Abstract
Cyclometalated Ir(III) complexes are of particular interest due to the wide tunability of their electronic structure via variation of their ligands. Here, a series of heteroleptic Ir-based photosensitizers with the general formula has been studied theoretically by means of an optimally-tuned long-range separated density functional. Focusing on the steady-state absorption spectra, correlations between the chemical modification of both ligand types with the natures of the relevant dark and bright electronic states are revealed. Understanding such correlations builds up a basis for the rational design of efficient photocatalytic systems.
1. Introduction
Exploring alternative renewable energy sources represents one of the most prominent tasks for natural sciences due to the world’s steadily growing energy consumption [1]. The conversion of solar energy into appropriate chemical forms and its storage, e.g., in form of hydrogen, attracts particular attention, as sun light meets all ecological and power requirements for renewable energy sources [2,3,4]. Although the history of sun-light driven conversion of water into hydrogen started with heterogeneous systems (see, e.g., [5,6]), homogeneous photocatalysis has been developed and extensively studied [7,8,9,10]. The key substance in the whole process is a photostable material, a photosensitizer (PS), capable to effectively absorb visible light and to live long enough in an excited state of charge-transfer nature in order to initiate separate oxidation and reduction half-reactions. The most popular PSs are noble-metal–organic complexes of ruthenium, platinum, rhenium, and iridium, although various organic dyes, porphyrins, manganese and copper complexes have been proposed as well (see reviews [9,10,11,12] and references therein). The rational design and further improvement of photocatalytic systems for hydrogen generation requires a mechanistic understanding of all underlying processes [13] and in particular, a detailed characterization of the excited electronic states of the PSs.
In the present article, we study a number of derivatives from the family of Ir(III)-based PSs with the general formula (sketched in Figure 1), where denotes (substituted) phenyl pyridine (ppy) ligands and stands for (substituted) bipyridine (bpy) ligands introduced by Bernhard et al. [14,15,16,17,18,19]. The unsubstituted is abbreviated as IrPS in the following. Electronic transitions and charge-transfer (CT) properties of Ir(III) PSs can be flexibly tuned by chemical modification of the ligands (for a review, see [13]). The parent IrPS and its derivatives were applied in the photocatalytic scheme for water reduction half-reaction developed by Beller et al. [20]. It comprises a heteroleptic Ir(III)-based PS, triethylamine as a sacrificial reductant, and a series of iron carbonyl compounds as water reduction catalysts. A number of catalytic, EPR, spectroscopic and theoretical studies of these systems has been already published [11,21,22,23,24,25,26,27,28,29,30,31,32,33]. Among these works, mostly IrPS was in the focus of joint experimental/theoretical studies for investigation of the underlying mechanisms. In this communication, previously described and synthesized Ir-based PSs are studied theoretically in detail. The aim is to reveal the influence of chemical modification by ligand variation onto the characteristics of the bright and dark states of these photosensitizers.
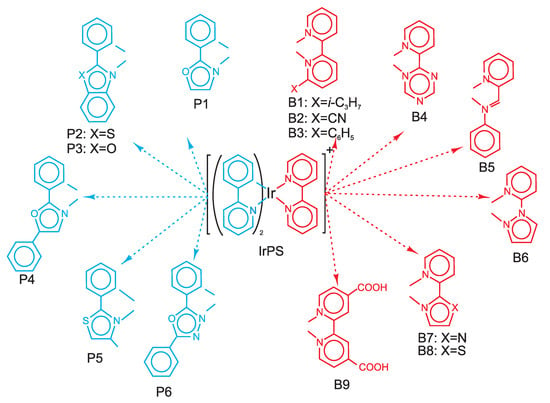
Figure 1.
Structure formulae and notations of all photosensitizers (PSs) studied in this work.
The paper is organized as follows. First, we analyze absorption spectra and discuss peculiarities of excited states structures; Second, we provide computational and experimental details. We close the discussion with an outlook and conclusions.
2. Results and Discussion
Previously, on the basis of a multi-spectroscopic/computational study, the ppy ligand has been identified as being relevant for the properties of the first bright transition in IrPS, whereas the bpy ligand has been found to be important as an electron reservoir storing the electron after photoreduction by a sacrificial reductant [27]. Thus, chemical modification of these ligands separately could shed light on how these properties can be manipulated, thus paving the way for a rational design of future PSs. The chemical structures of the derivatives of both ppy and bpy ligands is sketched in Figure 1. The parent complex is denoted as IrPS, whereas the PSs containing modified ppy and bpy ligands are denoted as Pn and Bn, respectively. For some cases, B1–B3 and B9, chemical derivation represents addition of some donor or acceptor groups to the original ligand. For most of the cases, however, the entire aromatic system of the ligand is modified by introducing heteroatoms and changing the size of the conjugated aromatic system. These derivatives were indicated as potentially perspective for photocatalytic applications, see Refs. [21,22,33].
As a first step, we have optimized the range-separation parameter. For all PSs studied in this work was found to be 0.17–0.19 bohr. For the parent IrPS, its complexes with silver clusters, triethylamine, and iron carbonyls, a similar value of about 0.18 bohr has been obtained previously [11,28,31]. LC-BLYP with optimal was shown to provide the best agreement with experimental spectra of the oxidized and reduced IrPS and, simultaneously, to increase the triplet stability of the ground state solution, which is essential for the accuracy of the excited states energies [31]. The obtained value is notably smaller than standard parameters (0.33 bohr [34] and 0.47 bohr [35]) obtained for training sets of atoms or small molecules and implemented as default values in quantum-chemical codes. The decrease of the optimal value with increasing system size has been previously found for numerous cases (see e.g., Refs. [36,37,38,39]), and rationalized by the fact that defines a characteristic distance for switching between short- and long-range parts of the Coulomb interaction. Thus, the values obtained here for different species were found to be very close to the parent IrPS case, since the characteristic extent of the electron density, conjugation length, and inter-electronic distances are similar. As a note in caution we add that one should not count on such similarity to hold true in general.
