
Insights into Molecular Beryllium–Silicon Bonds

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. NMR-Spectroscopy
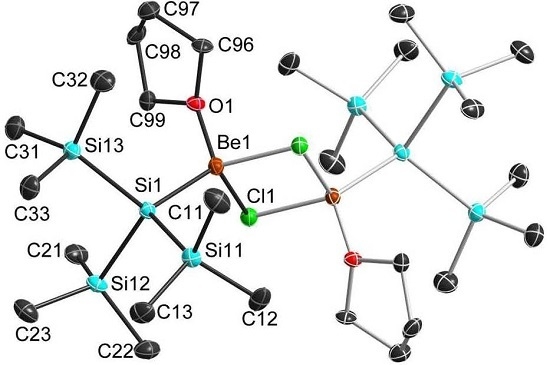
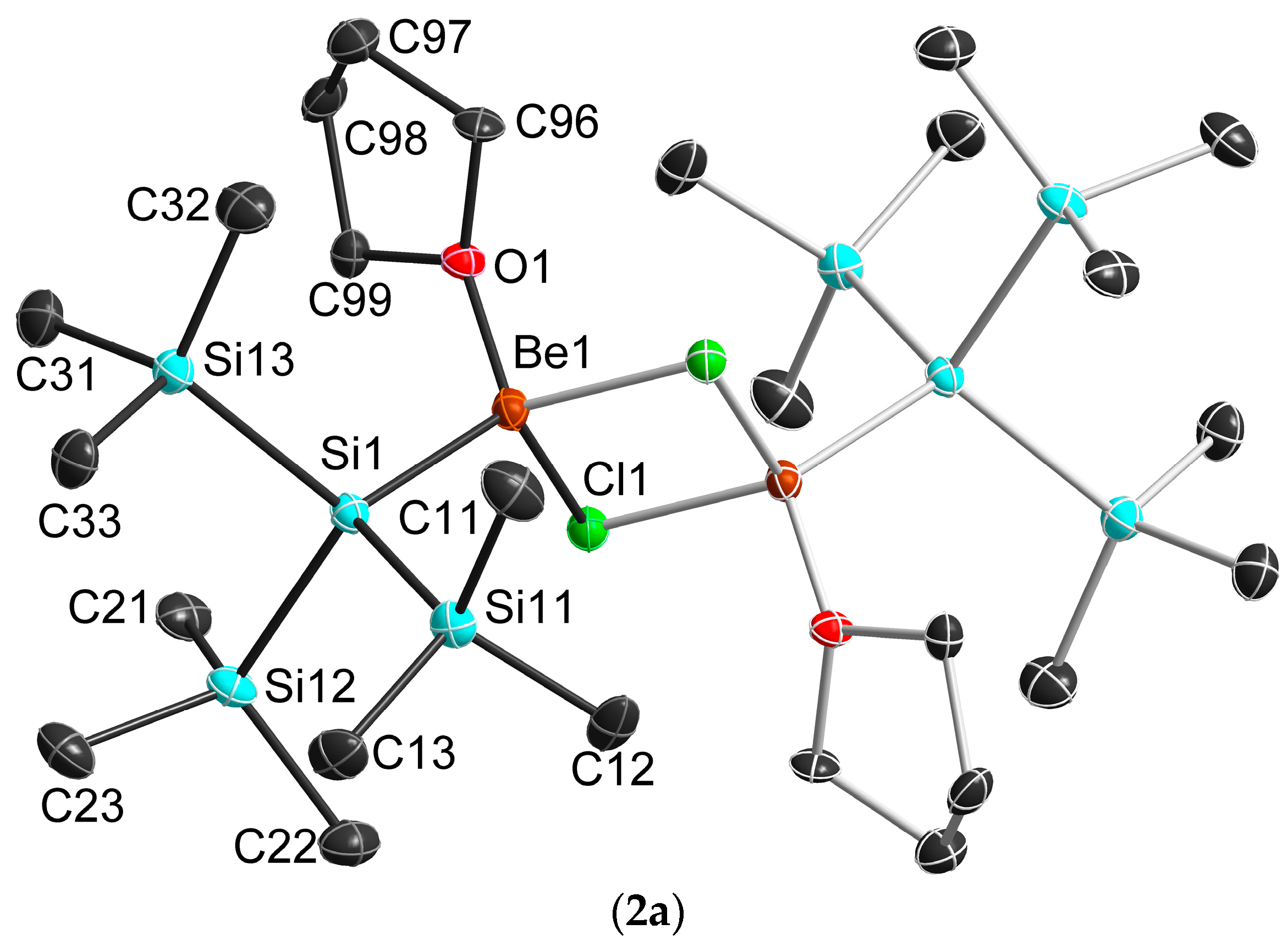
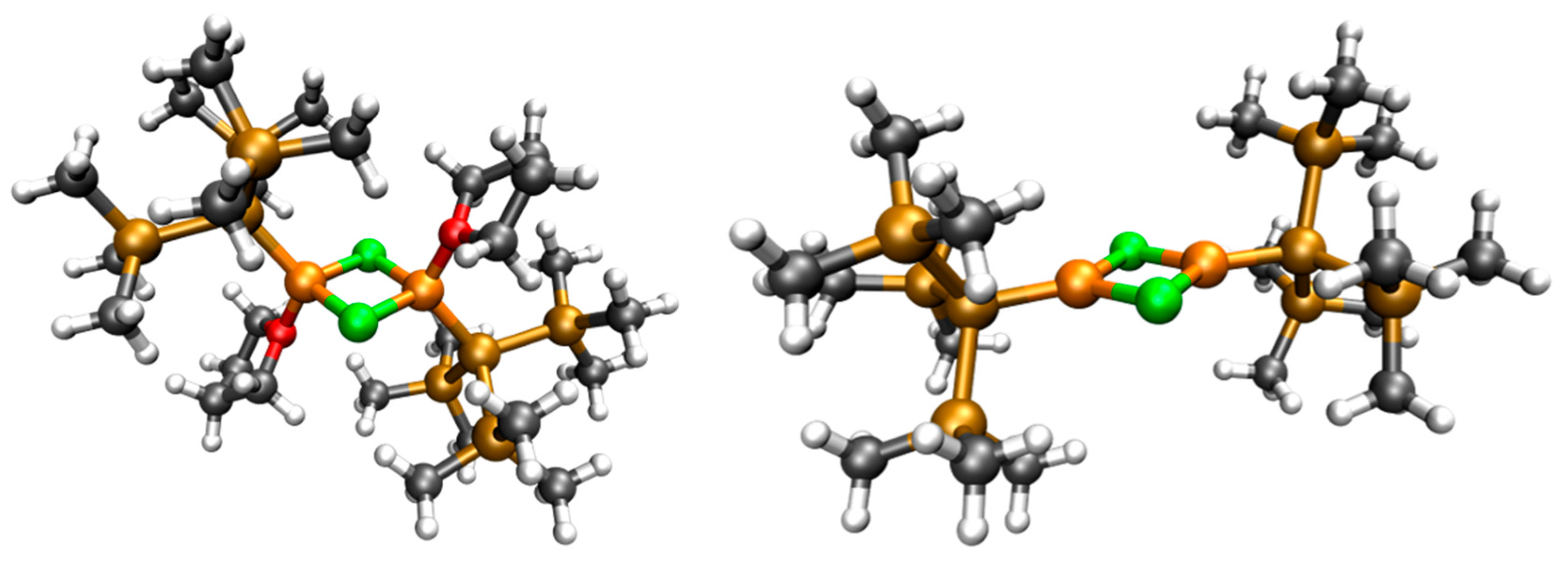
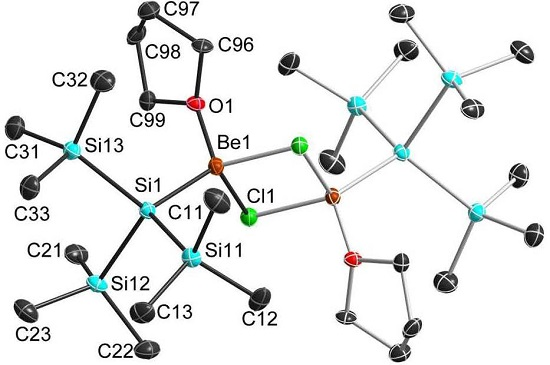
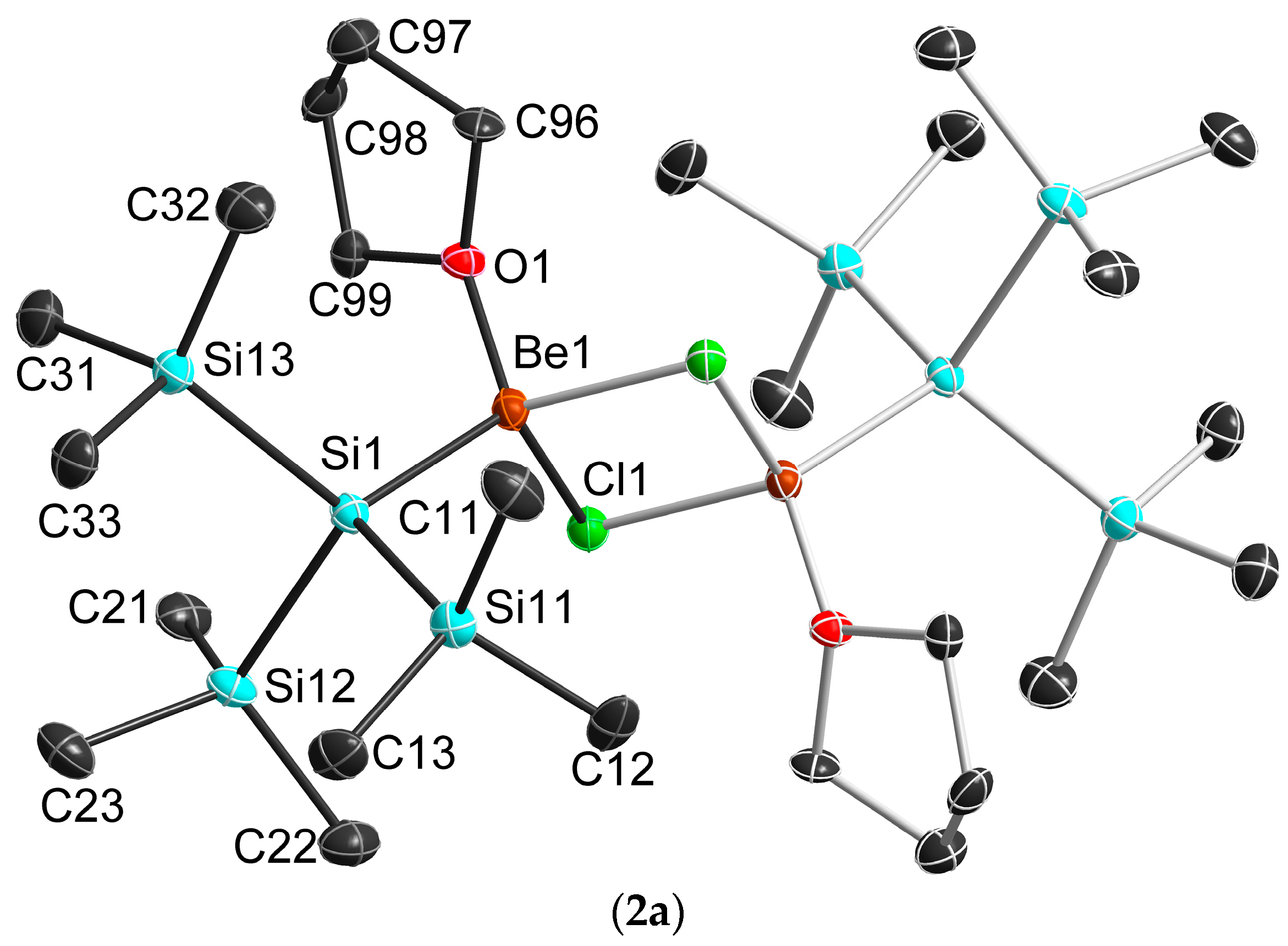
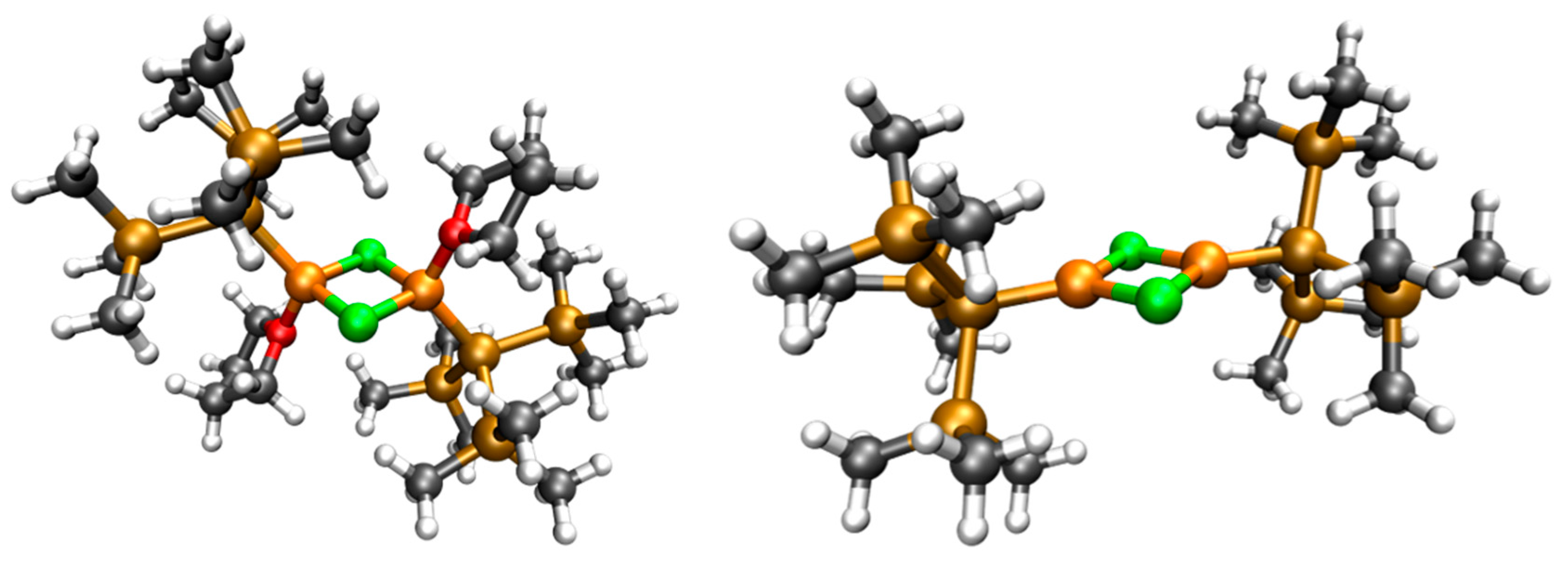
2.3. Solid State Structure
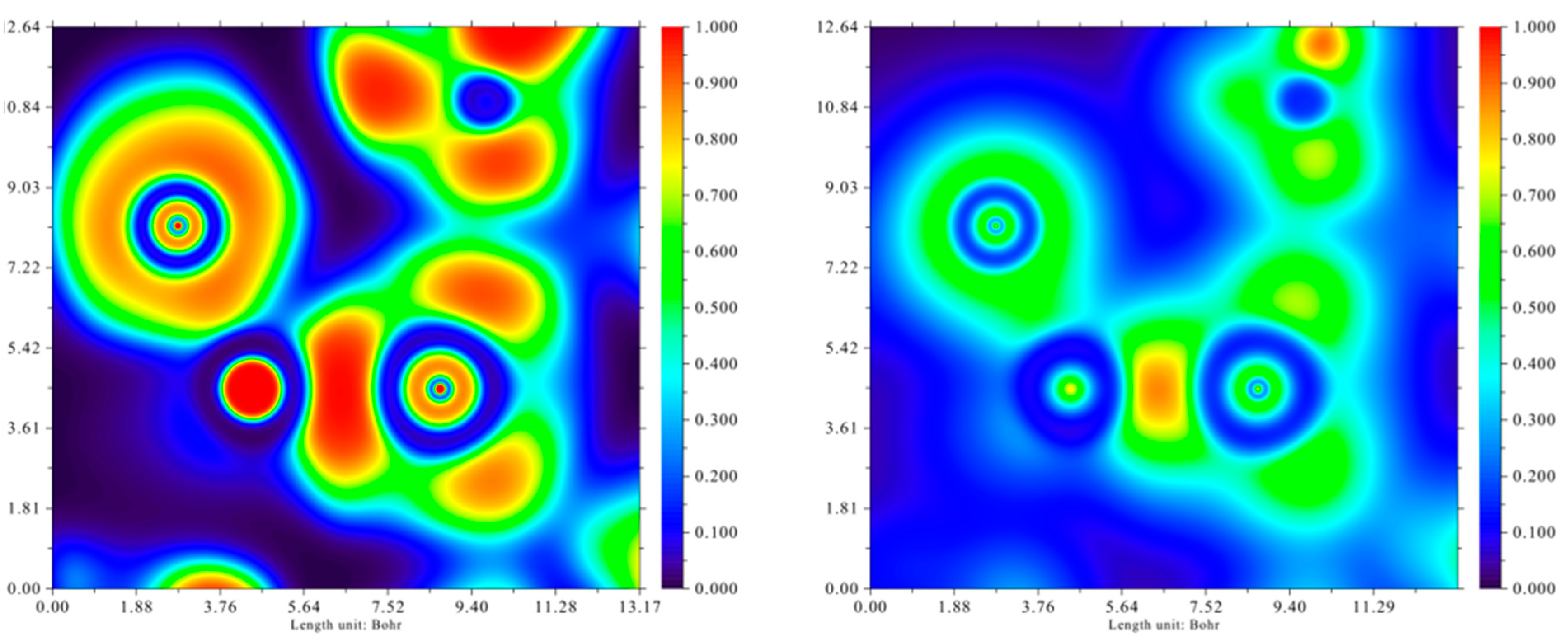
2.4. Quantum Chemical Calculations
3. Materials and Methods
General Procedure for the Synthesis of HypSiBeX∙(thf) (X = Cl 2a, I 4a)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Iversen, K.J.; Couchman, S.A.; Wilson, D.J.; Dutton, J.L. Modern organometallic and coordination chemistry of beryllium. Coord. Chem. Rev. 2015, 297–298, 40–48. [Google Scholar] [CrossRef]
- Arrowsmith, M.; Braunschweig, H.; Celik, M.A.; Dellermann, T.; Dewhurst, R.D.; Ewing, W.C.; Hammond, K.; Kramer, T.; Krummenacher, I.; Mies, J.; et al. Neutral zero-valent s-block complexes with strong multiple bonding. Nat. Chem. 2016, 8, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, M.; Hill, M.S.; Kociok-Kohn, G.; MacDougall, D.J.; Mahon, M.F. Beryllium-induced C–N bond activation and ring opening of an N-heterocyclic carbene. Angew. Chem. Int. Ed. Engl. 2012, 51, 2098–2100. [Google Scholar] [CrossRef] [PubMed]
