
Methanediide Formation via Hydrogen Elimination in Magnesium versus Aluminium Hydride Complexes of a Sterically Demanding Bis(iminophosphoranyl)methanediide


 and
and 
Abstract
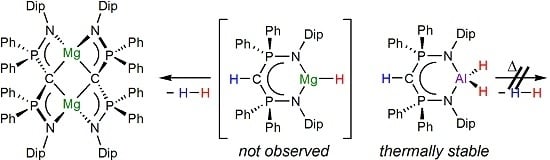
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
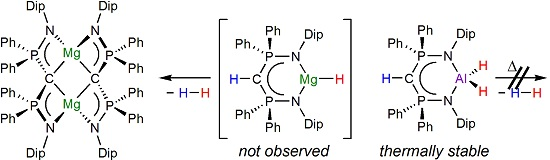
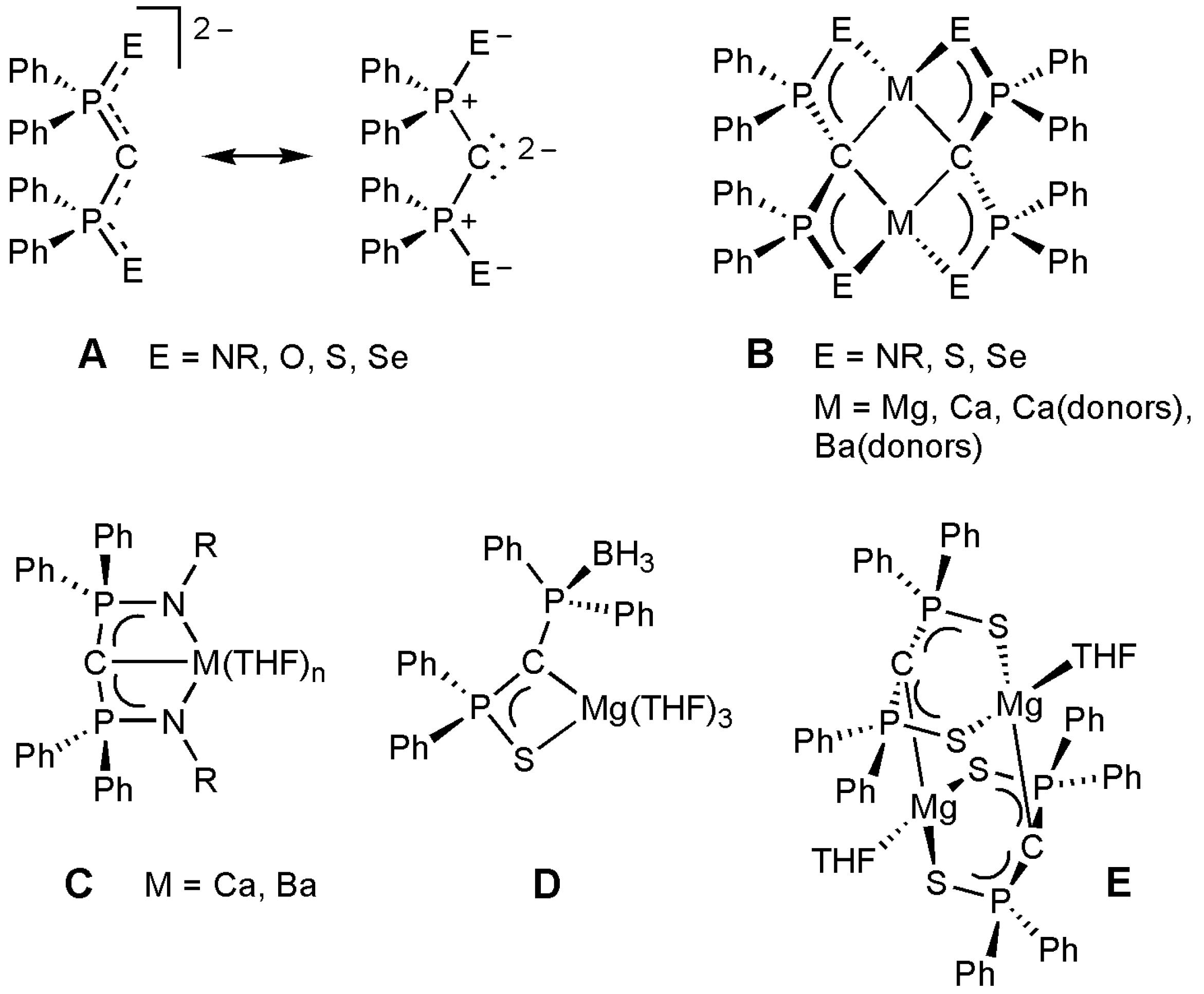
1. Introduction
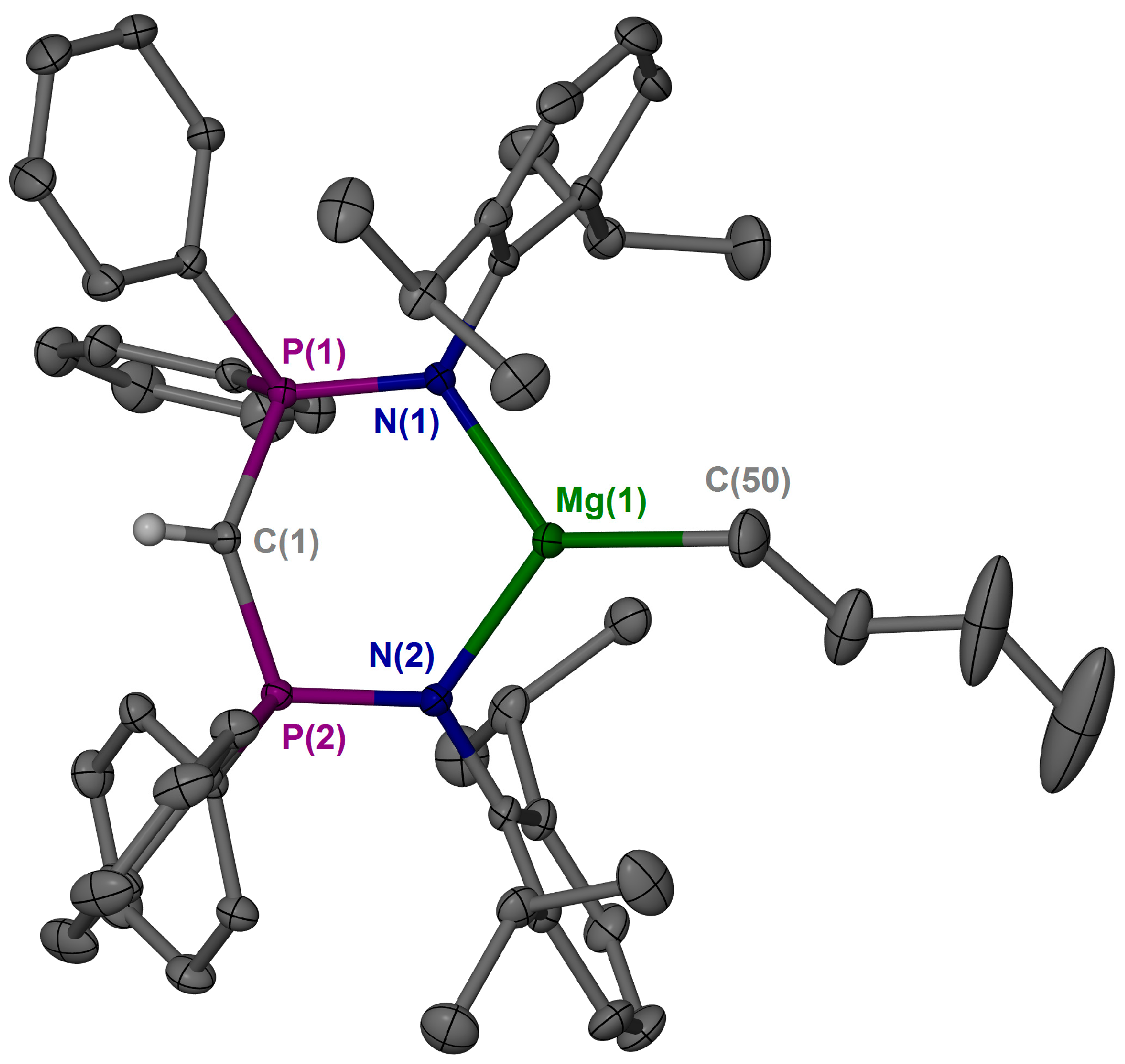
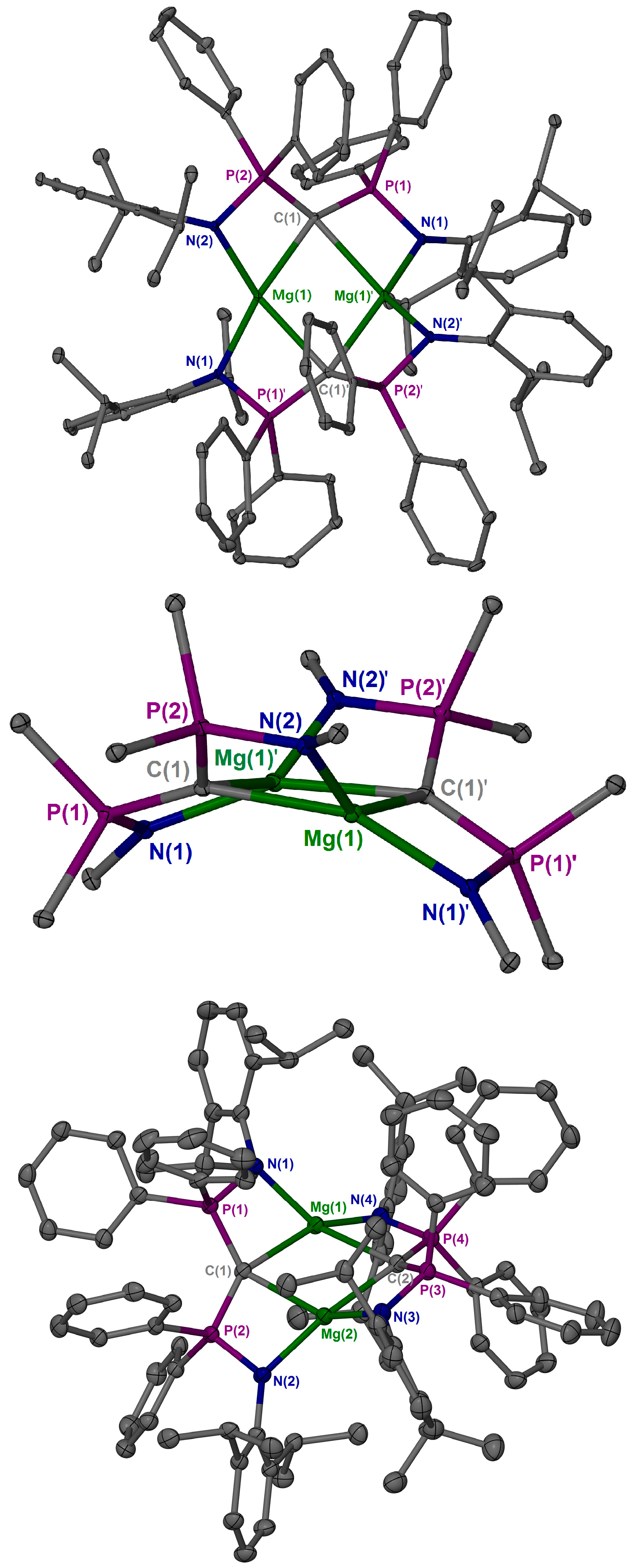
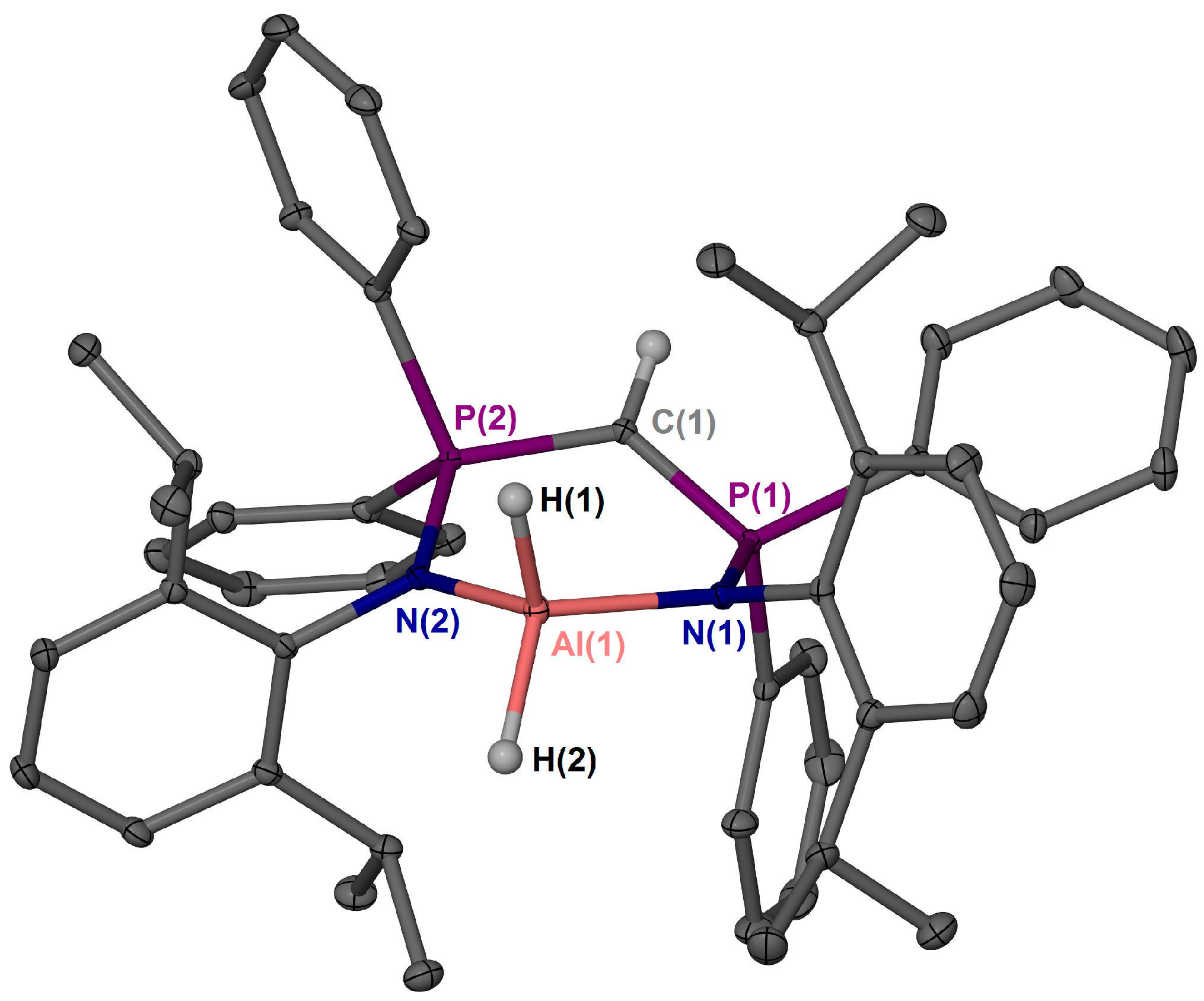
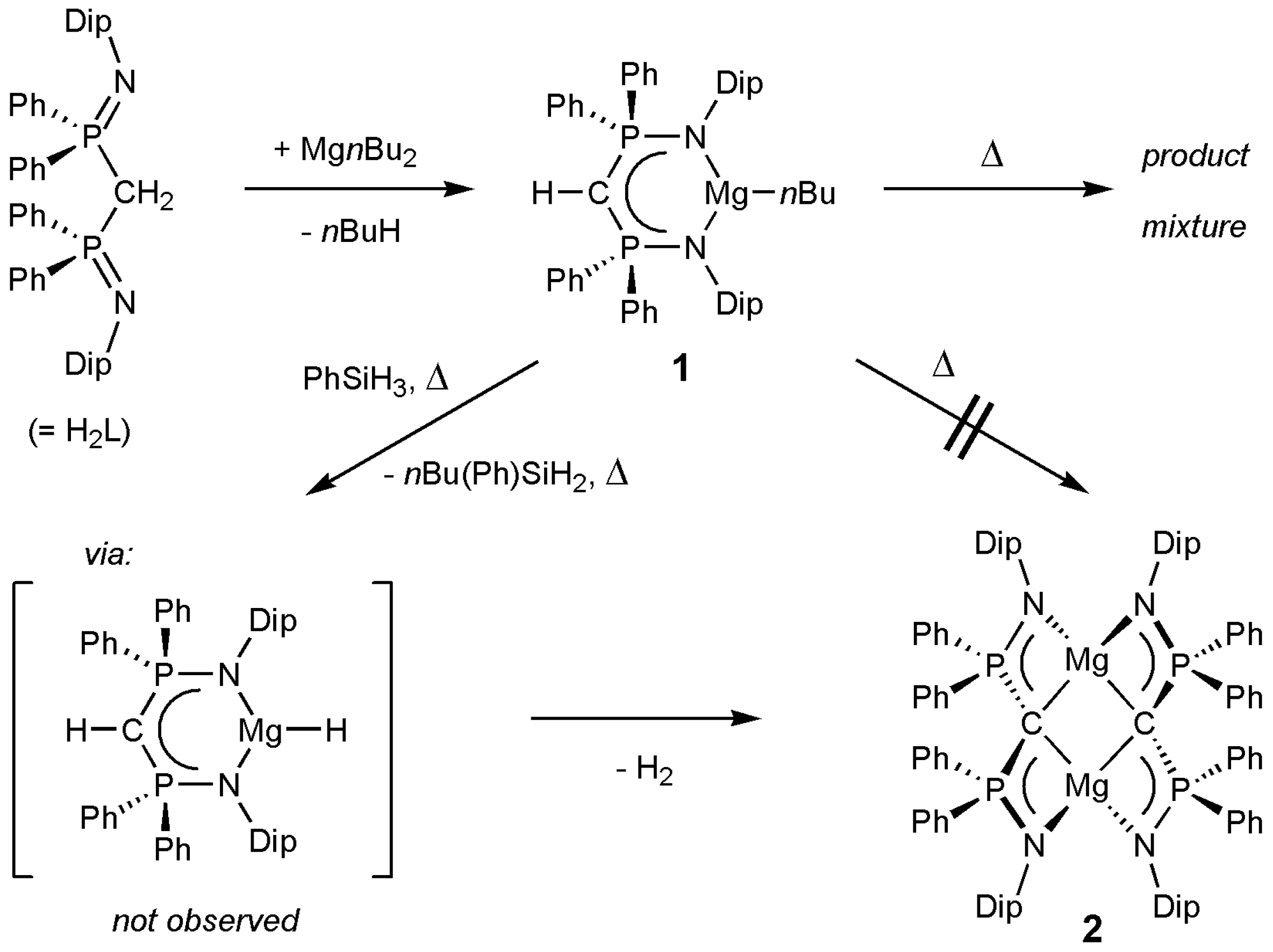
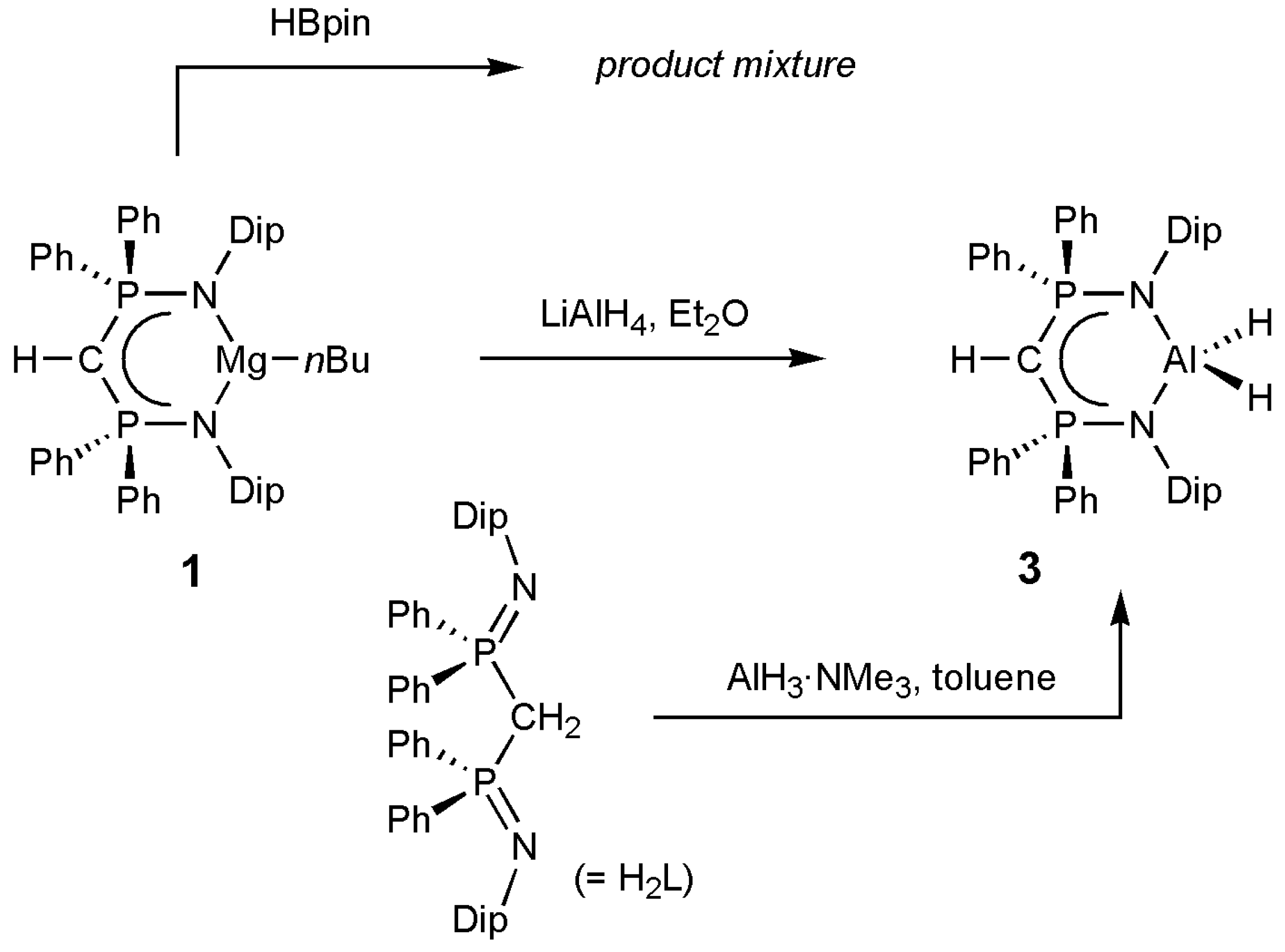
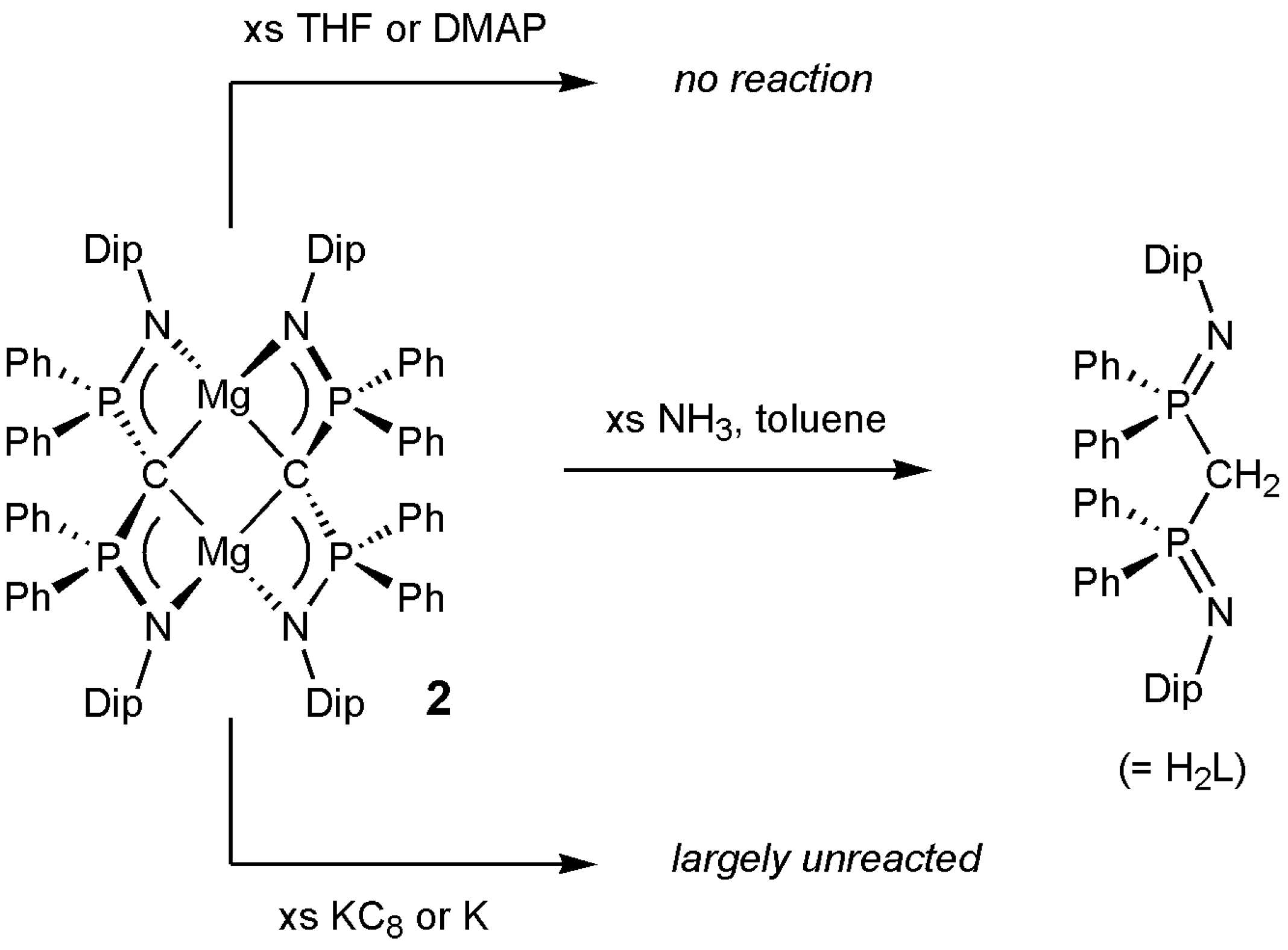
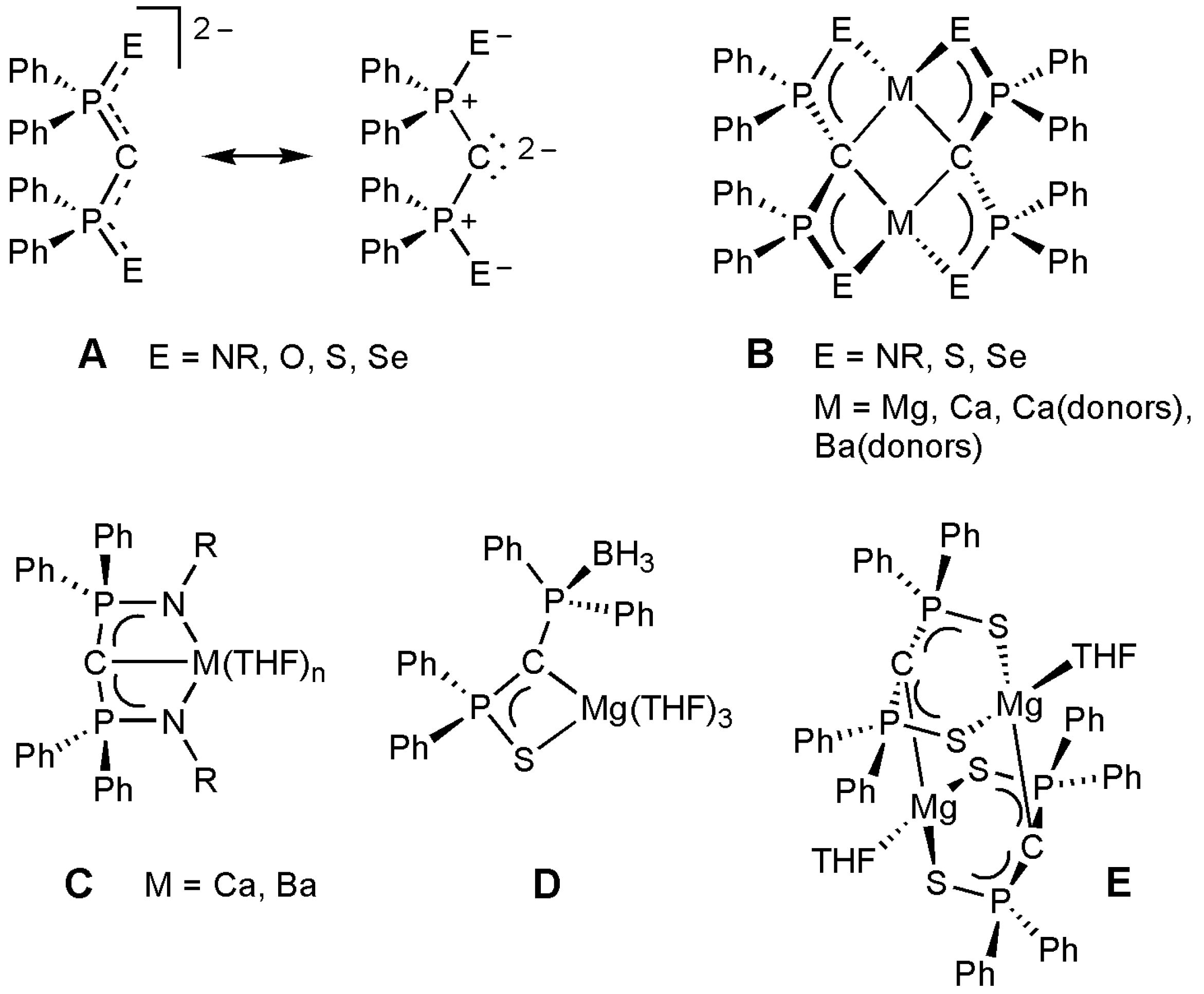
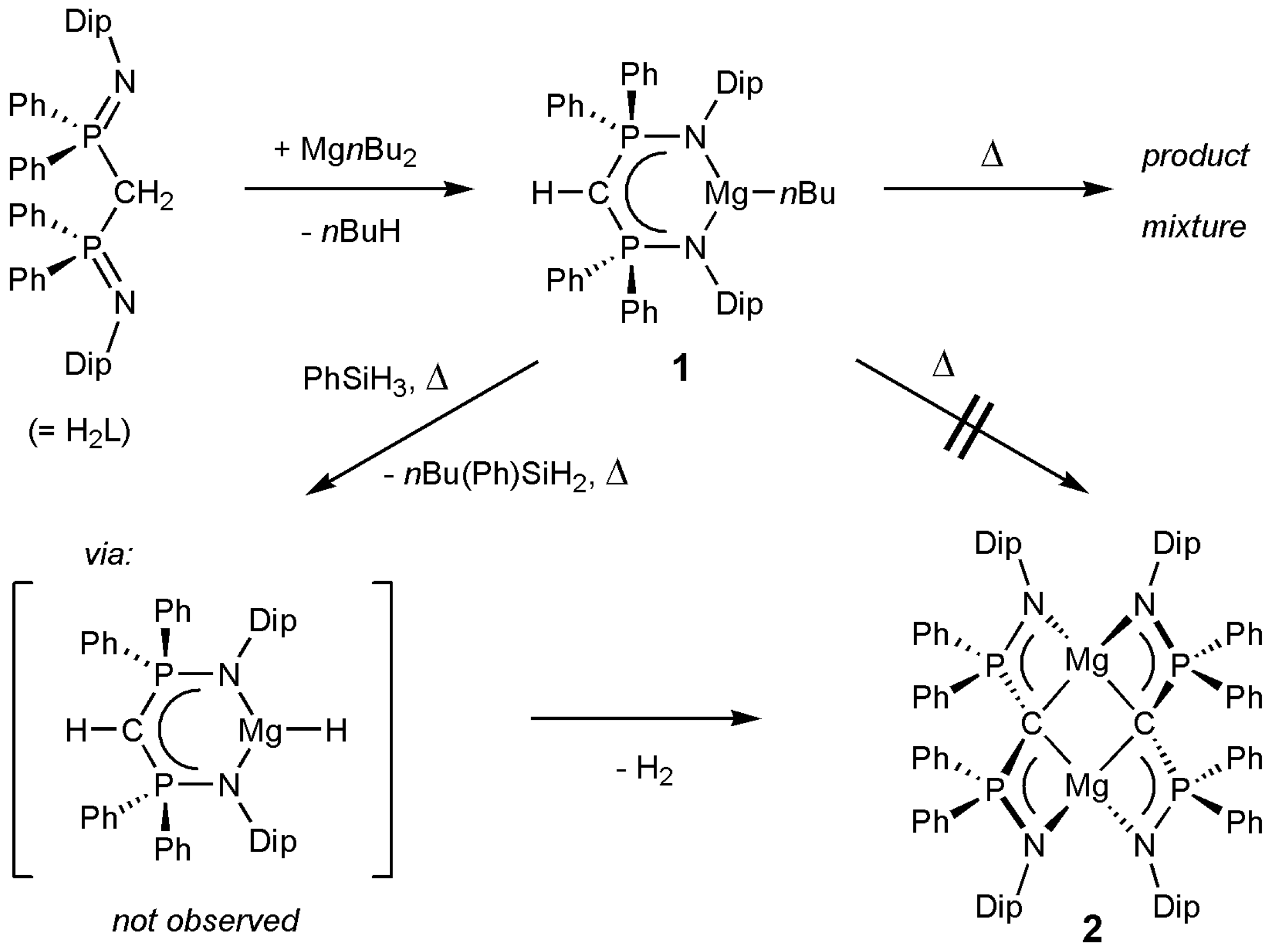
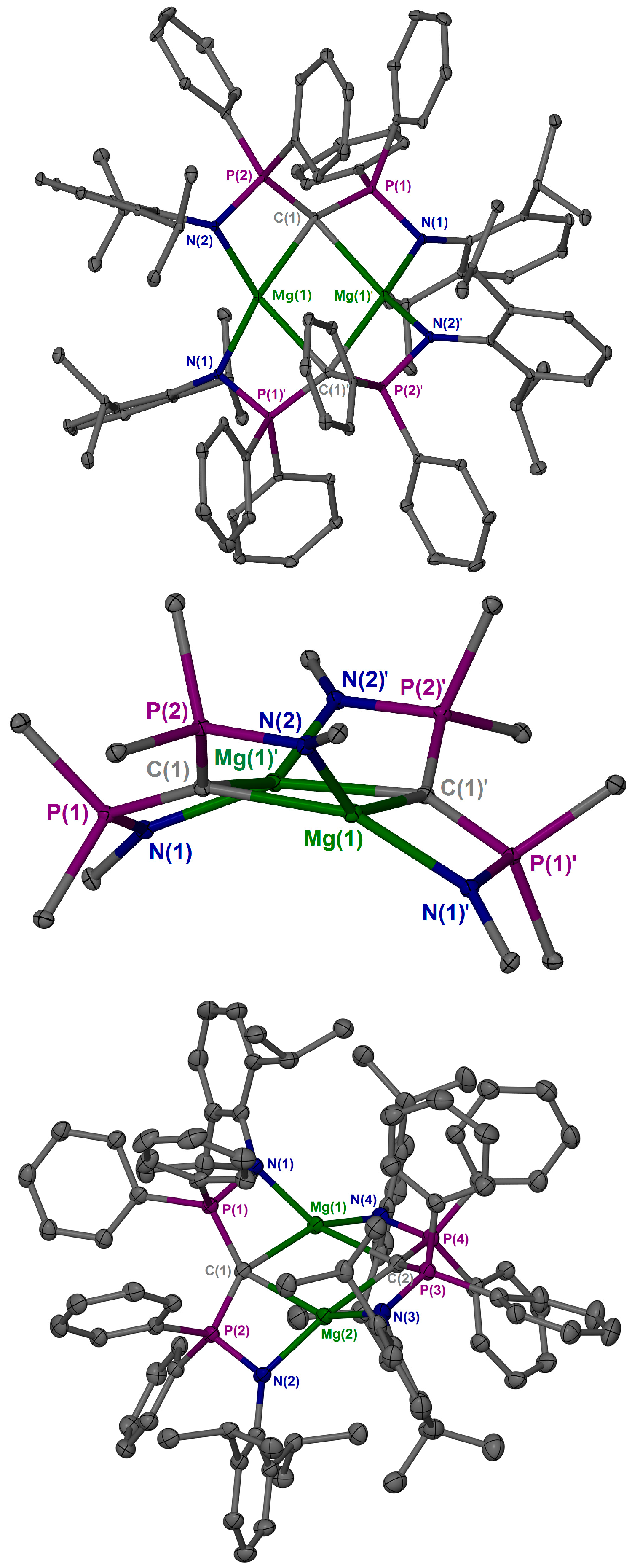
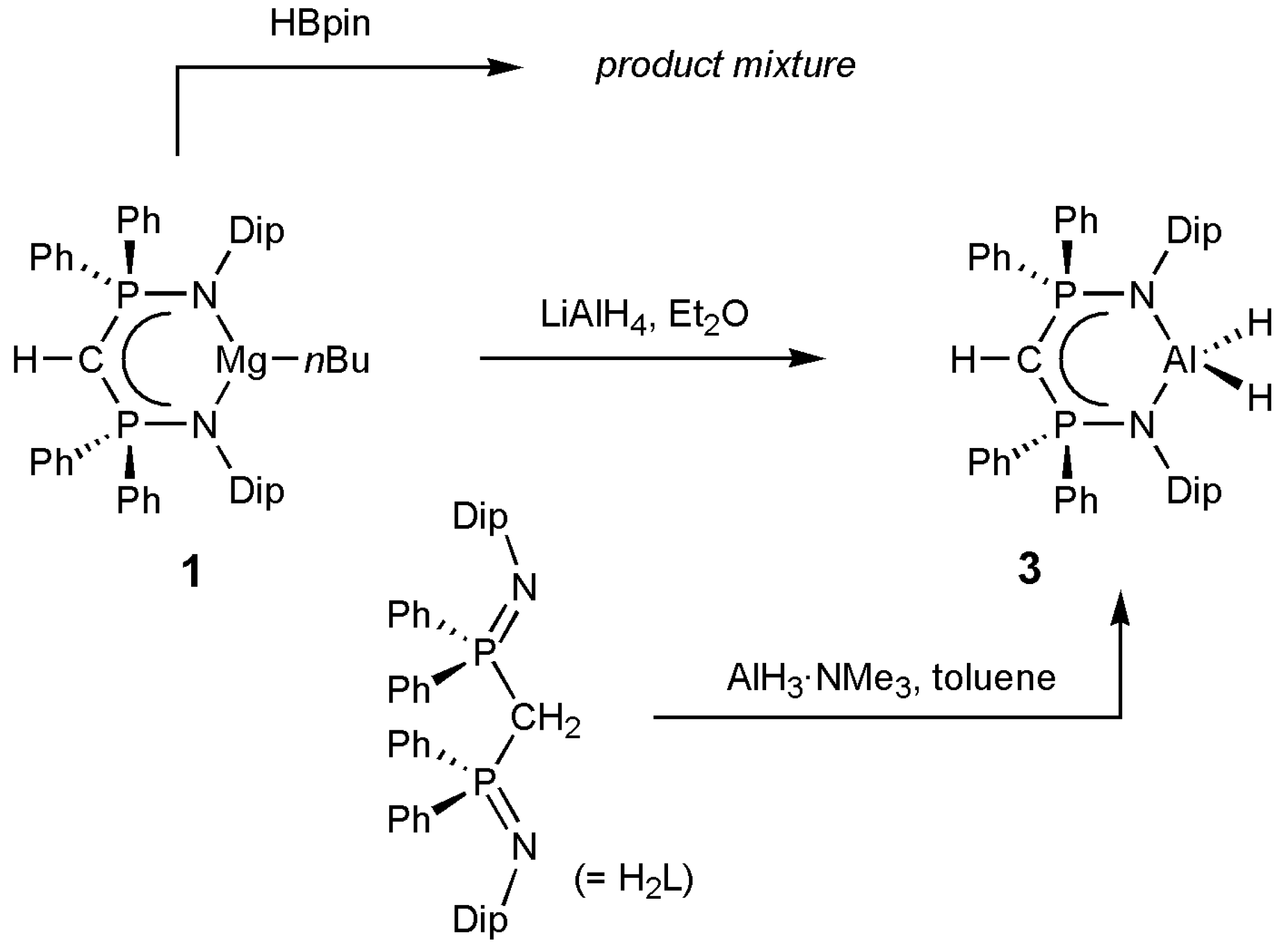
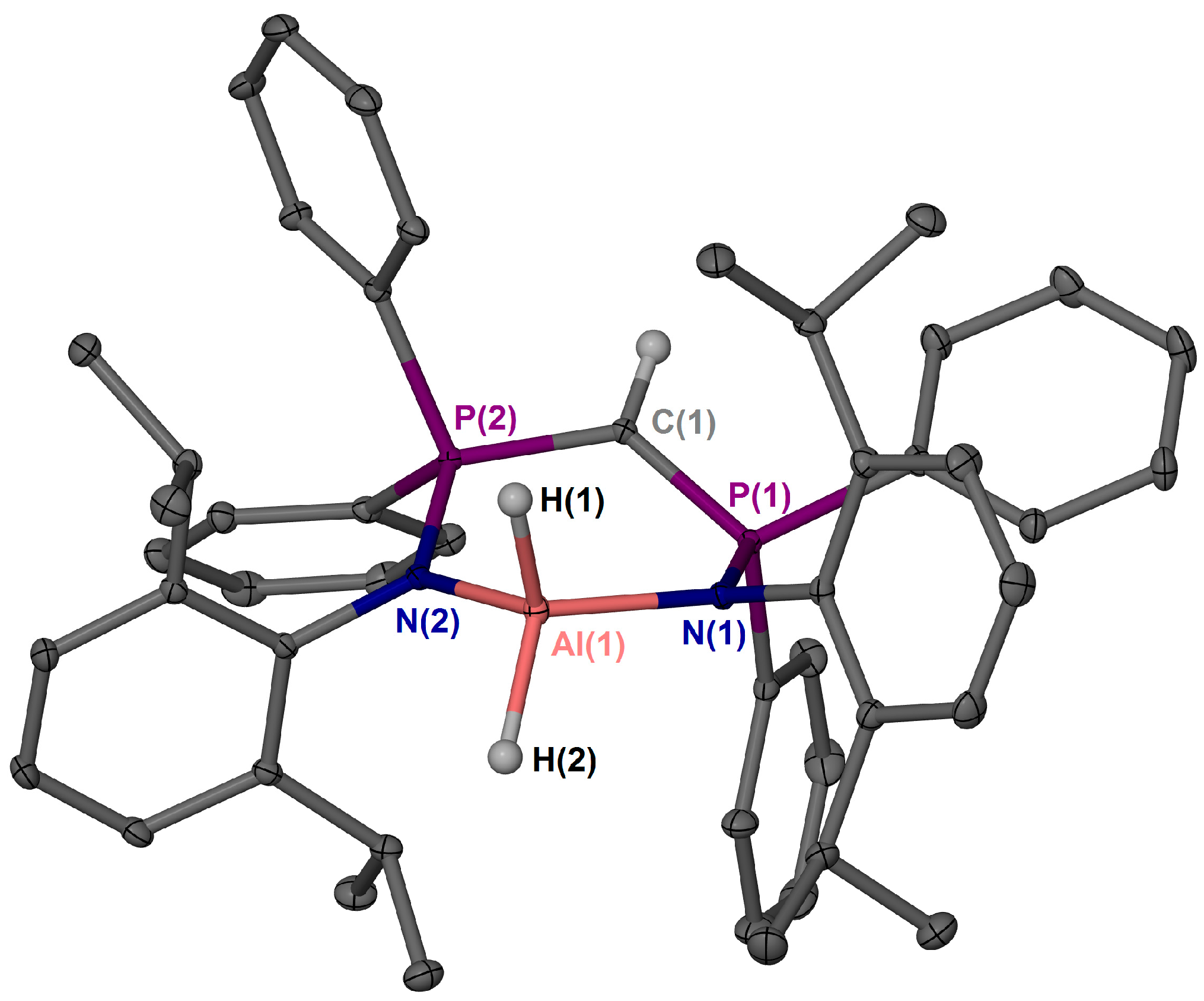
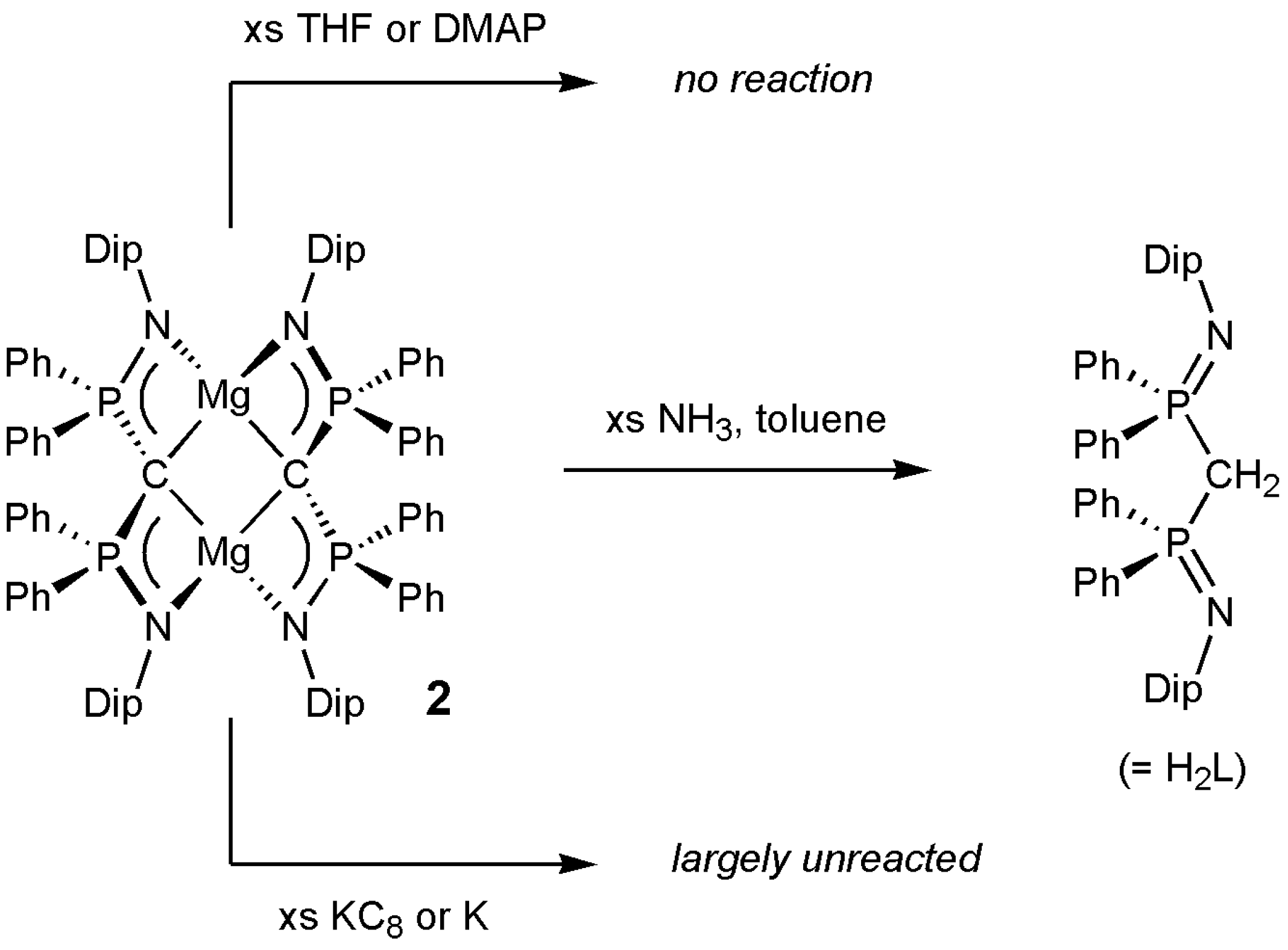
2. Results and Discussion
3. Experimental Section
3.1. General Considerations
3.2. Syntheses of Complexes 1–3
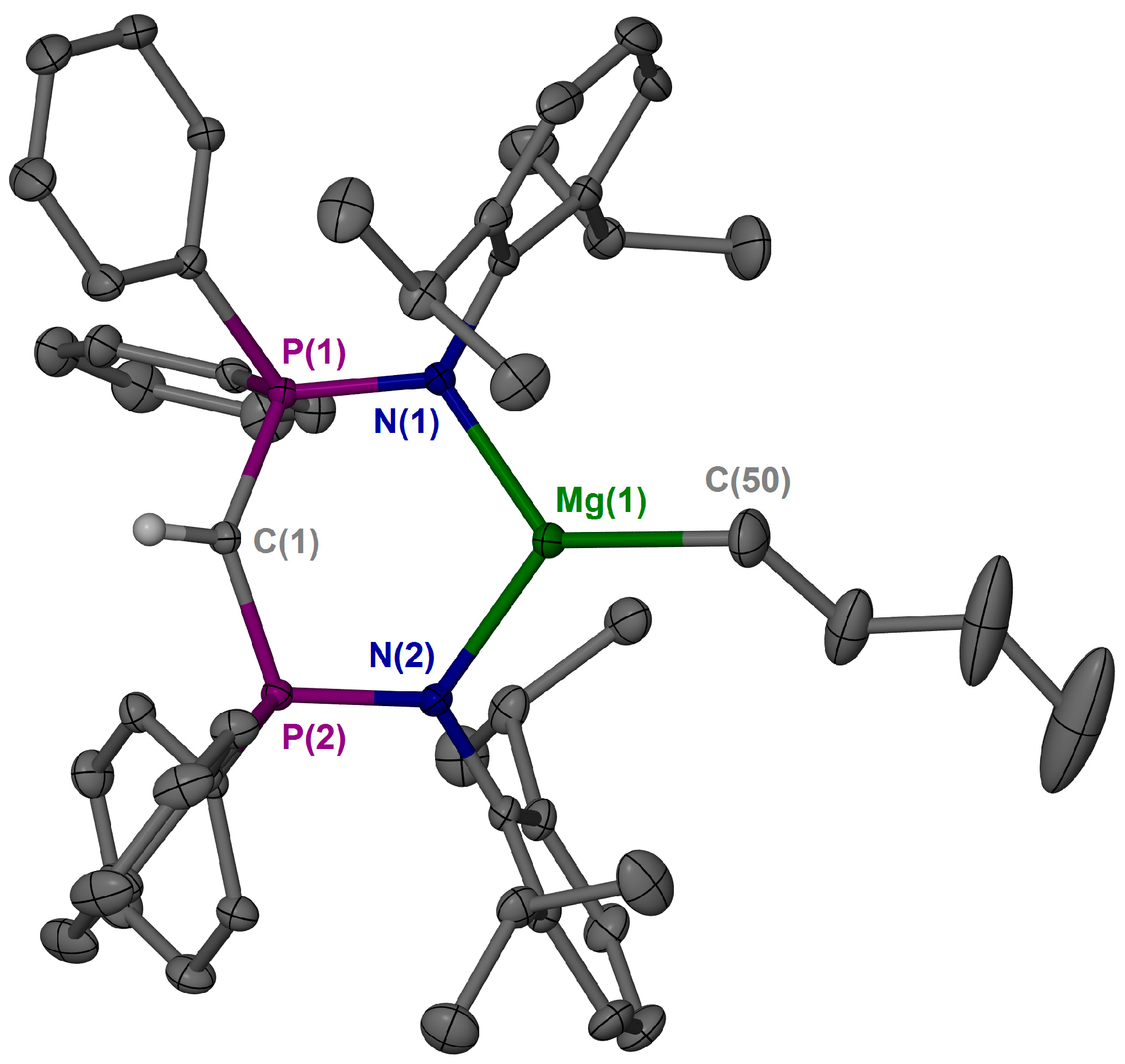
3.3. X-ray Crystallography
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Marek, I.; Normant, J.-F. Synthesis and reactivity of sp3-geminated organodimetallics. Chem. Rev. 1996, 96, 3241–3267. [Google Scholar] [CrossRef] [PubMed]
- Gessner, V.H.; Becker, J.; Feichtner, K.-S. Carbene complexes based on dilithium methandiides. Eur. J. Inorg. Chem. 2015, 1841–1859. [Google Scholar] [CrossRef]
