1. Introduction
Metals have been used in medicinal applications for more than 500 years [
1]. For example, the Egyptians used copper to sterilize water, gold was used in a variety of medicines in Arabia and China, and various iron remedies were used in Egypt around 1500 BC. At about the same time zinc was discovered to promote the healing of wounds. In the Renaissance era, mercury chloride was used as a diuretic and the nutritional essentiality of iron was discovered. However, in the last 100 years, the medicinal activity of inorganic compounds has been developed in a rational manner. Thus, in the early 1900s K[Au(CN)
2] was used for treating tuberculosis and various Sb compounds for leishmaniasis. In addition, the antibacterial activity of various gold salts and arsenic compounds were used for treating various diseases [
2].
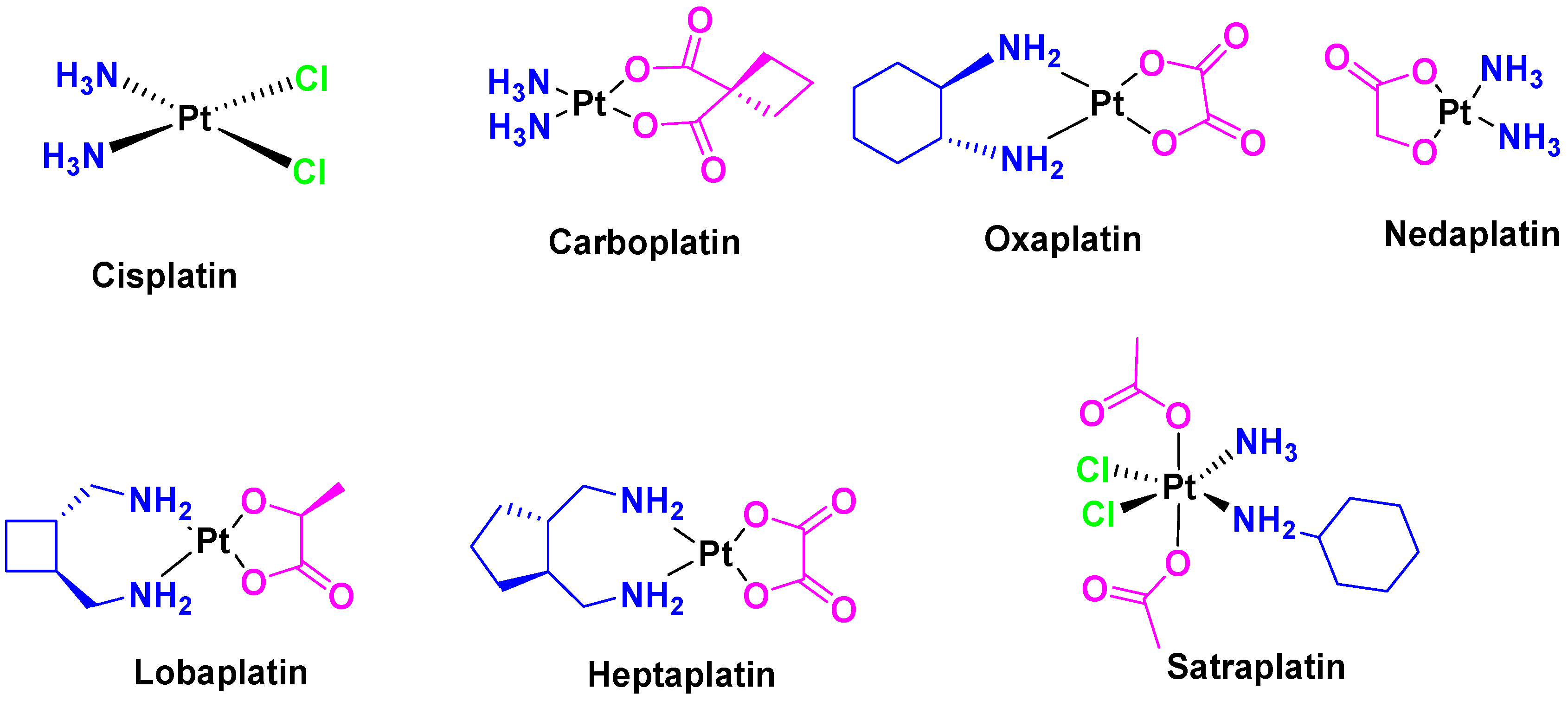
In the twentieth century, a very important therapeutic activity of metal complexes was discovered, namely their application for the treatment of cancer. Rosenberg’s serendipitous discovery of the anti-cancer action of cisplatin (
cis-[Pt(NH
3)
2Cl
2],
Figure 1) in the 1960s precipitated a widespread search for related complexes with similar or better activity [
3]. The observation of the cell division suppression by this compound was crucial for its development.
Until the turn of the century, cisplatin had been the most used drug in the world in therapy of cancer, administered alone or combined with other compounds. Researchers still had the expectation to develop alternative drugs to improve the potential and the effectiveness against cancer, and especially to overcome the undesirable effects of cisplatin, such as nephrotoxicity, neurotoxicity, ototoxicity, nausea and vomiting [
4].
In this context, an extensive study of other metal complexes with similar anti-cancer action was carried out by the scientific community. Thus, the first non-platinum complex to enter clinical trials was budotitane although its applications were limited due to its low solubility and liver toxicity [
5].
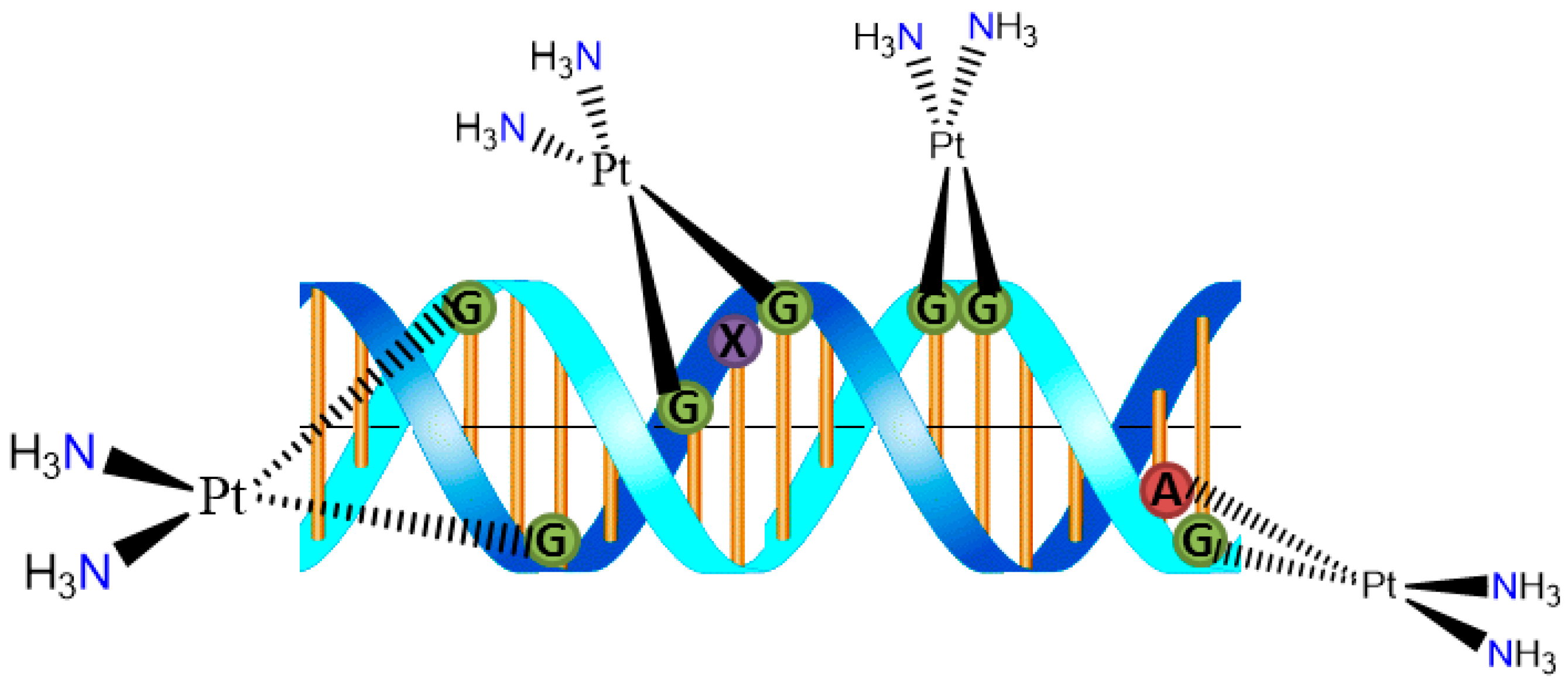
The cytotoxicity of cisplatin originates from its binding to DNA and the formation of covalent cross-links. The 1,2-intrastrand d(GpG) cross-link is the major adduct. Binding of cisplatin to DNA causes significant distortion of the helical structure and results in inhibition of DNA replication and transcription (
Figure 2) [
6]. The Pt
2+ unit covalently binds to deoxyribonucleic acid (DNA), particularly to the N7 of either guanine (G) or adenine (A) in the nucleotide sequences GG and AG to form interstrand cross-links [
7] The so-formed cisplatin-DNA unit activates a new cellular pathway which leads to transcription inhibition, cell-cycle arrest, DNA repair, and finally apoptosis [
8].