- Naglav, D.; Neumann, A.; Bläser, D.; Wölper, C.; Haack, R.; Jansen, G.; Schulz, S. Bonding situation in Be(N(SiMe3)2)2—An experimental and computational study. Chem. Commun. 2015, 51, 3889–3891. [Google Scholar] [CrossRef] [PubMed]
- Bayram, M.; Naglav, D.; Wölper, C.; Schulz, S. Synthesis and Structure of Bis(diphenylphosphinimino)methanide and Bis(diphenylphosphinimino)methanediide Beryllium Complexes. Organometallics 2016, 35, 2378–2383. [Google Scholar] [CrossRef]
- Buchner, M.R.; Müller, M.; Rudel, S.S. Beryllium Phosphine Complexes: Synthesis, Properties, and Reactivity of (PMe3)2BeCl2 and (Ph2PC3H6PPh2)BeCl2. Angew. Chem. Int. Ed. Engl. 2017, 56, 1130–1134. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Li, Y.; Zhu, G.; Su, H.; Chan, S.H.; Sun, Q. Be12O12 Nano-cage as a Promising Catalyst for CO2 Hydrogenation. Sci. Rep. 2017, 7, 40562. [Google Scholar] [CrossRef] [PubMed]
- Naglav, D.; Buchner, M.R.; Bendt, G.; Kraus, F.; Schulz, S. Off the Beaten Track—A Hitchhiker’s Guide to Beryllium Chemistry. Angew. Chem. Int. Ed. Engl. 2016, 55, 10562–10576. [Google Scholar] [CrossRef] [PubMed]
- Naglav, D.; Bläser, D.; Wölper, C.; Schulz, S. Synthesis and characterization of heteroleptic 1-tris(pyrazolyl)borate beryllium complexes. Inorg. Chem. 2014, 53, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Naglav, D.; Tobey, B.; Neumann, A.; Bläser, D.; Wölper, C.; Schulz, S. Synthesis, Solid-State Structures, and Computational Studies of Half-Sandwich Cp*BeX (X = Cl, Br, I) Compounds. Organometallics 2015, 34, 3072–3078. [Google Scholar] [CrossRef]