- Chivers, T.; Konu, J.; Thirumoorthi, R. PCP-bridged chalcogen-centred anions: Coordination chemistry and carbon-based reactivity. Dalton Trans. 2012, 41, 4283–4295. [Google Scholar] [CrossRef] [PubMed]
- Harder, S. Geminal dianions stabilized by phosphonium substituents. Coord. Chem. Rev. 2011, 255, 1252–1267. [Google Scholar] [CrossRef]
- Liddle, S.T.; Mills, D.P.; Wooles, A.J. Early metal bis(phosphorus-stabilized)carbene chemistry. Chem. Soc. Rev. 2011, 40, 2164–2176. [Google Scholar] [CrossRef] [PubMed]
- Liddle, S.T.; Mills, D.P.; Wooles, A.J. Bis(phosphorus-Stabilized)methanide and methandiide derivatives of group 1–5 and f-element metals. Organomet. Chem. 2010, 36, 29–55. [Google Scholar] [CrossRef]
- Panda, T.K.; Roesky, P.W. Main-group and transition-metal complexes of bis(phosphinimino)methanides. Chem. Soc. Rev. 2009, 38, 2782–2804. [Google Scholar] [CrossRef] [PubMed]
- Cantat, T.; Mézailles, N.; Auffant, A.; Le Floch, P. Bis-phosphorus stabilised carbene complexes. Dalton Trans. 2008, 15, 1957–1972. [Google Scholar] [CrossRef] [PubMed]
- Birchall, C.; Moxey, G.J.; McMaster, J.; Blake, A.J.; Lewis, W.; Kays, D.L. A monomeric, heterobimetallic complex with an unsupported Mg–Fe bond. Inorg. Chim. Acta 2017, 458, 97–100. [Google Scholar] [CrossRef]