Immediately after the initial elucidation of the cell death mechanism of cisplatin, other platinum analogues such as carboplatin [
9] and oxaliplatin [
10] (
Figure 3) were synthesized and approved by the FDA for use as anticancer drugs. In addition, some other compounds such as nedaplatin, lobaplatin, heptaplatin, and satraplatin (
Figure 3) are currently in clinical trial phase [
11].
One of the main disadvantages of cisplatin is that, in many cases, cancer cells acquire resistance to this drug, deactivating its effect against damaged cells. To overcome this, cisplatin can be combined with other chemotherapeutics agents like 5-fluorouracil, for example [
12].
Thus, cisplatin and its derivatives have been used for many years as chemotherapy agents in the treatment of cancer with excellent results against a wide variety of cell lines and tumors. However, because of the induction of drug-resistance of the tumors after treatment with cisplatin, the side-effects, the intrinsic toxicity of platinum, the limited bioavailability, and the solubility in physiological media of cisplatin-like compounds, it is of upmost importance to find alternative agents based on non-platinum metals-based systems with fewer side effects and improved cytotoxic and anticancer properties.
Thus, a wide variety of preclinical and clinical studies using anticancer metallodrugs have been reported using different elements such as gallium, titanium, palladium, gold, cobalt, ruthenium, and tin.
In this review, we describe the synthetic methods and preclinical studies in anticancer tests that our research group has carried out in the search for alternatives to cisplatin-like materials. As our work has mainly been based on the use of gallium, tin, and titanium compounds, we have divided this manuscript into three main parts, which cover specifically these metal complexes. In addition, a short section describes the latest results from our group using metallodrugs of other elements.
2. Gallium-Based Metallodrugs
Among the p-block metals, gallium has shown some clinical activity in the treatment of soft tissue tumors. Gallium(III) complexes present a special activity in anticancer therapy due to the analogy of the Ga(III) ion with the Fe(III) ion in ionic radius, electron affinity, electronegativity, coordination geometry, and Lewis base affinity [
13,
14]. These similarities suggest that the Ga(III) ion may follow an analogous biochemical pathway to that observed in iron metabolism. Gallium(III) is stable under biological conditions, while the oxidation state 2+ in gallium is energetically unfavorable and too reactive under physiological conditions to be stable. Hence, redox chemistry is therefore not possible for Ga(III) in biological media. This phenomenon enables the utilization of gallium(III) as a potential therapeutic agent and facilitates its study in biological conditions [
15].
The literature has described some interesting results of gallium(III) compounds in phase II clinical trials in the treatment of lymphomas and bladder carcinoma. In addition, the combination with other agents in the treatment of metastatic carcinoma of the urethelium and cisplatin-resistant ovarian cancer has delivered promising results [
16].
The mechanism of action of gallium(III) complexes in anticancer chemotherapy has been briefly studied. Ga
3+ ions usually compete with Fe
3+ for binding to transferrin to reach the intracellular medium. In this way, a cellular uptake of large amounts of gallium is achieved [
17]. Analyzing all the biochemical pathways of the gallium(III) ion, it seems clear that the enzyme ribonucleotide reductase is its biological target [
18]. Furthermore, the induction of apoptosis through activation of the proapoptotic factor BAX and caspase-3 can be considered as a possible mechanism of cell death. However, proteasome inhibition should not be ruled out [
19].
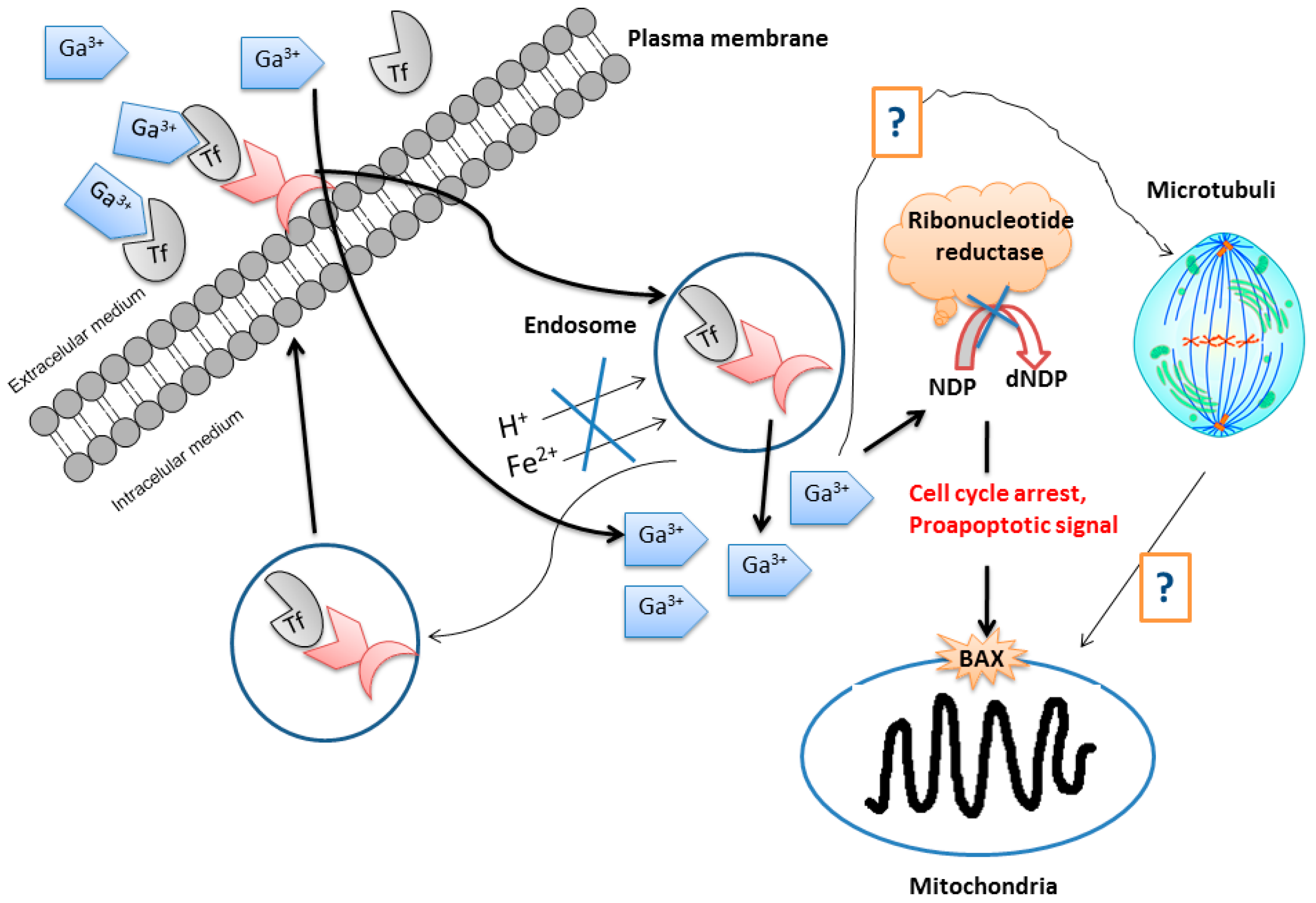
The simplest and most familiar gallium compound used as an anticancer drug is gallium nitrate. However, this compound is readily hydrolyzed in biological medium to give non-soluble gallium oxides which are able to block the absorption and membrane permeation of the gallium ion reducing its effectivity in cancer treatments. Other gallium(III) compounds containing caboxylato, thiolato, and alkoxo ligands have been tested as anticancer agents. In general, the cellular acquisition of gallium mainly occurs by transferrin-mediated uptake followed by accumulation in endosomes. After transport into the cytosol, gallium(III) binds to and inhibits the functioning of ribonucleotide reductase (RR, an enzyme recognized as the most significant intracellular target for antiproliferative activity of gallium). DNA replication activates cell cycle arrest and results finally in apoptosis through the mitochondrial pathway, from which gallium is liberated during, or before, passage across the intestinal epithelium to become in part bound to transferrin in blood (
Figure 4) [
20].