- Lerner, H.-W.; Scholz, S.; Bolte, M.; Wiberg, N.; Nöth, H.; Krossing, I. Synthesis and Structures of Alkaline-Earth Metal Supersilanides: tBu3SiMX and tBu3Si−M−SitBu3 (M = Be, Mg; X = Cl, Br). Eur. J. Inorg. Chem. 2003, 2003, 666–670. [Google Scholar] [CrossRef]
- Saulys, D.A.; Powell, D.R. Synthesis, Experimental/Theoretical Characterization, and Thermolysis Chemistry of CpBe(SiMe3), a Molecule Containing an Unprecedented Beryllium–Silicon Bond. Organometallics 2003, 22, 407–413. [Google Scholar] [CrossRef]
- Niemeyer, M.; Power, P.P. Synthesis, 9Be NMR Spectroscopy, and Structural Characterization of Sterically Encumbered Beryllium Compounds. Inorg. Chem. 1997, 36, 4688–4696. [Google Scholar] [CrossRef] [PubMed]
- Ruhlandt-Senge, K.; Bartlett, R.A.; Olmstead, M.M.; Power, P.P. Synthesis and Structural Characterization of the Beryllium Compounds [Be(2,4,6-Me3C6H2)2(OEt2)], [Be{O(2,4,6-t-Bu3C6H2}(OEt2)], and [Be{S(S(2,4,6-t-Be3C6H2)}2(thf)]2(THF)]PhMe and Determination of the Structure of [BeCl2(OEt2)2]. Inorg. Chem. 1993, 32, 1724–1728. [Google Scholar] [CrossRef]
- Han, R.; Parkin, G. [Tris(3-tert-butylpyrazolyl)hydroborato]beryllium hydride: Synthesis, structure, and reactivity of a terminal beryllium hydride complex. Inorg. Chem. 1992, 31, 983–988. [Google Scholar] [CrossRef]