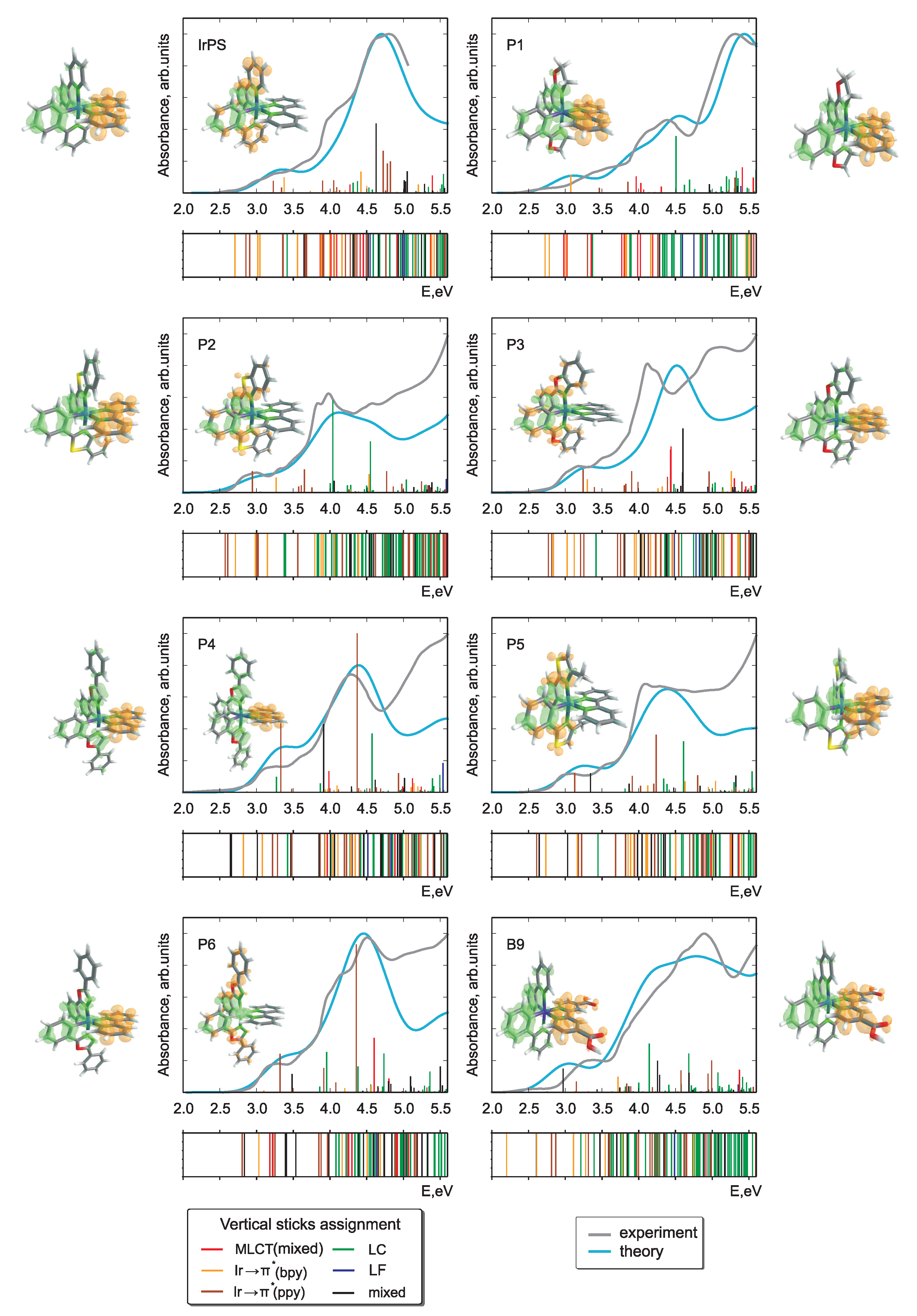
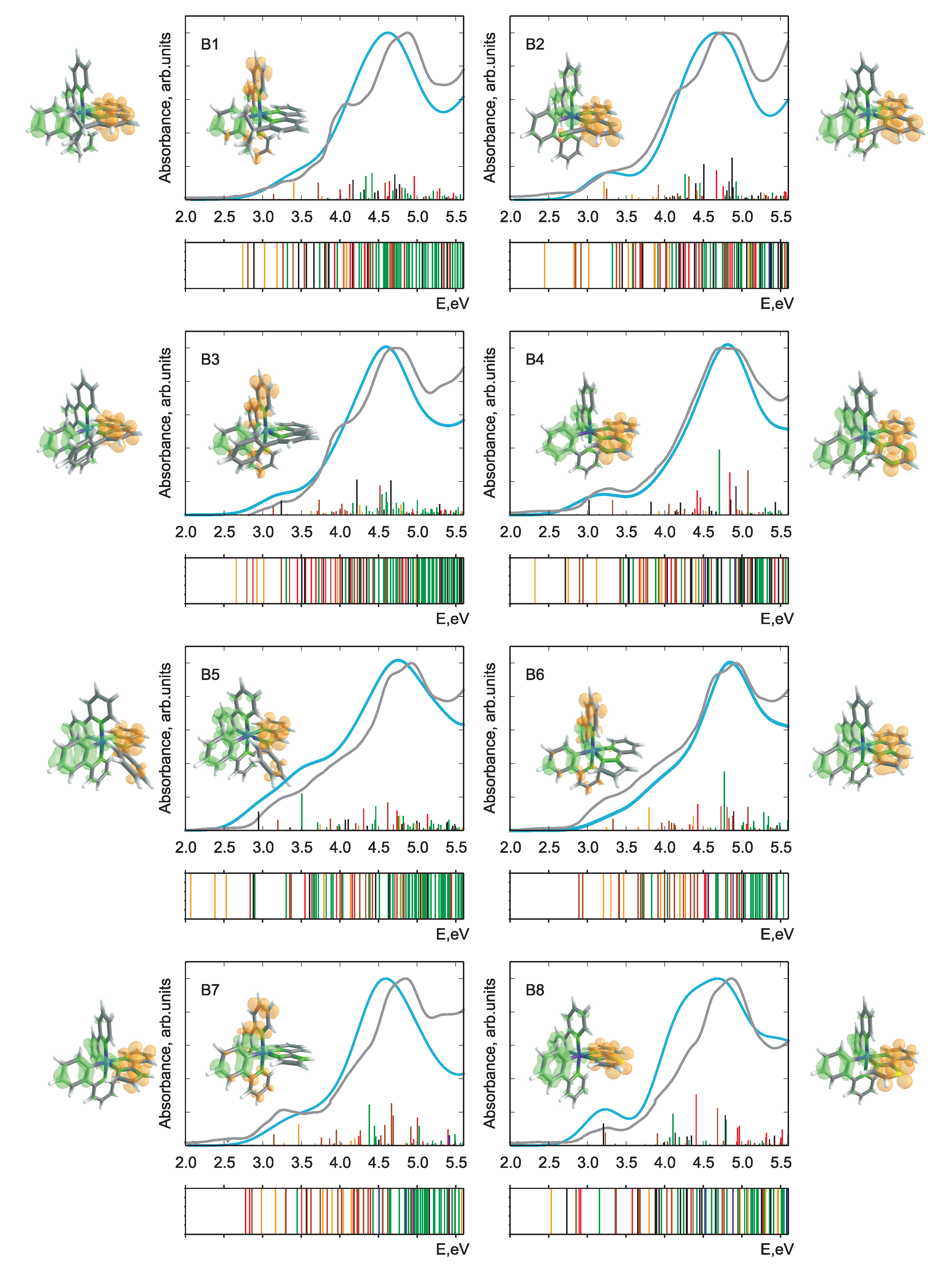
In Figure 2 and Figure 3, the calculated and experimental absorption spectra are presented for Pn and Bn derivatives. The respective stick spectra are also shown, with the sticks’ colors denoting transitions of different character. The assignment was done in a fully automatized way on the basis of an analysis of configuration interaction coefficients and orbital contributions stemming from different moieties. Here, we distinguish between ligand-centered, LC, (both intraligand and ligand-to-ligand charge-transfer excitations are included), iridium ligand-field, LF, and three types of metal-to-ligand charge-transfer, MLCT, transitions. The latter includes Ir (ppy), Ir (bpy), and mixed MLCT transitions denoted collectively as MLCT(mixed). In addition, there is a number of transitions (denoted as mixed) having a very complicated character which cannot be classified into the above-mentioned groups. Such an assignment was done to simplify the analysis in terms of ligands acquiring an electron in course of photoexcitation to MLCT