x] species could be obtained from thf, we exploited the well-known affinity of 18-crown-6 for the potassium ion. Treatment of
1 with 18-crown-6 and crystallisation from a
n-hexane/toluene solution, gave monomeric [K(DFForm)(18-crown-6)] (
3). The structure was determined by X-ray crystallography, where the data were solved and refined in the monoclinic space group
P2
1/
n, with two molecules occupying the asymmetric unit (only one is depicted in
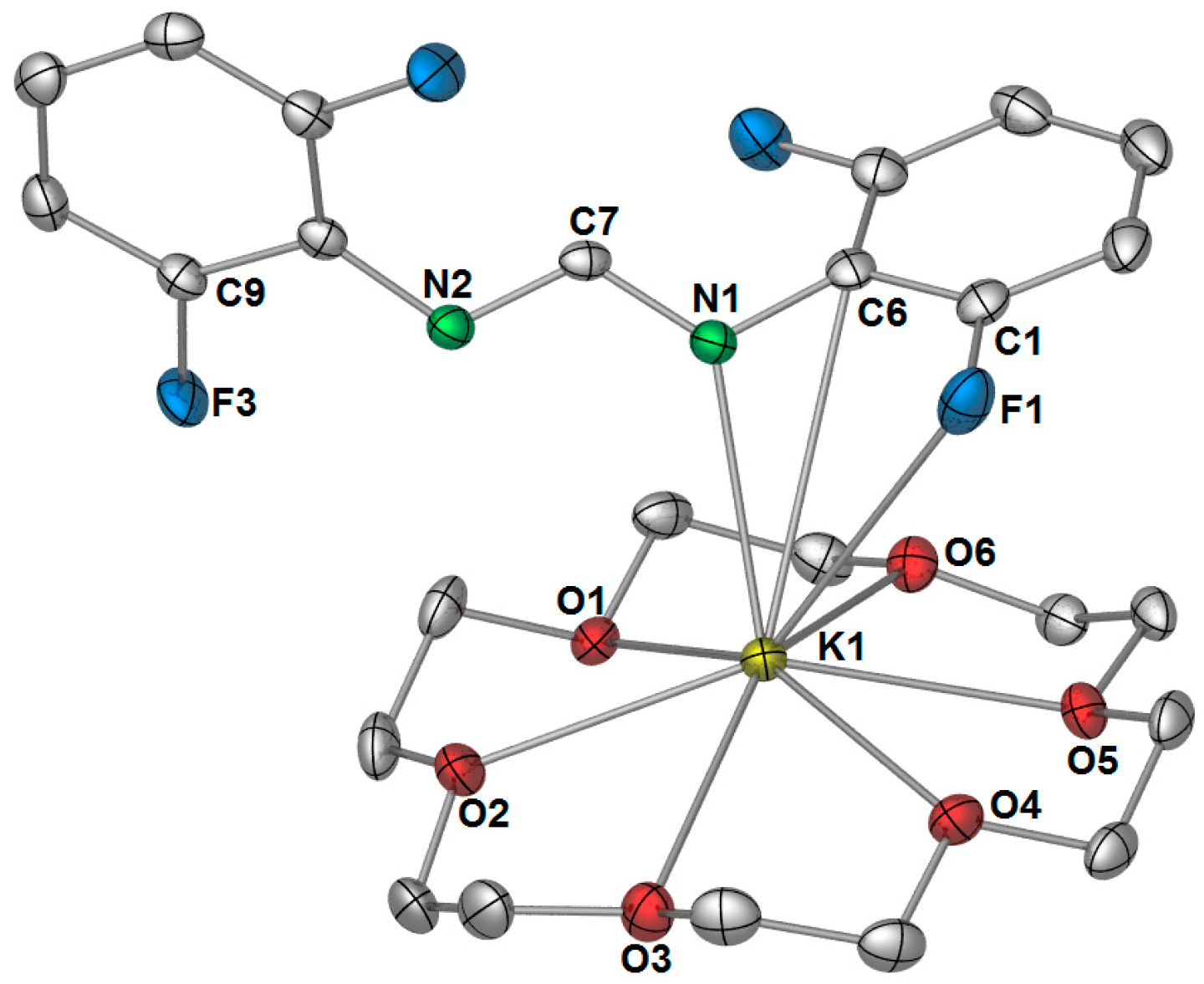
Figure 6). The most surprising feature of this structure is the κ(
N,
C,
F) coordination of the DFForm ligand to the potassium centre, as opposed to the expected κ(
N,
N′) coordination that is observed in [K(
p-TolForm)(18-crown-6)] [
32], and in several other C{NCXN}
−C based ligand systems [
18], such as [K(pyr)(18-crown-6)] (pyr = 1,3,4,6,7,8-hexahydro-2
H-pyrimido[1,2-a]pyrimidide) [
37]. The
ipso carbon–potassium bond length (
Figure 6) lies in the expected range for such interactions. For example, the
ipso-carbon potassium interactions observed in the bimetallic 2,6-diphenylphenolate complex [KCa(OAr
Ph)
3], has a K–C
ipso bond lengths of 3.391(6) Å [
38] and the K–C
ipso bond length in the phenylthiolato complex [K
2Fe(SPh)
4] is 3.477 (5) Å [
39]. Despite the non-binding of N2, there is still charge delocalisation across the NCN bite, as there is only a slight shortening of the free C7–N2 bond, making it far too long for a formal double bond (e.g., DFForm(CPh
3): C=N: 1.2762(12) Å) [
17].