- Arrowsmith, M.; Hill, M.S.; Kociok-Kohn, G.; MacDougall, D.J.; Mahon, M.F.; Mallov, I. Three-coordinate beryllium beta-diketiminates: Synthesis and reduction chemistry. Inorg. Chem. 2012, 51, 13408–13418. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, M.; Crimmin, M.R.; Hill, M.S.; Kociok-Kohn, G. Beryllium derivatives of a phenyl-substituted beta-diketiminate: A well-defined ring opening reaction of tetrahydrofuran. Dalton Trans. 2013, 42, 9720–9726. [Google Scholar] [CrossRef] [PubMed]
- Naglav, D.; Tobey, B.; Dzialkowski, K.; Jansen, G.; Neumann, A.; Bläser, D.; Wölper, C.; Schulz, S. Synthesis and Characterization of Heteroleptic Diiminophosphinate Beryllium Complexes. Unpublished results. 2017. [Google Scholar]
- Chmely, S.C.; Hanusa, T.P.; Brennessel, W.W. Bis(1,3-trimethylsilylallyl)beryllium. Angew. Chem. Int. Ed. Engl. 2010, 49, 5870–5874. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr. Sect. A Found. Crystallogr. 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Lorenz, V.; Fischer, A.; Edelmann, F.T. Silsesquioxane chemistry, The first beryllium silsesquioxane: Synthesis and structure of [Cy7Si7O12BeLi]2 2THF. Inorg. Chem. Commun. 2000, 3, 292–295. [Google Scholar] [CrossRef]
- Lerner, W.; Goethe-Universität Frankfurt, Frankfurt, Germany. Supply of the value for the second independent molecule. Personal Communication, 2017. [Google Scholar]