states. Since the singlet-triplet intensities were not calculated, the energetic positions of the triplet states are presented as bars below each spectrum with the same color code as for singlet states. It should be noted that the nature of the lowest-lying triplet states is of primary importance for light-emitting devices and photocatalytic reactions. However, the main focus in this communication is put on the absorption properties, where the properties of singlet states are of primary interest [40,41,42].
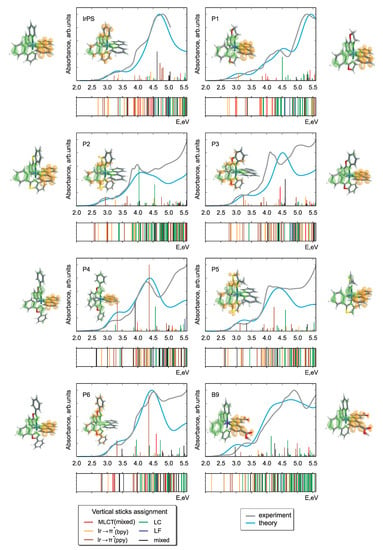
Figure 2.
Theoretical singlet-singlet and experimental absorption spectra of IrPS and all its Pn-derivatives (one bpy-derivative is shown as well). The calculated singlet-triplet transitions are presented as bars below the respective graphs. The color of the stick spectra denote different types of electronic transitions, see text. Also shown are transition density differences for the first dark (aside) and first bright (inset) singlet-singlet transitions. The green and orange colors denote the areas of electron lack and gain upon the respective excitation.