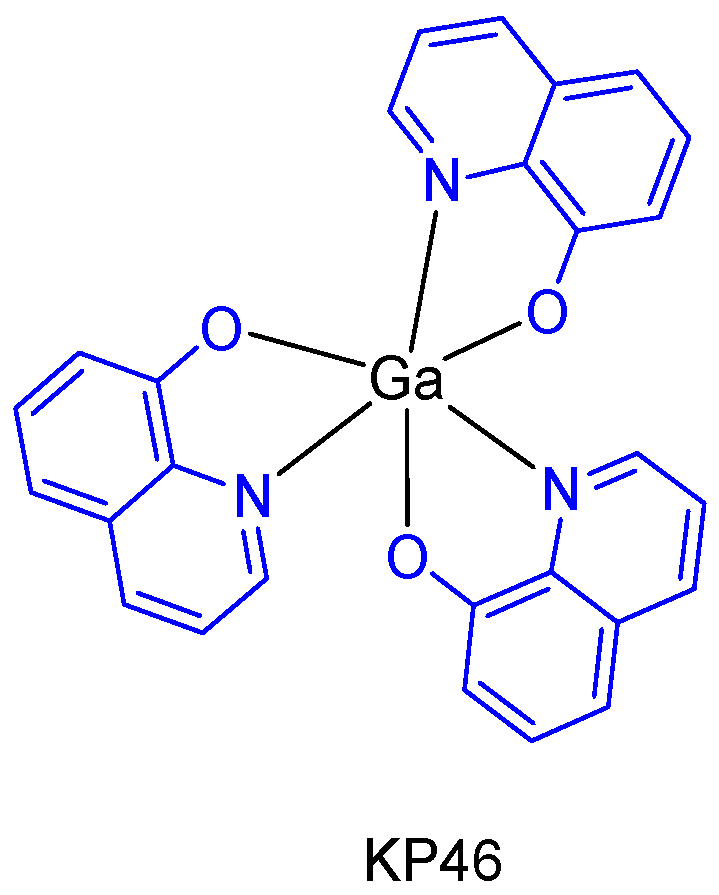
The metallodrug, KP46 (tris(8-hydroxyquinolinato)gallium(III)) (
Figure 5), contains the metal chelating agent 8-hydroxyquinoline, which itself has anticancer properties [
21]. Due to its well-defined toxicological and pharmacokinetic advantages, KP46 not only enables higher and well tolerable tissue gallium concentrations to be established, but also inhibitory effects on cell growth proliferation in vitro and in vivo superior to gallium salts (with IC
50 values typically in the low micromolar range). In addition, an oral formulation of KP46 (IT-235 from the companies Niikipharma and Intezyne Technologies) showed a novel pattern of cytotoxicity with synergism across a broad range of antitumor agents targeting the endoplasmic reticulum in multiple tumor types [
22].
Similar gallium(III) complexes to KP46, including the ligand 7-chloroquinoline, were synthesized by other groups and showed not only a very high cytotoxic activity in vitro but also antimalarial properties [
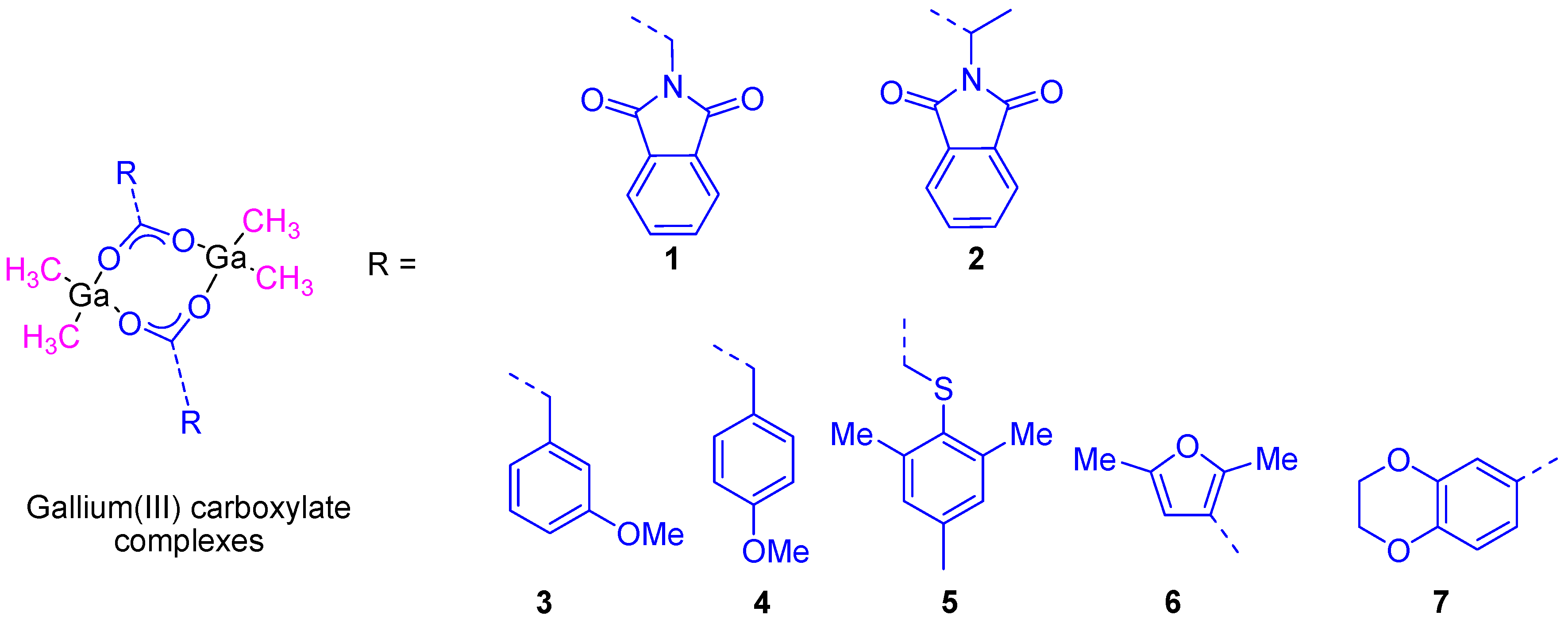
23]. Bearing in mind the promising properties of gallium compounds our group embarked on the preparation of several gallium compounds with different ligands. Thus, as the literature had shown an interesting and synergistic relation between gallium complexes and aminoacid derivatives such as glycine and
DL-alanine in cancer cell death, we decided to synthesize two gallium complexes based on
N-phthaloyl derivatives of neutral aminoacids, namely [Me
2Ga(µ-O
2CCH
2N(CO)
2C
6H
4)]
2 (
1) and
RS-[Me
2Ga(µ-O
2CCHMeN(CO)
2C
6H
4)]
2 (
2) (
Figure 6). The formation of a single diastereoisomer
RS was observed in the crystal structure determined by X-ray diffraction studies. The high solubility and stability of both compounds in DMSO and mixtures of DMSO/water, make them good candidates for anticancer tests.