- Treutler, O.; Ahlrichs, R. Efficient molecular numerical integration schemes. J. Chem. Phys. 1995, 102, 346–354. [Google Scholar] [CrossRef]
- Eichkorn, K.; Treutler, O.; Öhm, H.; Häser, M.; Ahlrichs, R. Auxiliary basis sets to approximate Coulomb potentials. Chem. Phys. Lett. 1995, 240, 283–290. [Google Scholar] [CrossRef]
- Eichkorn, K.; Weigend, F.; Treutler, O.; Ahlrichs, R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor. Chim. Acta 1997, 97, 119–124. [Google Scholar] [CrossRef]
- Von Arnim, M.; Ahlrichs, R. Performance of parallel TURBOMOLE for density functional calculations. J. Comput. Chem. 1998, 19, 1746–1757. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Chem. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. PCCP 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Kollwitz, M.; Gauss, J. A direct implementation of the GIAO-MBPT(2) method for calculating NMR chemical shifts. Application to the naphthalenium and anthracenium ions. Chem. Phys. Lett. 1996, 260, 639–646. [Google Scholar] [CrossRef]
- Davidson, E.R. Electronic Population Analysis of Molecular Wavefunctions. J. Chem. Phys. 1967, 46, 3320–3324. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classifaction of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Schmider, H.; Becke, A. Chemical content of the kinetic energy density. J. Mol. Struct. THEOCHEM 2000, 527, 51–61. [Google Scholar] [CrossRef]