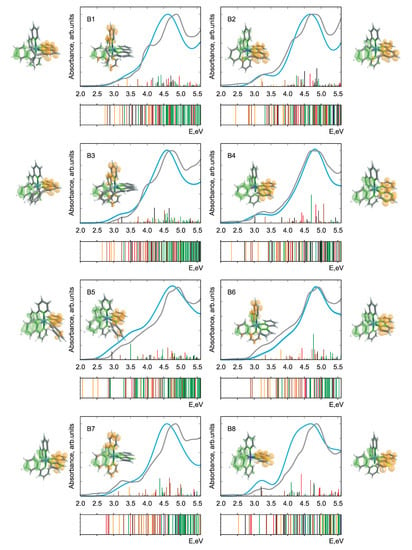
Figure 3.
Theoretical and experimental absorption spectra of all Bn-derivatives of the parent IrPS. For notation see caption of Figure 2.
Overall, we note that there is a fairly good agreement of the calculated spectra with experiment. This holds true despite the fact that singlet-triplet contributions are omitted, vibronic structure of the transitions is not included, and a single width for broadening was applied without attempting to fit to the experiment. Thus, one may conclude that the optimally-tuned range-separation functional provides accurate information on the excited states, building a basis for further detailed analysis. This is remarkable since in the SCF procedure energies of only the HOMO and LUMO are self-consistently adjusted. Nevertheless, such tuning leads to a reliable prediction also of higher-lying transitions involving deeper occupied and higher unoccupied orbitals.
The photochemical fate of the photosensitizer is determined first of all by the character of the lowest bright transition as well as by lower-lying dark states. Concerning the performance, the first bright transition is of particular importance because the sun’s spectrum has minute overlap with the high-energy part of the absorption spectrum. Thus, one of the conditions to improve the efficiency of PSs is to ensure a notable spectral overlap, what requires shifting of the bright transitions below 3.7 eV. However, this energy should be larger than the theoretical limit of 1.23 eV required to reduce HO to hydrogen in course of the subsequent steps of the catalytic reaction. Difference densities for the mentioned transitions are shown in Figure 2 and Figure 3. More detailed information on the natures of first dark and first bright singlet-singlet transitions is presented in Table 1.

Table 1.
Energies and assignment of the first dark and first bright singlet-singlet transitions for all compounds. The character is given in terms of leading orbitals with respect to highest occupied molecular orbitals (HOMO) (H) and lowest unoccupied molecular orbitals (LUMO) (L).
Regardless of the chemical derivation, the HOMO and LUMO orbitals always correspond to d and *(bpy) MOs, respectively. The first dark transition can be unambiguously assigned to a HOMO-LUMO transition in all cases (see Table 1 and compare density difference plots given as the right-most and left-most columns in Figure 2 and Figure 3), whereas the bright state shows more variability (see density differences in the insets in Figure 2 and Figure 3). Although for the parent IrPS the d*(ppy) transition is the lowest bright one, the modification of both ligands can change its nature. For about half of the derivatives, the excitation ends up at a *(bpy) orbital and the donor orbitals change to d or d of Ir, (ppy) or even to mixture of both cases. Albeit the fact that the molecules are in general asymmetric, i.e., no symmetry-based selection rules can be applied, the dark states correspond to transitions between orbitals lying in the same plane, as, e.g., d*(bpy). In turn, bright states correspond to transitions where donor and acceptor orbitals are not coplanar. For instance, this holds true for the transitions d*(ppy), d*(bpy), and (ppy) *(bpy).
The nature of the relevant transitions can be roughly correlated with the type of the chemical modification of the original ligands. Apart from adding side donor or acceptor groups (–i-CH, –Ph, –COOH, –CN) to the initial bpy or ppy ligands, the aromatic system is modified by introducing heteroatoms. The sp pyridinic nitrogen atoms slightly increase the aromatic stabilization energy, whereas the sp pyrolic nitrogen or sp oxygen and sulfur heteroatoms decrease it. For the Pn series, this implies that apart from P2 and P3 the energy of the d*(ppy) transition shifts to the blue and may become higher than the bright d*(bpy) ones. In such situations, the nature of the lowest bright transitions changes. For the Bn series, the introduction of acceptor groups or the increase of aromaticity of the “bpy” ligand leads to a change of character of the first bright transition to d*(bpy), see Table 1. The only exception is B8.
The energies of the bright states slightly vary in the series of PSs. For most species, a slight red shift up to 0.3 eV with respect to the parent IrPS can be observed. Exceptions being B6, P4, and P6 where a small blue shift up to 0.1 eV occurs. This implies that the spectral overlap with the sun light should be enhanced upon certain chemical modifications, but a dramatic increase of the absorption efficiency cannot be achieved without a principal change of the class of PSs. The transition energies of the lowest dark state mostly vary with changing the bpy ligand, dropping by up to 0.06 eV in case of B5, although the derivation of the ppy ligand may also influence its energy as in case of P6. The relative intensities of the two considered transitions for each compound vary by a factor from 6 to 270.
Interestingly, the modification of one ligand may notably influence the transitions involving other parts of the complex. This can be caused by general changes in the orbital shapes due to the changing donor or acceptor character of the ligands. As an illustration one can look at the character of doubly-occupied 5d-orbitals of IrPS. In Table 2, the percentage of localization on the central Ir-atom is presented. The extent of d-character lies between 35% and 67% pointing to the partial charge delocalization even without electronic excitation. For some cases, even 4 or 5 orbitals in the occupied subspace near the HOMO can be ascribed as d-ones mixed with some orbitals of the ligands.
Table 2.