Compounds
1 and
2 were tested as anticancer agents against four human tumor cell lines: 8505C anaplastic thyroid cancer, A253 head and neck carcinoma, A549 lung carcinoma, A2780 ovarian cancer, and DLD-1 colon carcinoma. Comparing the results of cytotoxicity with gallium(III) nitrate, the compounds
1 and
2 presented a higher antiproliferative effect. Both complexes present similar IC
50 values in all the studied cell lines (
Table 1) [
24].
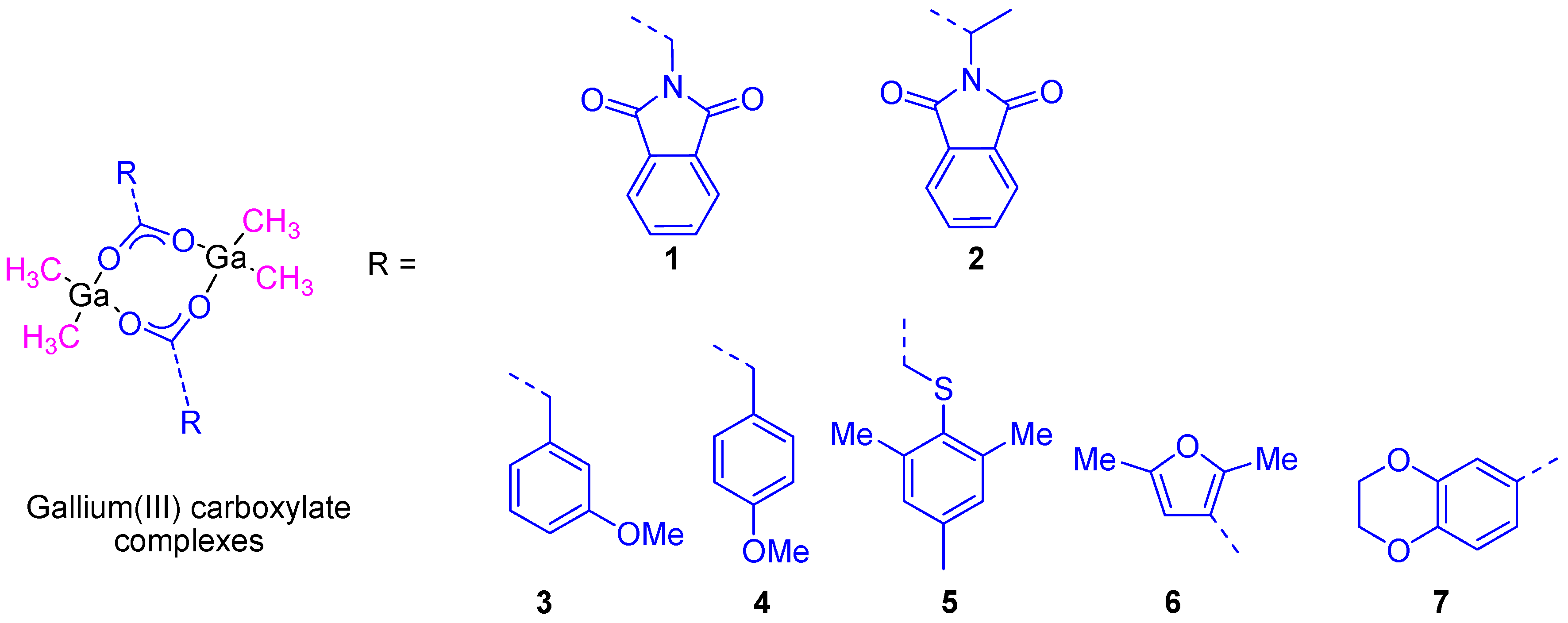
In a second study, additional organometallic gallium(III) compounds (
3–
7) containing phenyl, thiophenyl, furane, and benzodioxane carboxylato ligands (
Figure 6) were synthesized and characterized. The cytotoxic study of all of these compounds showed a dose-dependent antiproliferative effect towards different cancer cell lines such as 8505C, A253, A549, A2780, and DLD-1. The cytotoxic activity of all the studied compounds was much higher than that presented by gallium(III) nitrate. From all the reported complexes,
7 (containing the benzodioxane carboxylate ligand) presented the highest cytotoxicity against A253 cells with the lowest IC
50 value of 6.6 ± 0.2 µM [
25].