- Thirumoorthi, R.; Chivers, T. Potassium and magnesium complexes of the (iminophosphoranyl)(selenophosphoranyl)methanide ligand [CH(PPh2Se)(PPh2NSiMe3)]−. Eur. J. Inorg. Chem. 2015, 2188–2192. [Google Scholar] [CrossRef]
- Xie, H.; Mou, Z.; Liu, B.; Li, P.; Rong, W.; Li, S.; Cui, D. Phosphinimino-amino magnesium complexes: Synthesis and catalysis of heteroselective ROP of rac-lactide. Organometallics 2014, 33, 722–730. [Google Scholar] [CrossRef]
- Heuclin, H.; Fustier-Boutignon, M.; Ho, S.Y.; Le Goff, X.; Carenco, S.; So, C.; Mézailles, N. Synthesis of phosphorus(V)-stabilized geminal dianions. The cases of mixed P=X/P→BH3 (X = S, O) and P=S/SiMe3 derivatives. Organometallics 2013, 32, 498–508. [Google Scholar] [CrossRef]
- Thirumoorthi, R.; Chivers, T. Alkali metal, magnesium, and zinc complexes of bis(chalcogenophosphinoyl)methanide ligands. Eur. J. Inorg. Chem. 2012, 3061–3069. [Google Scholar] [CrossRef]
- Marks, S.; Kuzdrowska, M.; Roesky, P.W.; Annunziata, L.; Guillaume, S.M.; Maron, L. Organometallic strontium borohydrides: Synthesis, X-ray structures, catalytic polymerization of ε-caprolactone, and density functional calculations. ChemPlusChem 2012, 77, 350–353. [Google Scholar] [CrossRef]
- Leung, W.; Wan, C.; Mak, T.C.W. Synthesis and structure of magnesium and group 13 metal bis(thiophosphinoyl)methanediide complexes. Organometallics 2010, 29, 1622–1628. [Google Scholar] [CrossRef]