Percentage of localization of electron density on d orbitals of Ir for all compounds. In parenthesis, the character is given in terms of leading orbitals with respect to HOMO (H).
The variation of CT character can be most easily seen on the example of triplet states given as bars under the respective spectra in Figure 2 and Figure 3. For most derivatives, a high density of the LC states is observed for energies above 4 eV. However, for P2, B5, and B9 they systematically appear at notably lower energies. LC states in general can be expected to produce charge separation in the molecule insufficient to initiate further photocatalytic redox reactions. Such changes can also explain the strong variation of transition intensities mentioned above.
Note also that the environment, due to, e.g., solvatochromic and rigidochromic effects, could also influence the relative positions of LC and MLCT states, see Ref. [43]. Although for emission from a single or a small group of states this can have a pronounced effect, we found that the general shape of the absorption spectrum does not change much when e.g., comparing acetonitrile with tetrahydrofuran for the case of IrPS (data not shown). The positions of individual lines can vary somewhat but due to their large number and significant broadening this influence is mitigated.
3. Computational Details
Density functional theory (DFT) together with the long-range corrected functional LC-BLYP [34,44,45] was chosen as the main theoretical approach. Although DFT is a well-known approach for investigation of ground and excited-state properties of relatively large systems [46] the CT properties of metal–organic complexes were shown to be strongly dependent on the exchange-kernel [25,47]. In case of widely used local and hybrid functionals, DFT suffers from incorrect description of long-range CT states [48,49], which are of primary interest for photocatalytic applications of metal–organic complexes, since the catalytic cycle necessarily includes intra- or inter-molecular charge separation. The problem might be solved by using long-range corrected functionals, where exact Hartree-Fock exchange is used at long distances providing the correct asymptotic behavior [50,51,52,53,54,55]. This scheme introduces an additional parameter, , determining the range of switching between short- and long-range exchange contributions. In general, this parameter depends on the electronic density and, thus, is system-dependent. A fully ab initio procedure for tuning the range-separation parameter is provided by the SCF method. It ensures that Koopmans’ theorem is fulfilled for the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO) [53,56,57]. The details of the particular implementation of the protocol applied here can be found elsewhere [31].
All PSs have been studied using the following computational strategy. First, the ground state geometry in vacuum has been optimized with the standard value of the range-separation parameter (0.47 bohr). For this geometry, the optimal tuning procedure has been applied. Second, the geometry has been re-optimized with the corresponding optimal value of and including the solvent (THF) via the polarizable continuum model (PCM). All minimal configurations have been checked for the absence of imaginary frequencies by harmonic frequency analysis. For the calculations of the lowest 200 singlet and 200 triplet states, the standard linear-response time-dependent DFT (TDDFT) formalism was utilized. However, the intensities of the spin-forbidden singlet-triplet transitions were not calculated, although Ir has large spin-orbit coupling constant. Recently, it was shown that singlet/triplet mixing in similar Ir photosensitizers has only a minor impact on the absorption spectra and results in small red shifts of the bands in the visible range and a decrease of intensities as triplet states possess small oscillator strengths [40,41,42]. As we have not aimed at fitting spectral widths for various PSs and transitions of different nature, a Gaussian broadening with a universal parameter (0.25 eV) has been applied to all stick-spectra. For DFT/TDDFT calculations no symmetry restrictions were applied. All calculations have been performed using the LANL2DZ ECP basis set for Ir and the 6-31G(d) basis set for all other atoms. Solvent effects on transitions have been included within the PCM approach [58]. All calculations were done with the Gaussian09 suite of programs [59]. Pre- and post-processing of data has been done with homemade programs.
4. Experimental Details
Steady-state UV/Vis absorption spectra of Pn compounds were recorded with a Specord 50 spectrophotometer (Analytik Jena, Jena, Germany) at room temperature. The photosensitizers were dissolved with a concentration of about 10 M in tetrahydroforan of Uvasol-quality (Merck, Darmstadt, Germany). The solutions were filled in a fused silica cuvette with an optical path length of 1 cm. Experimental spectra of B1–B8 and B9 in acetonitrile were taken from Refs. [21,33], respectively.
5. Conclusions
In the present work, we have explored ground and excited state properties of a series of Ir-based photosensitizers, which are derivatives of the parent IrPS extensively investigated before in catalytic, spectroscopic, and theoretical studies [11,21,22,23,24,25,26,27,28,29,30,31,32,33]. To facilitate an accurate description of the excited states, the optimally-tuned range-separated density functional approach has been chosen. This method has demonstrated a very good agreement of calculated absorption spectra with experiments and thus provides a good basis for theoretical analysis. This holds true especially for CT excitations, where conventional density functionals are known to fail. Chemical modification of the parent IrPS demonstrates a number of trends, with some of them being non-trivial. For instance, modification of one ligand can notably influence the transitions involving other parts of the photosensitizer. In general, the most important bright singlet-singlet transition is of Ir *(ppy) type and chemical derivation may shift it both to the red (what is a favorable scenario to enhance absorption efficiency due to the better overlap with the sun spectrum) or to the blue. In the latter case the nature of the lowest bright transition may change.
To summarize, we have demonstrated that the chosen method can provide an in-depth understanding of the correlations between chemical structure and photochemical properties of photosensitizers. This is an important prerequisite for the rational design of efficient photocatalytic systems.