- Kumberger, O.; Schmidbaur, H. Warum ist Beryllium so toxisch? Chem. Unserer Zeit 1993, 27, 310–316. [Google Scholar] [CrossRef]
- Gutekunst, G.; Brook, A.G. Tris(trimethylsilyl)silyllithium·3 thf: A stable crystalline silyllithium reagent. J. Organomet. Chem. 1982, 225, 1–3. [Google Scholar] [CrossRef]
- Brauer, G. Handbuch der Präparativen Anorganischen Chemie, 3rd ed.; Ferdinand Enke: Stuttgart, Germany, 1975; Volume 1. [Google Scholar]
- Sheldrick, G.M. Phase annealing in SHELX-90: Direct methods for larger structures. Acta Crystallogr. A Found. Crystallogr. 1990, 46, 467–473. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Hubschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
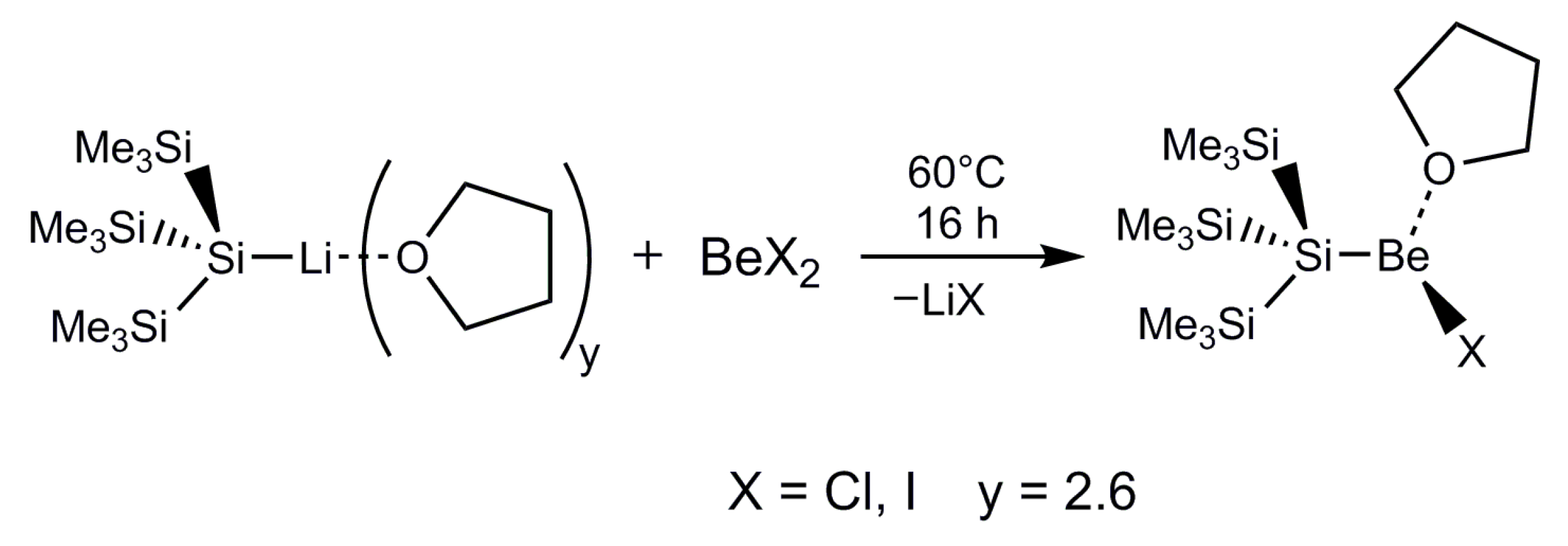{kind=link}
| Compound | 9Be NMR Shifts (δ in ppm) | Solvent | Coordination Mode | Coordination Number | Literature |
|---|---|---|---|---|---|
| (Me3Si)3SiBeCl∙(thf) | 2.45 | thf-d8 | tetrahedral | 4 | this work |
| (Me3Si)3SiBeI∙(thf) | −0.92 | thf-d8 | tetrahedral | 4 | this work |
| BeCl2(OEt2)2 | 2.6 | C6D6 | tetrahedral | 4 | [13] |
| BeBr2(OEt2)2 | 3.0 | C6D6 | tetrahedral | 4 | [13] |
| CpBeSiMe3 | −27.70 | C6D6 | aromatic (η5) | 6 | [12] |
| CpBe(SiMe2SiMe3) | −27.20 | C6D6 | aromatic (η5) | 6 | [12] |
| TpBeF | 4.54 | thf-d8/C7D8 3:5 | tetrahedral | 4 | [9] |