- Guo, J.; Lee, J.; Foo, M.; Lau, K.; Xi, H.; Lim, K.H.; So, C. Synthesis and characterization of magnesium and aluminum bis(phosphoranyl)methanediide complexes. Organometallics 2010, 29, 939–944. [Google Scholar] [CrossRef]
- Wiecko, M.; Marks, S.; Panda, T.K.; Roesky, P.W. Bis(phosphinimino)methanides as ligands in divalent ytterbium and strontium chemistry—Synthesis and structure. Z. Anorg. Allg. Chem. 2009, 635, 931–935. [Google Scholar] [CrossRef]
- Orzechowski, L.; Harder, S. Syntheses, structures, and reactivity of barium carbene complexes with chelating bis-iminophosphorano arms. Organometallics 2007, 26, 5501–5506. [Google Scholar] [CrossRef]
- Orzechowski, L.; Jansen, G.; Harder, S. Synthesis, structure, and reactivity of a stabilized calcium carbene: R2CCa. J. Am. Chem. Soc. 2006, 128, 14676–14684. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.A.; Hill, M.S.; Hitchcock, P.B. Synthesis and M–Cγ hemilability of group 2 bis(phosphinimino)methanides. Organometallics 2006, 25, 394–402. [Google Scholar] [CrossRef]
- Panda, T.K.; Zulys, A.; Gamer, M.T.; Roesky, P.W. Bis(phosphinimino)methanides as ligands in divalent lanthanide and alkaline earth chemistry—Synthesis, structure, and catalysis. J. Organomet. Chem. 2005, 690, 5078–5089. [Google Scholar] [CrossRef]