Acknowledgments
We are grateful to Nils Rockstroh for experimental absorption spectra of B1–B8 and Stefanie Tschierlei for providing experimental spectrum for B9. We would also like to thank Felix Gärtner, Daniela Cozzula, Stefania Denurra, Sebastian Losse, Gopinatan Anilkumar, Henrik Junge and Matthias Beller from Leibniz Institute for Catalysis for synthesis of the studied PSs.
Author Contributions
Olga S. Bokareva and Tobias Möhle performed theoretical computations, Antje Neubauer measured absorption spectra, Sergey I. Bokarev, Oliver Kühn and Stefan Lochbrunner designed and coordinated the project. All authors were involved in writing the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schiermeier, Q.; Tollefson, J.; Scully, T.; Witze, A.; Morton, O. Energy alternatives: Electricity without carbon. Nature 2008, 454, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Esswein, A.J.; Nocera, D.G. Hydrogen production by molecular photocatalysis. Chem. Rev. 2007, 107, 4022–4047. [Google Scholar] [CrossRef] [PubMed]
- Blankenship, R.E.; Tiede, D.M.; Barber, J.; Brudvig, G.W.; Fleming, G.; Ghirardi, M.; Gunner, M.R.; Junge, W.; Kramer, D.M.; Melis, A.; et al. Comparing photosynthetic and photovoltaic efficiencies and recognizing the potential for improvement. Science 2011, 332, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Hambourger, M.; Moore, G.F.; Kramer, D.M.; Gust, D.; Moore, A.L.; Moore, T.A. Biology and technology for photochemical fuel production. Chem. Soc. Rev. 2009, 38, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Burschka, J.; Pellet, N.; Moon, S.J.; Humphry-Baker, R.; Gao, P.; Nazeeruddin, M.K.; Grätzel, M. Sequential deposition as a route to high-performance perovskite-sensitized solar cells. Nature 2013, 499, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.A.; Bahnemann, D.W. Photochemical splitting of water for hydrogen production by photocatalysis: A review. Sol. Ener. Mat. Sol. Cells 2014, 128, 85–101. [Google Scholar] [CrossRef]
- Wang, M.; Na, Y.; Gorlov, M.; Sun, L. Light-driven hydrogen production catalysed by transition metal complexes in homogeneous systems. Dalton Trans. 2009, 33, 6458–6467. [Google Scholar] [CrossRef] [PubMed]
- Teets, T.S.; Nocera, D.G. Photocatalytic hydrogen production. Chem. Comm. 2011, 47, 9268–9274. [Google Scholar] [CrossRef] [PubMed]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-sensitized solar cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef] [PubMed]
- Eckenhoff, W.T.; Eisenberg, R. Molecular systems for light driven hydrogen production. Dalton Trans. 2012, 41, 13004–13021. [Google Scholar] [CrossRef] [PubMed]
- Bokarev, S.I.; Bokareva, O.S.; Kühn, O. A theoretical perspective on charge transfer in photocatalysis. The example of Ir-based systems. Coord. Chem. Rev. 2015, 304–305, 133–145. [Google Scholar] [CrossRef]
- Junge, H.; Rockstroh, N.; Fischer, S.; Brückner, A.; Ludwig, R.; Lochbrunner, S.; Kühn, O.; Beller, M. Light to Hydrogen: Photocatalytic Hydrogen Generation from Water with Molecularly-Defined Iron Complexes. Inorganics 2017, 5, 14. [Google Scholar] [CrossRef]
- You, Y.; Nam, W. Photofunctional triplet excited states of cyclometalated Ir(III) complexes: Beyond electroluminescence. Chem. Soc. Rev. 2012, 41, 7061–7084. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, J.I.; Hudson, W.R.; Lowry, M.S.; Anderson, T.H.; Bernhard, S. Discovery and high-throughput screening of heteroleptic iridium complexes for photoinduced hydrogen production. J. Am. Chem. Soc. 2005, 127, 7502–7510. [Google Scholar] [CrossRef] [PubMed]
- Lowry, M.S.; Goldsmith, J.I.; Slinker, J.D.; Rohl, R.; Pascal, R.A.; Malliaras, G.G.; Bernhard, S. Single-Layer Electroluminescent Devices and Photoinduced Hydrogen Production from an Ionic Iridium(III) Complex. Chem. Mater. 2005, 17, 5712–5719. [Google Scholar] [CrossRef]
- Tinker, L.L.; McDaniel, N.D.; Curtin, P.N.; Smith, C.K.; Ireland, M.J.; Bernhard, S. Visible light induced catalytic water reduction without an electron relay. Chem. Eur. J. 2007, 13, 8726–8732. [Google Scholar] [CrossRef] [PubMed]
- Cline, E.D.; Adamson, S.E.; Bernhard, S. Homogeneous catalytic system for photoinduced hydrogen production utilizing iridium and rhodium complexes. Inorg. Chem. 2008, 47, 10378–10388. [Google Scholar] [CrossRef] [PubMed]
- Tinker, L.L.; Bernhard, S. Photon-driven catalytic proton reduction with a robust homoleptic iridium(III) 6-phenyl-2,2′-bipyridine complex ([Ir(C–N–N)2)]+). Inorg. Chem. 2009, 48, 10507–10511. [Google Scholar] [CrossRef] [PubMed]