| TpBeCl | 4.95 | thf-d8 | tetrahedral | 4 | [9] |
| TpBeBr | 5.15 | thf-d8/C6D6 1:3 | tetrahedral | 4 | [9] |
| TpBeI | 4.66 | thf-d8 | tetrahedral | 4 | [9] |
| TptBuBeCl | 2.7 | C6D6 | tetrahedral | 4 | [15] |
| TptBuBeBr | 2.4 | C6D6 | tetrahedral | 4 | [15] |
| TptBuBeI | 1.3 | C6D6 | tetrahedral | 4 | [15] |
| Cp*BeCl | −14.88 | C6D6 | aromatic (η5) | 6 | [10] |
| Cp*BeBr | −14.81 | C6D6 | aromatic (η5) | 6 | [10] |
| Cp*BeI | −15.78 | C6D6 | aromatic (η5) | 6 | [10] |
| DDPBeCl | 12.2 | C6D6 | trigonal planar | 3 | [16,17] |
| DDPBeI | 13.4 | C6D6 | trigonal planar | 3 | [16,17] |
| Ph2P(NDipp)2BeCl | 11.36 | C6D6 | trigonal planar | 3 | [18] |
| Ph2P(NDipp)2BeBr | 11.94 | C6D6 | trigonal planar | 3 | [18] |
| Ph2P(NDipp)2BeI | 11.53 | C6D6 | trigonal planar | 3 | [18] |
| TerphenylBeCl∙(Et2O) | 12.8 | C6D6 | trigonal planar | 3 | [13] |
| TerphenylBeBr∙(Et2O) | 13.4 | C6D6 | trigonal planar | 3 | [13] |
| [1,3-(SiMe3)2C3H3]2Be∙(Et2O) | 18.2 | C6D6 | trigonal planar | 3 | [19] |
| Be[N(SiMe3)2]2 | 12.3 | C6D6 | linear | 2 | [4] |
| Be[N(SiMe3)2]2 | 9.6 | thf-d8 | linear | 2 | [4] |
| Be[N(SiMe3)2]2 | 12.4 | tol-d8 | linear | 2 | [4] |
| 1b 1 | 2a | 2b 1 | 3b 1 | 4b 1 | |
|---|---|---|---|---|---|
| Be–X | 1.614 | 2.101 1 | 2.112 | 2.292 | 2.531 |
| Be–Si | 2.249 | 2.239(5) | 2.222 | 2.213 | 2.203 |
| Be–O | 1.670 | 1.654(5) | 1.670 | 1.655 | 1.649 |