- Wei, P.; Stephan, D.W. Magnesium complexes of bis(phosphinimine)methane and -methanide ligands. Organometallics 2003, 22, 601–604. [Google Scholar] [CrossRef]
- Hill, M.S.; Hitchcock, P.B. Bis(phosphinimino)methyl derivatives of Ca, Sr and Ba: Facile access to heavier alkaline earth organometallic chemistry. Chem. Commun. 2003, 1758–1759. [Google Scholar] [CrossRef]
- Al-Benna, S.; Sarsfield, M.J.; Thornton-Pett, M.; Ormsby, D.L.; Maddox, P.J.; Brès, P.; Bochmann, M. Sterically hindered iminophosphorane complexes of vanadium, iron, cobalt and nickel: A synthetic, structural and catalytic study. J. Chem. Soc. Dalton Trans. 2000, 4247–4257. [Google Scholar] [CrossRef]
- Green, S.P.; Jones, C.; Stasch, A. Stable magnesium(I) compounds with Mg–Mg bonds. Science 2007, 318, 1754–1757. [Google Scholar] [CrossRef] [PubMed]
- Stasch, A.; Jones, C. Stable dimeric magnesium(I) compounds: From chemical landmarks to versatile reagents. Dalton Trans. 2011, 40, 5659–5672. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, M.; Maitland, B.; Kociok-Köhn, G.; Stasch, A.; Jones, C.; Hill, M.S. Mononuclear three-coordinate magnesium complexes of a highly sterically encumbered β-diketiminate ligand. Inorg. Chem. 2014, 53, 10543–10552. [Google Scholar] [CrossRef] [PubMed]
- Balasanthiran, V.; Chisholm, M.H.; Choojun, K.; Durr, C.B.; Wambua, P.M. BDI*MgX(L) where X = nBu and OtBu and L = THF, py and DMAP. The rates of kinetic exchange of L where BDI* = CH{C(tBu)N-2,6-iPr2C6H3}2. Polyhedron 2016, 103, 235–240. [Google Scholar] [CrossRef]