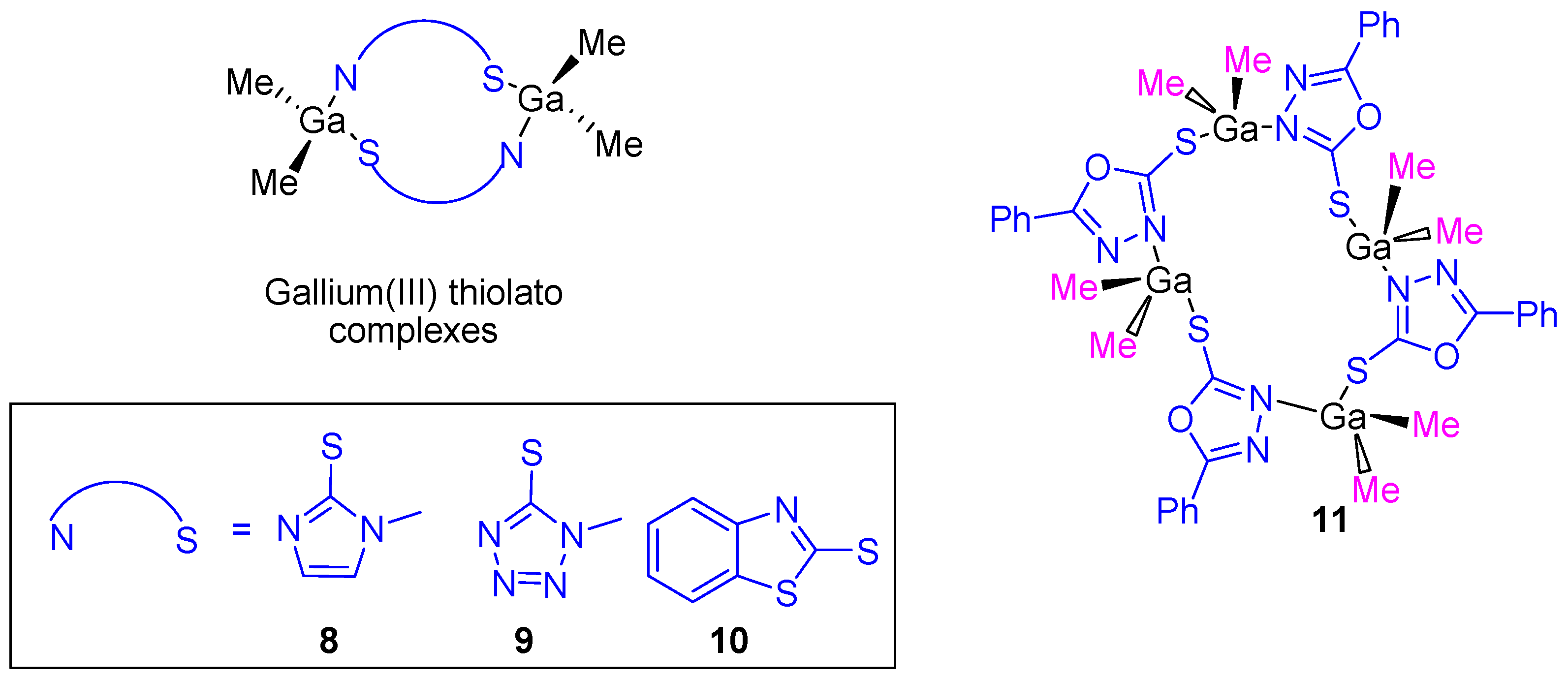
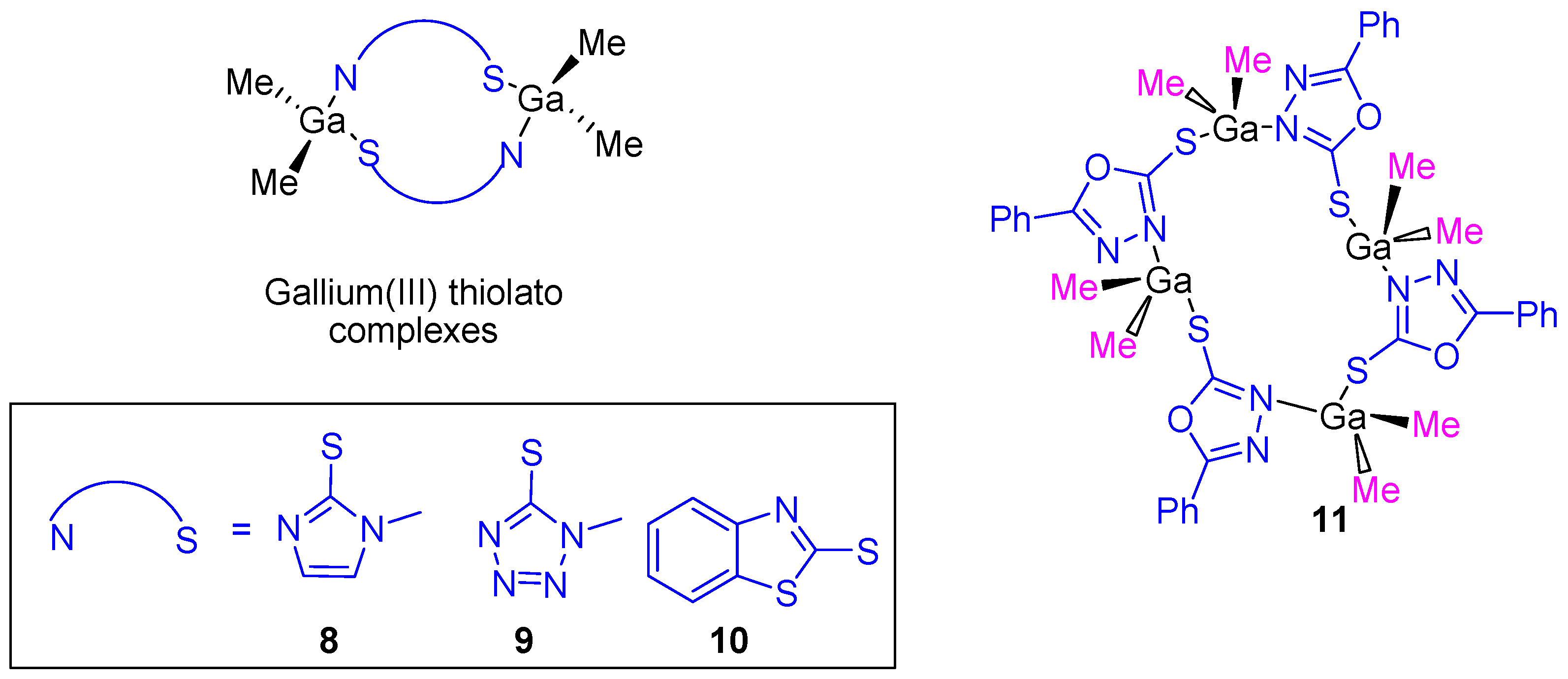
After the cytotoxic studies using the gallium(III) carboxylate complexes, our research group prepared dinuclear and tetranuclear organometallic gallium(III) compounds containing heterocyclic thiolato ligands (
Figure 7). These compounds were synthesized by a simple protonolysis reaction of trimethylgallium and the thiol group of mercapto-substituted imidazole, tetrazole, benzothiazole or phenyl-oxadiazol heterocycles (
Figure 7). All the compounds were characterized by NMR, IR, and UV–Vis spectroscopy and X-ray diffraction studies confirmed the formation of dinuclear or tetranuclear complexes.
Thiolate complexes
8–
11 were tested against the same cancer cell lines used on carboxylate gallium compounds (
1–
7) and again a dose-dependent antiproliferative effect on all cancer cell lines (
Table 1) was observed. The cytotoxicity of gallium(III) heterocyclic thiolato complexes is much higher than that of gallium nitrate, while being in the same range as that of cisplatin. An especially high cytotoxic activity was observed for
11 with an IC
50 value against DLD-1 of 5.49 ± 0.16 µM which is similar to that observed for cisplatin (5.14 ± 0.12 µM). After selectivity tests of gallium compounds
8–
11 and cisplatin on WWO70327 human fibroblasts, gallium(III) complexes were shown to be much more selective to cancer cells than cisplatin, indicating, therefore their potential applicability in anticancer therapy [
26]. In the apoptosis studies, after 24 h exposure to IC
90 concentrations of compounds
8–
11, typical DNA ladders in DLD-1 cell line were observed which indicated the induction of apoptosis promoted by the gallium compounds. In addition, compounds
8–
11 showed binding affinity to FS-DNA (confirmed by UV spectroscopy in simulated physiological medium) but not to plasmid pBR322 DNA.
Following the interesting results observed for carboxylate and thiolate gallium(III) complexes (
1–
11), additional biological studies were carried out on a series of cancer cell lines, HN (soft palate), Cal27 and Cal33 (tongue), FaDu (hypopharynx), and A253 (Submandibular duct) (
Table 1) [
27]. Gallium(III) complexes
3,
6, and
8 induced cell death mediated apoptosis. Cal27 and FaDu cells were treated for 24 h with IC
90 concentration of the complexes and DNA appeared as characteristic ladder-like fragments suggesting an apoptotic cell death promotion. In contrast to the Cal27 cell line, there was a slight translation of FaDu cells from the G1 phase to the apoptotic phase (Sub-G1) after treatment with compounds
3,
6, and
8, which indicates that apoptosis caused by these compounds on FaDu cells may be due to interference caused in the G1 phase of the cell cycle [
28].