3. Materials and Methods
3.1. General Experimental Details
All reactions were undertaken using Schlenk line and glove box techniques. Solvents (thf, toluene, hexane, C
6D
6, thf-d
8) were purified by distillation over sodium or sodium benzophenone, and were degassed prior to use. NMR experiments were recorded on a Bruker Avance 300 spectrometer or a Bruker AVII+400 machine (Billerica, MA, USA).
1H NMR resonances were referenced to tetramethylsilane by way of the residual
1H resonance of C
6D
6 (and
19F coupled unless specified otherwise).
19F-NMR data were
1H decoupled (unless specified otherwise) and referenced to external CFCl
3. Microanalyses were performed by the elemental analysis service of London Metropolitan University or by an Elementar Vario Micro cube (Elementar, Langenselbold, Germany) by Wolfgang Bock of Tübingen University. IR spectra were recorded on a Perkin–Elmer 1600 Fourier transform infrared spectrometer (
ῦ = 4000–500 cm
−1), as either mulls in sodium-dried Nujol, or a Nicolet 6700 FTIR spectrometer (Thermo Nicolet, Madison, WI, USA) or using a DRIFT chamber with dry KBr/sample mixtures and KBr windows. K[(NSiMe
3)
2] was purchased from Sigma-Aldrich (St Louis, MO, USA) and used as received.
N,
N′-bis(2,6-difluorophenyl)formamidine (DFFormH) was synthesised by a published procedure [
40], 18-crown-6 was purchased from Sigma-Aldrich and used as received.
[K(DFForm)]∞ (1): K[(NSiMe3)2] (0.25 g, 1.3 mmol) and DFFormH (0.33 g, 1.2 mmol) were each dissolved in toluene and combined with stirring, immediately forming a white, poorly soluble powder. The supernatant solution was decanted and the resulting powder was dried in vacuo. After the addition of thf, the powder dissolved, and then the solution was concentrated and layered with n-hexane. Colourless white block crystals grew overnight and were suitable for X-ray diffraction, revealing the composition [K(DFForm)]∞ (1, Yield = 0.30 g, 80%).1H NMR (C6D6, 400 MHz, 25 °C): δ 8.88 (br s, 1H, NCHN), 6.68 (m, 4 H, Ar-H(3,5)), 6.63 (m, 2 H, Ar-H(4)).19F NMR (C6D6, 25 °C): δ −127.2 (br s). 1H NMR (thf-d8, 400 MHz, 25 °C): δ 8.92 (p 5JH–F: 3.09 Hz, 1 H, NCHN), 6.68 (m, 4 H, Ar-H(3,5)), 6.46 (m, 2 H, Ar-H(4)). 1H NMR (thf-d8, 400 MHz, 25 °C, 19F decoupled): δ 8.89 (s, 1 H, NCHN), 6.68 (m, 4 H, Ar-H(3,5)), 6.46 (m, 2 H, Ar-H(4)).19F NMR (thf-d8, 25 °C): δ −127.8 (s). 19F NMR (thf-d8, 25 °C, F–H coupled) −127.8 (br s). IR (DRIFT): ν 1612 (m), 1562 (vs), 1513 (s), 1477 (s), 1464 (s), 1395 (w), 1326 (m), 1287 (w), 1254 (m), 1231 (m), 1199 (s), 1062 (w), 1005 (m), 984 (s), 954 (w), 922 (w), 828 (w), 779 (m), 766 (m). Elemental analysis (C13H7F4KN2, 306.31 g·mol−1): calcd.: C 50.97, H 2.30, N 9.15, found: C 50.81, H 2.34, N 9.07.