- Jones, C.; Bonyhady, S.J.; Nembenna, S.; Stasch, A. New routes to soluble magnesium amidoborane complexes. Eur. J. Inorg. Chem. 2012, 2596–2601. [Google Scholar] [CrossRef]
- Xie, H.; Hua, X.; Liu, B.; Wu, C.; Cui, D. Phosphinimino-amino supported complex: Synthesis, polymerization of ethylene and dearomatisation of pyridine. J. Organomet. Chem. 2015, 798, 335–340. [Google Scholar] [CrossRef]
- Green, S.P.; Jones, C.; Stasch, A. Stable adducts of a dimeric magnesium(I) compound. Angew. Chem. Int. Ed. 2008, 47, 9079–9083. [Google Scholar] [CrossRef] [PubMed]
- Bonyhady, S.J.; Jones, C.; Nembenna, S.; Stasch, A.; Edwards, A.J.; McIntyre, G.J. β-Diketiminate-stabilized magnesium(I) dimers and magnesium(II) hydride complexes: Synthesis, characterization, adduct formation, and reactivity studies. Chem. Eur. J. 2010, 16, 938–955. [Google Scholar] [CrossRef] [PubMed]
- Sindlinger, C.P.; Stasch, A. Aluminium complexes of a sterically demanding bis(iminophosphorane)methandiide. Aust. J. Chem. 2013, 66, 1219–1225. [Google Scholar] [CrossRef]
- Arrowsmith, M.; Hadlington, T.J.; Hill, M.S.; Kociok-Köhn, G. Magnesium-catalysed hydroboration of aldehydes and ketones. Chem. Commun. 2012, 48, 4567–4569. [Google Scholar] [CrossRef] [PubMed]