Finally, gallium(III) complexes
1,
3–
8, and
11 were also tested against CT26CL25, HCT116, and SW480 colon cancer cell lines using CV and MTT assays. Compounds
1 and
3–
8 affect mitochondrial function, while gallium(III) complex
11 activates different cell death pathways and presents an activity 1.7–3.0 times higher than the other organogallium(III) complexes. In addition,
11 induces caspase independent apoptosis with a strong blockage of first and second division inhibition of CT26CL25 cell proliferation [
29].
In view of the biological tests carried out for the organogallium(III) compounds reported by our group, one can envisage that these compounds may be suitable alternatives to KP46 which finished phase I trials with the outcome of promising tolerability and evidence of clinical activity in renal cell carcinoma. However, we have observed that gallium(III) complexes present a limited selectivity on cancer cells. Only in some studies have we observed selectivity when comparing their action against cancer cells with fibroblasts. Thus, the research in this area should be directed to the preparation of new gallium(III) compounds with recognizable fragments to different overexpressed targets in cancer cells to improve the selectivity and cancer cell uptake. In addition, as gallium(III) compounds present water solubility issues, formulation of these compounds with encapsulating agents (such as chitosan or analogues) may increase the solubility or dispersability in water and the cell permeation ability, and should, therefore, be of current interest for the application of these compounds in animal tests. Finally, bearing in mind that our group has not carried out in vivo studies, a complete investigation on the toxicity in animals should be undertaken to determine their potential use in humans.
3. Tin-Based Metallodrugs
The therapeutic properties of triphenyltin acetate in mice tumors was observed in the early 1970s [
30], and this discovery triggered a very wide study of other organotin compounds against different cancer cells [
31,
32]. In this context, a recent study carried out by our group using very simple tricyclohexyltin(IV) compounds demonstrated the potential of tin compounds to overcome multidrug resistance as these metallodrugs are not substrates of the Pgp protein in K562 (leukemia), PANC-1 (pancreatic carcinoma), LN-229 and U87 (multiform glioblastoma) [
33].
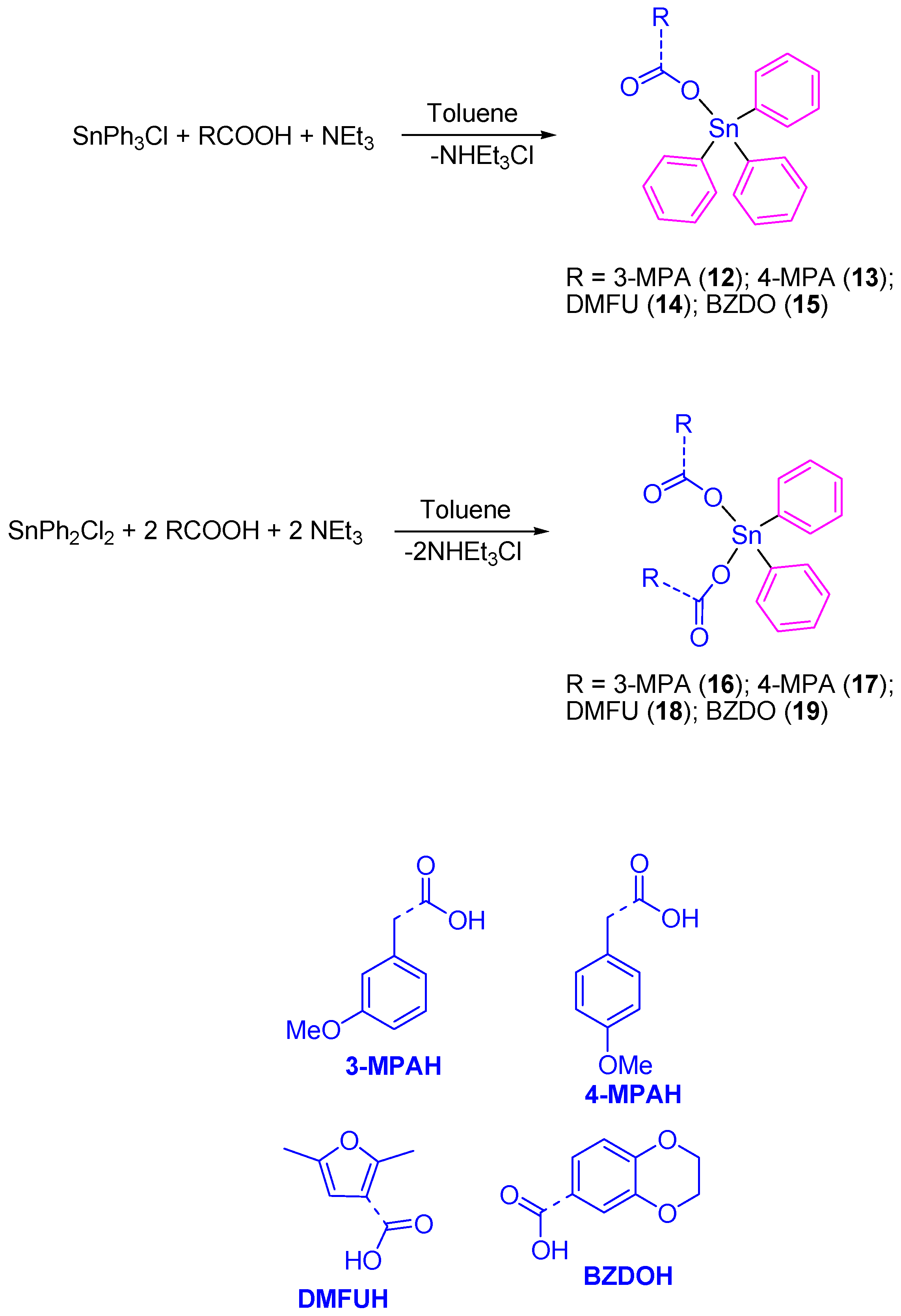
Our research group prepared a series of Sn(IV) compounds namely [SnPh
3(3-MPA)] (
12), [SnPh
3(4-MPA)] (
13), [SnPh
3(DMFU)] (
14), [SnPh
3(BZDO)] (
15), [SnPh
2(3-MPA)
2] (
16), [SnPh
2(4-MPA)
2] (
17), [SnPh
2(DMFU)
2] (
18), and [SnPh
2(BZDO)
2] (
19) by the reaction of the carboxylic acids 3-methoxyphenylacetic acid (3-MPAH), 4-methoxyphenylacetic acid (4-MPAH), 2,5-dimethyl-3-furoic acid (DMFUH) or 1,4-benzodioxane-6-carboxylic acid (BZDOH) with triphenyltin(IV) chloride or diphenyltin(IV) dichloride, respectively, in the presence of triethylamine (
Scheme 1).
All the tin(IV) compounds
12–
19 were characterized by multinuclear NMR spectroscopy, mass spectrometry, and IR, and were tested against human adenocarcinoma (HeLa), human myelegenous leukemia (K562), and human malignant melanoma (Fem-x) using MTT-based assays. The carboxylic acids showed no antipoliferative effect under physiological conditions, however, tin(IV) compounds (
12–
19) showed a dose-dependent antipoliferative effect toward all cell lines and on human PBMC and stimulated PBMC (
Table 2). The cytotoxic activity of the compounds was several times higher than that of cisplatin. Notably, compound
14 presented from 30 to 112 times higher activity than that recorded for cisplatin. In this study, we observed that triphenyltin(IV) derivatives presented lower IC
50 values against all the studied cancer cell lines than their corresponding diphenyltin(IV) counterparts [
34].