[K2(DFForm)2(thf)2]∞ (2): 1 (0.10 g, 0.32 mmol) was dissolved in minimal thf and concentrated in vacuo, giving colourless crystals that were not suitable for X-ray diffraction. The concentrated solution was stored at −35 °C, where large colourless block crystals grew of [K2(DFForm)2(thf)2] (2), suitable for X-ray diffraction. Upon exposure to vacuum, the crystals fractured and a white powder was obtained, likely consisting of a mixture of 1 and 2. (Yield = 0.11 g, 89%). 1H NMR (C6D6, 400 MHz, 25 °C, formation of insoluble white powder upon solvent addition, giving a large excess of thf in solution): δ 1.42 (m, 232 H, thf-β-CH2), 3.57 (m, 232 H, thf-α-CH2), 6.36 (m, 2 H, Ar-H4) 6.67 (m, 4 H, Ar-H(3,5)), 8.88 (br s, 1 H, NCHN). 19F NMR (C6D6, 25 °C): δ −127.1 (br s). IR (DRIFT): ν 1613 (m), 1564 (vs), 1551 (vs), 1514 (m), 1477 (vs), 1464 (vs), 1395 (w), 1325 (s), 1254 (m), 1231 (w), 1200 (s), 1062 (w), 1005 (w), 984.6 (s), 955 (w), 922 (w), 827 (w), 799 (w), 766 (m), 742 (w), 716 (m). Elemental analysis calcd (%) (for C34H30F8K2N4O2, 756.81 g·mol−1, pre-dried powder under vacuum). C 53.95, H 4.00, N 7.40, found: C 49.64, H 2.74, N 8.44. When the crystals were dried by slow evaporation in a glove box, a composition of [K2(DFForm)2(thf)1.6] was supported, calcd. (C58.4H40.8F8K2N4O1.6, 727.96 g·mol−1): C 53.46, H 3.71, N 7.69 found: C 53.08, H 3.66, N 7.35.
[K(DFForm)(18-crown-6)] (3): If 1 (~0.10 g, 0.32 mmol) was crystallised from toluene/hexane solutions in the presence of one equivalent of 18-crown-6 (~0.09 g, 0.34 mmol), pale yellow block crystals of [K(DFForm)(18-crown-6)] (3) developed. (Yield = ~0.07 g, 34%). 1H NMR (C6D6, 300 MHz 303.2 K): δ 9.15 (s, 1 H, NCHN), 6.84 (m, 4 H, Ar-H(3,5)), 6.42 (m, 2 H, Ar-H(4)), 3.22 (br s, 24 H, 18-crown-6). 19F NMR (C6D6, 303.2 K): δ = −125.81 (br s). IR (Nujol): ῦ = 1588 (vs), 1540 (vs), 1259 (vs), 1193 (m), 1096 (s), 1004 (s), 956 (m), 818 (m). Elemental analysis returned poor C H N values. (C25H31F4KN2O6, 570.62 g·mol−1): calcd. C 52.62, H 5.47, N 4.91, found: C 46.87, H 4.93, N 5.21.
3.2. X-ray Crystallography
All compounds were examined on a “Bruker APEX-II CCD” diffractometer at 100.15 or 150.15 K, mounted on a fibre loop in Paratone-N. Absorption corrections were completed using Apex II program suite [
41]. Structural solutions were obtained by charge flipping (
1,
2,
3) [
42] methods, and refined using full matrix least squares methods against
F2 using SHELX2013 [
43], within the OLEX 2 graphical interface [
43]. CCDC numbers:
1 (1540263),
2 (1540264),
3 (1540265).
[K(DFForm)]∞ (1): C13H7F4KN2 (M = 306.31 g/mol): triclinic, space group P-1 (no. 2), a = 7.4437(2) Å, b = 7.5357(2) Å, c = 11.8891(3) Å, α = 100.6590(10)°, β = 101.9020(10)°, γ = 101.1070(10)°, V = 622.56(3) Å3, Z = 2, T = 100(2) K, μ(MoKα) = 0.466 mm−1, Dcalc = 1.634 g/cm3, 10877 reflections measured (5.66° ≤ 2Θ ≤ 60.48°), 3657 unique (Rint = 0.0152, Rsigma = 0.0174) which were used in all calculations. The final R1 was 0.0279 (>2σ(I)) and wR2 was 0.0734 (all data). Note: NCHN hydrogen atom manually assigned from identified Q peak.
[K2(DFForm)2(thf)2]∞ (2): C34H30F8K2N4O2 (M = 756.82 g/mol): monoclinic, space group P21 (no. 4), a = 7.55040(10) Å, b = 19.8885(3) Å, c = 11.6408(2) Å, β = 105.4479(6)°, V = 1684.90(4) Å3, Z = 4, T = 100.1 K, μ(MoKα) = 0.364 mm−1, Dcalc = 1.492 g/cm3, 17983 reflections measured (3.62° ≤ 2Θ ≤ 60.66°), 8299 unique (Rint = 0.0136, Rsigma = 0.0201), which were used in all calculations. The final R1 was 0.0262 (>2σ(I)) and wR2 was 0.0652 (all data).
2[K(DFForm)(18-crown-6)] (3): C50H62F8K2N4O12 (M = 1141.23): note: two molecules present in the asymmetric unit. monoclinic, space group P21/n (no. 14), a = 10.9773(4) Å, b = 15.3047(5) Å, c = 31.3860(10) Å, β = 93.674(2)°, V = 5262.1(3) Å3, Z = 4, T = 123.15 K, μ(MoKα) = 0.273 mm−1, Dcalc = 1.441 g/mm3, 78411 reflections measured (2.6 ≤ 2Θ ≤ 56.76), 13119 unique (Rint = 0.0369, Rsigma = 0.0266) which were used in all calculations. The final R1 was 0.0321 (I > 2σ(I)) and wR2 was 0.1146 (all data).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}