| X–Be–X | 90.60 | 96.5(2) | 97.4 | 98.2 | 99.0 |
| Be–X–Be | 89.4 | 83.5(2) | 82.6 | 81.8 | 81.0 |
| Si–Be–X1 | 118.0 | 116.0(2) | 114.6 | 113.6 | 112.0 |
| Si–Be–X2 | 123.5 | 118.5(2) | 118.9 | 118.0 | 116.8 |
| Si–Be–O | 113.4 | 116.4(3) | 115.7 | 117.1 | 118.5 |
| Be–X–X–Be | 180.0 | 180.0(3) | 179.6 | 180.0 | 180.0 |
| X–X–Be–Si | 129.1 | 126.2(4) | 136.7 | 125.5 | 123.8 |
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| a | – | 2.45 | – | −0.92 |
| b | 6.90 | 10.62 | 12.14 | 15.93 |
| c | 14.01 | 25.53 | 29.16 | 24.78 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naglav, D.; Tobey, B.; Dzialkowski, K.; Jansen, G.; Wölper, C.; Schulz, S. Insights into Molecular Beryllium–Silicon Bonds. Inorganics 2017, 5, 22. https://doi.org/10.3390/inorganics5020022
Naglav D, Tobey B, Dzialkowski K, Jansen G, Wölper C, Schulz S. Insights into Molecular Beryllium–Silicon Bonds. Inorganics. 2017; 5(2):22. https://doi.org/10.3390/inorganics5020022
Chicago/Turabian StyleNaglav, Dominik, Briac Tobey, Kevin Dzialkowski, Georg Jansen, Christoph Wölper, and Stephan Schulz. 2017. "Insights into Molecular Beryllium–Silicon Bonds" Inorganics 5, no. 2: 22. https://doi.org/10.3390/inorganics5020022
APA StyleNaglav, D., Tobey, B., Dzialkowski, K., Jansen, G., Wölper, C., & Schulz, S. (2017). Insights into Molecular Beryllium–Silicon Bonds. Inorganics, 5(2), 22. https://doi.org/10.3390/inorganics5020022