In addition to this study, an analogous triphenyltin(IV) compound containing the 2,6-dimethoxynicotinate ligand was tested against HeLa, K562, Fem-x, and on human peripheral blood mononuclear cells (PMBC) showing a high activity against all evaluated cancer cell lines with a moderate selectivity on K562 compared to unstimulated PBMC [
35].
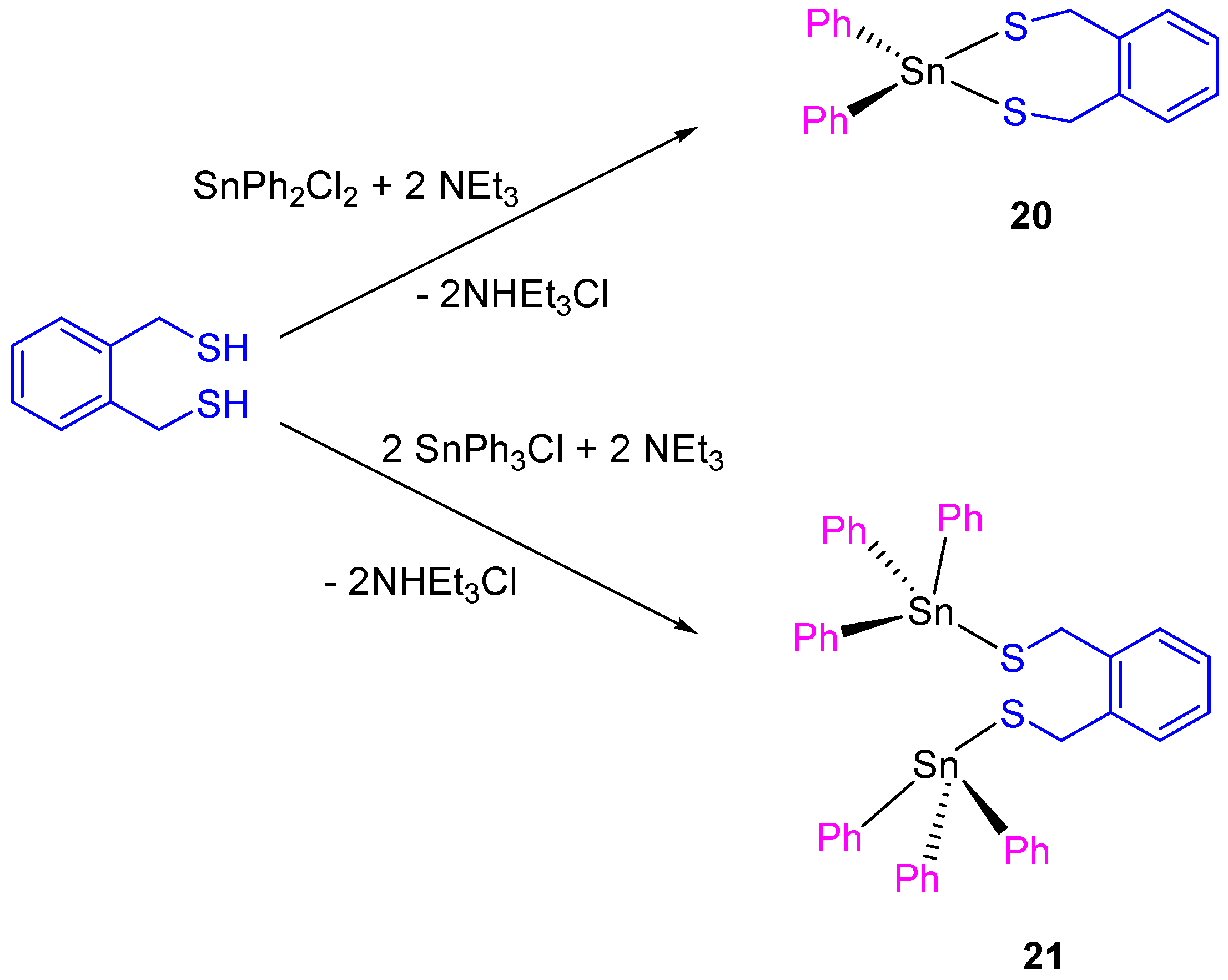
Following the work on tin(IV) compounds, additional thiolate complexes containing α,α’-dimercapto-
o-xylene ligand were synthesized (
Scheme 2). The compounds
20 and
21 showed a good activity against different cancer cell lines, with IC
50 values between 9.7 ± 0.2 and 21.1 ± 1.1 µM (
Table 2).
The dinuclear tin(IV) compound
21 is more cytotoxic than
20. This result was expected as compound
21 presents two SnPh
3 units which are normally associated with increase of cytotoxic activity due to the interaction of SnPh
3+ moieties with protein kinases DNA. The cytotoxic activity of
20 and
21 was lower than that reported for carboxylate tin(IV) complexes (
12–
19) [
36]. However, a more in depth study of compound
21 against HeLa and Fem-x cell showed the induction of an apoptotic cell death [
37].
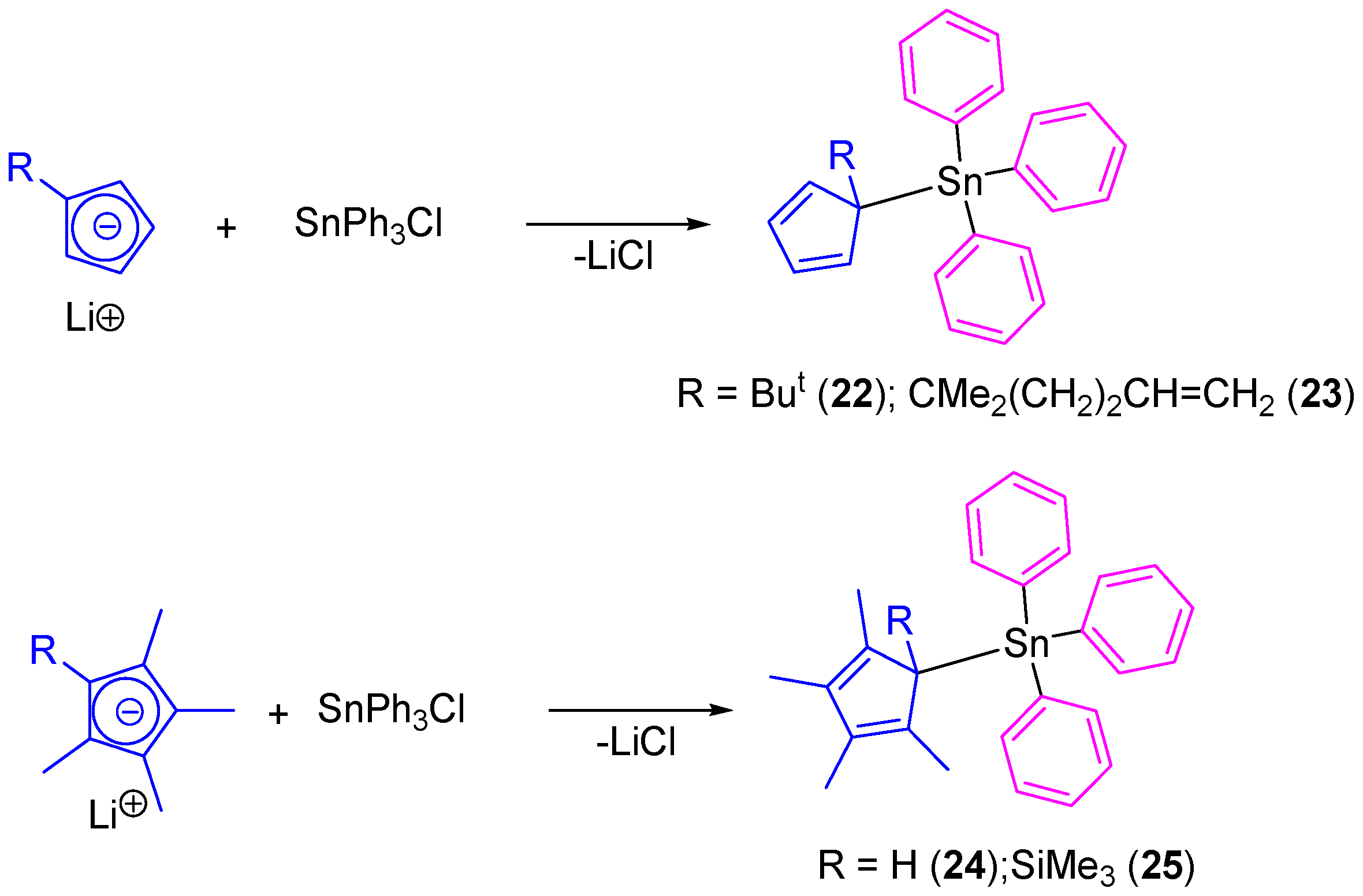
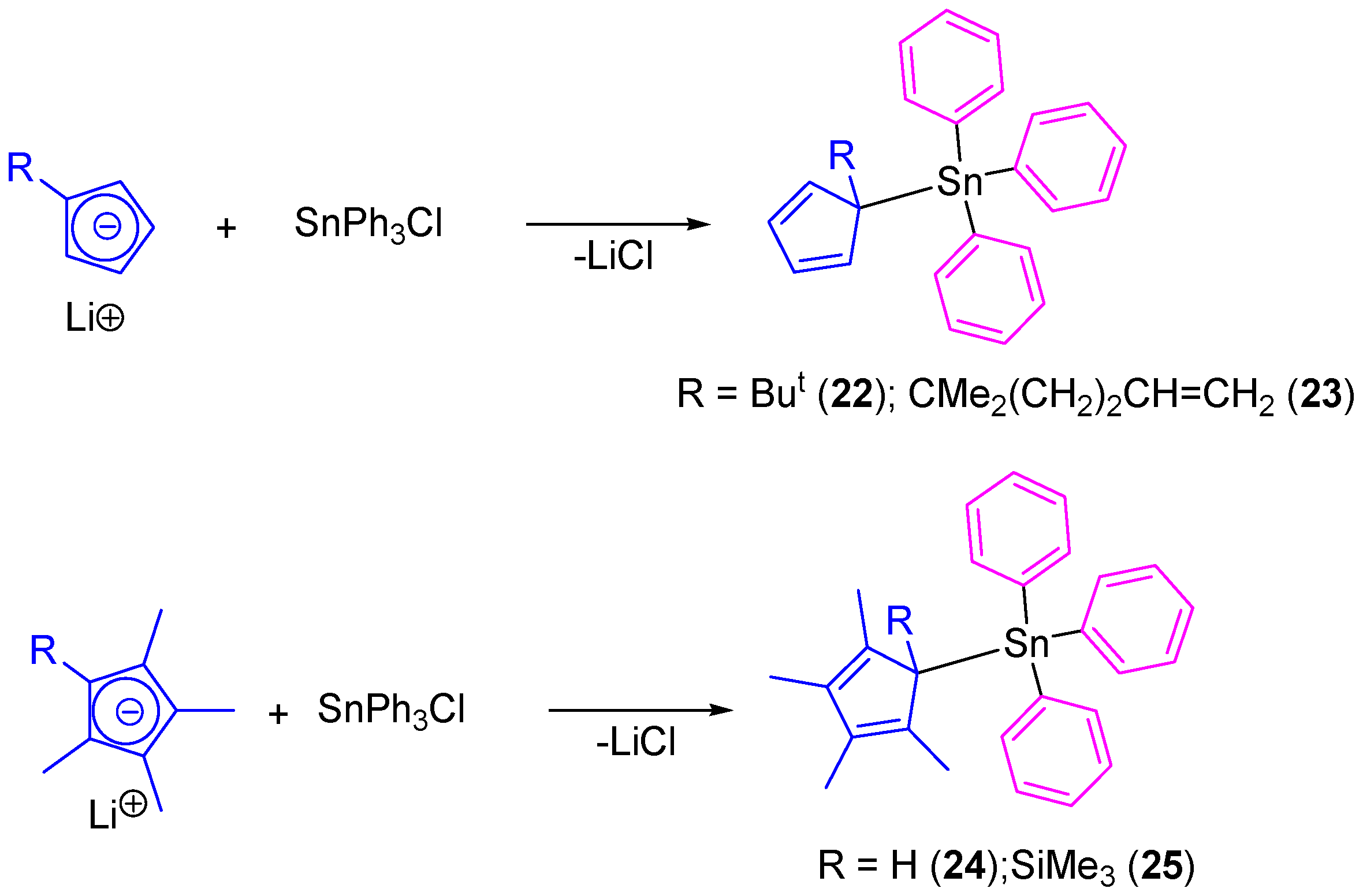
In another study, our group prepared tetraorganotin(IV) compounds containing cyclopentadienyl ligands (
22–
25) which were prepared by the simple transmetallation reaction of lithium cyclopentadienide derivatives with SnPh
3Cl (
Scheme 3) [
38]. All the compounds were isolated as single isomers even though the position of the double bonds makes possible the formation of a mixture of positional isomers.
Tin(IV) complexes (
22–
25) were tested in vitro against 8505C, A253, A549, A2780, and DLD-1 cell lines by using the sulforhodamine-B microculture colorimetric assay (
Table 3) [
39]. All the compounds showed a dose-dependent antiproliferative effect toward cell lines and presented lower IC