- Lalrempuia, R.; Stasch, A.; Jones, C. An extremely bulky tris(pyrazolyl)methanide: A tridentate ligand for the synthesis of heteroleptic magnesium(II) and ytterbium(II) alkyl, hydride, and iodide complexes. Chem. Asian J. 2015, 10, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Harder, S. Molecular early main group metal hydrides: Synthetic challenge, structures and applications. Chem. Commun. 2012, 48, 11165–11177. [Google Scholar] [CrossRef] [PubMed]
- Grochalla, W.; Edwards, P.P. Thermal decomposition of the non-interstitial hydrides for the storage and production of hydrogen. Chem. Rev. 2004, 104, 1283–1315. [Google Scholar] [CrossRef] [PubMed]
- Harder, S.; Spielmann, J.; Intemann, J.; Bandmann, H. Hydrogen storage in magnesium hydride: The molecular approach. Angew. Chem. Int. Ed. 2011, 50, 4156–4160. [Google Scholar] [CrossRef] [PubMed]
- CrystalClear-SM Expert v2.1; Rigaku Americas: The Woodlands, TX, USA; Rigaku Corporation: Tokyo, Japan, 2013.
- Cowieson, N.P.; Aragao, D.; Clift, M.; Ericsson, D.J.; Gee, C.; Harrop, S.J.; Mudie, N.; Panjikar, S.; Price, J.R.; Riboldi-Tunnicliffe, A.; et al. MX1: A bending-magnet crystallography beamline serving both chemical and macromolecular crystallography communities at the Australian Synchrotron. J. Synchrotron Radiat. 2015, 22, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Cryst. 