- Curtin, P.N.; Tinker, L.L.; Burgess, C.M.; Cline, E.D.; Bernhard, S. Structure-activity correlations among iridium(III) photosensitizers in a robust water-reducing system. Inorg. Chem. 2009, 48, 10498–10506. [Google Scholar] [CrossRef] [PubMed]
- Gärtner, F.; Sundararaju, B.; Surkus, A.E.; Boddien, A.; Loges, B.; Junge, H.; Dixneuf, P.H.; Beller, M. Light-driven hydrogen generation: Efficient iron-based water reduction catalysts. Angew. Chem. Int. Ed. 2009, 48, 9962–9965. [Google Scholar] [CrossRef] [PubMed]
- Gärtner, F.; Cozzula, D.; Losse, S.; Boddien, A.; Anilkumar, G.; Junge, H.; Schulz, T.; Marquet, N.; Spannenberg, A.; Gladiali, S.; et al. Synthesis, characterisation and application of iridium(III) photosensitisers for catalytic water reduction. Chem. Eur. J. 2011, 17, 6998–7006. [Google Scholar] [CrossRef] [PubMed]
- Gärtner, F.; Boddien, A.; Barsch, E.; Fumino, K.; Losse, S.; Junge, H.; Hollmann, D.; Brückner, A.; Ludwig, R.; Beller, M. Photocatalytic hydrogen generation from water with iron carbonyl phosphine complexes: Improved water reduction catalysts and mechanistic insights. Chemistry 2011, 17, 6425–6436. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, D.; Gärtner, F.; Ludwig, R.; Barsch, E.; Junge, H.; Blug, M.; Hoch, S.; Beller, M.; Brückner, A. Insights into the mechanism of photocatalytic water reduction by DFT-supported in situ EPR/Raman spectroscopy. Angew. Chem. Int. Ed. 2011, 50, 10246–10250. [Google Scholar] [CrossRef] [PubMed]
- Gärtner, F.; Denurra, S.; Losse, S.; Neubauer, A.; Boddien, A.; Gopinathan, A.; Spannenberg, A.; Junge, H.; Lochbrunner, S.; Blug, M.; et al. Synthesis and characterization of new iridium photosensitizers for catalytic hydrogen generation from water. Chem. Eur. J. 2012, 18, 3220–3225. [Google Scholar] [CrossRef] [PubMed]
- Bokarev, S.I.; Bokareva, O.S.; Kühn, O. Electronic excitation spectrum of the photosensitizer [Ir(ppy)2(bpy)]+. J. Chem. Phys. 2012, 136, 214305. [Google Scholar] [CrossRef] [PubMed]
- Bokareva, O.S.; Bokarev, S.I.; Kühn, O. Electronic excitation spectra of the [Ir(ppy)2(bpy)]+ photosensitizer bound to small silver clusters Agn (n = 1–6). Phys. Chem. Chem. Phys. 2012, 14, 4977–4984. [Google Scholar] [CrossRef] [PubMed]
- Bokarev, S.I.; Hollmann, D.; Pazidis, A.; Neubauer, A.; Radnik, J.; Kühn, O.; Lochbrunner, S.; Junge, H.; Beller, M.; Brückner, A. Spin density distribution after electron transfer from triethylamine to an [Ir(ppy)2(bpy)]+ photosensitizer during photocatalytic water reduction. Phys. Chem. Chem. Phys. 2014, 16, 4789–4796. [Google Scholar] [CrossRef] [PubMed]
- Bokareva, O.S.; Kühn, O. DFT-D investigation of the interaction between Ir (III) based photosensitizers and small silver clusters Agn (n = 2–20, 92). Chem. Phys. 2014, 435, 40–48. [Google Scholar] [CrossRef]
- Neubauer, A.; Grell, G.; Friedrich, A.; Bokarev, S.I.; Schwarzbach, P.; Gärtner, F.; Surkus, A.E.; Junge, H.; Beller, M.; Kühn, O.; et al. Electron- and Energy-Transfer Processes in a Photocatalytic System Based on an Ir(III)-Photosensitizer and an Iron Catalyst. J. Phys. Chem. Lett. 2014, 5, 1355–1360. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Hollmann, D.; Tschierlei, S.; Karnahl, M.; Rockstroh, N.; Barsch, E.; Schwarzbach, P.; Luo, S.P.; Junge, H.; Beller, M.; et al. Death and Rebirth: Photocatalytic Hydrogen Production by a Self-Organizing Copper–Iron System. ACS Catal. 2014, 4, 1845–1849. [Google Scholar] [CrossRef]
- Bokareva, O.S.; Grell, G.; Bokarev, S.I.; Kühn, O. Tuning Range-Separated Density Functional Theory for Photocatalytic Water Splitting Systems. J. Chem. Theor. Comp. 2015, 11, 1700–1709. [Google Scholar] [CrossRef] [PubMed]
- Bokareva, O.S.; Kühn, O. Quantum chemical study of the electronic properties of an Iridium-based photosensitizer bound to medium-sized silver clusters. Chem. Phys. 2015, 457, 1–6. [Google Scholar] [CrossRef]
- Tschierlei, S.; Neubauer, A.; Rockstroh, N.; Karnahl, M.; Schwarzbach, P.; Junge, H.; Beller, M.; Lochbrunner, S. Ultrafast excited state dynamics of iridiumIII complexes and their changes upon immobilisation onto titanium dioxide layers. Phys. Chem. Chem. Phys. 2016, 18, 10682–10687. [Google Scholar] [CrossRef] [PubMed]
- Tawada, Y.; Tsuneda, T.; Yanagisawa, S.; Yanai, T.; Hirao, K. A long-range-corrected time-dependent density functional theory. J. Chem. Phys. 2004, 120, 8425–8433. [Google Scholar] [CrossRef] [PubMed]
- Song, J.W.; Hirosawa, T.; Tsuneda, T.; Hirao, K. Long-range corrected density functional calculations of chemical reactions: Redetermination of parameter. J. Chem. Phys. 2007, 126, 154105. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.; Eisenberg, H.; Kronik, L.; Baer, R. Fundamental Gaps in Finite Systems from Eigenvalues of a Generalized Kohn-Sham Method. Phys. Rev. Lett. 2010, 105, 266802. [Google Scholar] [CrossRef] [PubMed]
- Refaely-Abramson, S.; Baer, R.; Kronik, L. Fundamental and excitation gaps in molecules of relevance for organic photovoltaics from an optimally tuned range-separated hybrid functional. Phys. Rev. B 2011, 84, 075144. [Google Scholar] [CrossRef]
- Körzdörfer, T.; Sears, J.S.; Sutton, C.; Brédas, J.L. Long-range corrected hybrid functionals for π-conjugated systems: Dependence of the range-separation parameter on conjugation length. J. Chem. Phys. 2011, 135, 204107. [Google Scholar] [CrossRef] [PubMed]
- Salzner, U.; Baer, R. Koopmans’ springs to life. J. Chem. Phys. 2009, 131, 231101. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Minaev, B.; Ågren, H.; Tian, H. Theoretical Study of Phosphorescence of Iridium Complexes with Fluorine-Substituted Phenylpyridine Ligands. Eur. J. Inorg. Chem. 2011, 2011, 2517–2524. [Google Scholar] [CrossRef]
- Minaev, B.; Baryshnikov, G.; Agren, H. Principles of phosphorescent organic light emitting devices. Phys. Chem. Chem. Phys. 2014, 16, 1719–1758. [Google Scholar] [CrossRef] [PubMed]
- Brahim, H.; Daniel, C. Structural and spectroscopic properties of Ir(III) complexes with phenylpyridine ligands: Absorption spectra without and with spin–orbit-coupling. Comp. Theor. Chem. 2014, 1040–1041, 219–229. [Google Scholar] [CrossRef]
- Colombo, M.; Hauser, A.; Güdel, H. Competition between ligand centered and charge transfer lowest excited states in bis cyclometalated Rh3+ and Ir3+ complexes. In Electronic and Vibronic Spectra of Transition Metal Complexes I Topics in Current Chemistry; Springer: Heidelberg, Germany, 1994; Volume 171, pp. 143–171. [Google Scholar]
- Iikura, H.; Tsuneda, T.; Yanai, T.; Hirao, K. A long-range correction scheme for generalized-gradient- approximation exchange functionals. J. Chem. Phys. 2001, 115, 3540–3544. [Google Scholar] [CrossRef]
- Chiba, M.; Tsuneda, T.; Hirao, K. Excited state geometry optimizations by analytical energy gradient of long-range corrected time-dependent density functional theory. J. Chem. Phys. 2006, 124, 144106. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J.A. General performance of density functionals. J. Phys. Chem. A 2007, 111, 10439–10452. [Google Scholar] [CrossRef] [PubMed]
- Laurent, A.D.; Jacquemin, D. TD-DFT benchmarks: A review. Int. J. Quant. Chem. 2013, 113, 2019–2039. [Google Scholar] [CrossRef]
- Dreuw, A.; Head-Gordon, M. Single-reference ab initio methods for the calculation of excited states of large molecules. Chem. Rev. 2005, 105, 4009–4037. [Google Scholar] [CrossRef] [PubMed]
- Peach, M.J.G.; Helgaker, T.; Salek, P.; Keal, T.W.; Lutnaes, O.B.; Tozer, D.J.; Handy, N.C. Assessment of a Coulomb-attenuated exchange-correlation energy functional. Phys. Chem. Chem. Phys. 2006, 8, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Gerber, I.C.; Ángyán, J.G. Hybrid functional with separated range. Chem. Phys. Lett. 2005, 415, 100–105. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density Functional for Spectroscopy: No Long-Range Self-Interaction Error, Good Performance for Rydberg and Charge-Transfer States, and Better Performance on Average than B3LYP for Ground States. J. Phys. Chem. A 2006, 110, 13126–13130. [Google Scholar] [CrossRef] [PubMed]
- Peach, M.J.G.; Benfield, P.; Helgaker, T.; Tozer, D.J. Excitation energies in density functional theory: An evaluation and a diagnostic test. J. Chem. Phys. 2008, 128, 44118. [Google Scholar] [CrossRef] [PubMed]
- Livshits, E.; Baer, R. A well-tempered density functional theory of electrons in molecules. Phys. Chem. Chem. Phys. 2007, 9, 2932–2941. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Phys. Chem. 2008, 128, 084106. [Google Scholar] [CrossRef] [PubMed]
- Mori-Sánchez, P.; Cohen, A.J.; Yang, W. Self-interaction-free exchange-correlation functional for thermochemistry and kinetics. J. Chem. Phys. 2006, 124, 91102. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.; Kronik, L.; Baer, R. Reliable Prediction of Charge Transfer Excitations in Molecular Complexes Using Time-Dependent Density Functional Theory. J. Am. Chem. Soc. 2009, 131, 2818–2820. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.; Kronik, L.; Baer, R. Prediction of charge-transfer excitations in coumarin-based dyes using a range-separated functional tuned from first principles. J. Chem. Phys. 2009, 131, 244119. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision C.1; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).