2015, C71, 9–18. [Google Scholar]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sindlinger, C.P.; Lawrence, S.R.; Cordes, D.B.; Slawin, A.M.Z.; Stasch, A. Methanediide Formation via Hydrogen Elimination in Magnesium versus Aluminium Hydride Complexes of a Sterically Demanding Bis(iminophosphoranyl)methanediide. Inorganics 2017, 5, 29. https://doi.org/10.3390/inorganics5020029
Sindlinger CP, Lawrence SR, Cordes DB, Slawin AMZ, Stasch A. Methanediide Formation via Hydrogen Elimination in Magnesium versus Aluminium Hydride Complexes of a Sterically Demanding Bis(iminophosphoranyl)methanediide. Inorganics. 2017; 5(2):29. https://doi.org/10.3390/inorganics5020029
Chicago/Turabian StyleSindlinger, Christian P., Samuel R. Lawrence, David B. Cordes, Alexandra M. Z. Slawin, and Andreas Stasch. 2017. "Methanediide Formation via Hydrogen Elimination in Magnesium versus Aluminium Hydride Complexes of a Sterically Demanding Bis(iminophosphoranyl)methanediide" Inorganics 5, no. 2: 29. https://doi.org/10.3390/inorganics5020029
APA StyleSindlinger, C. P., Lawrence, S. R., Cordes, D. B., Slawin, A. M. Z., & Stasch, A. (2017). Methanediide Formation via Hydrogen Elimination in Magnesium versus Aluminium Hydride Complexes of a Sterically Demanding Bis(iminophosphoranyl)methanediide. Inorganics, 5(2), 29. https://doi.org/10.3390/inorganics5020029