50 values than those observed for cisplatin against the same cell lines. From all the series of cyclopentadienyl-substituted tin compounds,
24 (which contains the tetramethylcyclopentadienyl moiety) presented the highest cytotoxic activities against all the studied cancer cell lines with IC
50 values between 0.037 and 0.085 µM (from 17 to 104 times higher than cisplatin). Compounds
22 and
23 presented similar activities (0.042–0.103 µM and 0.061–0.119 µM) while
25 had a lower cytotoxicity with IC
50 values between 0.163 and 0.384 µM.
In a subsequent study, a series of rare ionic triphenyltin(IV) chloride carboxylate complexes (
26–
28,
Figure 8) was synthesized and tested against 8505C, A253, A549, A2780, and DLD-1 cell lines (
Table 3). All the ionic tin(IV) compounds presented anticancer activities up to 50 times more active than those of cisplatin (for example in DLD-1 complex
26 has an IC
50 value of 0.103 µM compared to that of cisplatin of 5.14 µM). Therefore, from this series,
26 seems to be very promising for future applications in clinical trials due to its high solubility, high activity and its capacity to induce a clean apoptotic cell death. This compound affected the G1 and G2/M phases of the cell cycle. Its apoptotic action seems to be related to the interaction between SnPh
3+ moieties with protein kinases and DNA [
40]. In addition, the apoptotic properties of compound
26 and the interaction with caspases 2, 3, and 8 were studied in DLD-1 cells with only caspase 8 being found to be upregulated after 4 h. However, cells treated for 6 h showed an additional activation of caspase 2 and 8, which was in contrast with the results observed when treating the cells with cisplatin which only showed activation of caspase 8 and 9. These results suggest that
26 promotes a faster activation of apoptosis and that this was achieved in the DLD-1 cell line in a different way to that observed for cisplatin. Respectively, cisplatin promotes apoptosis by both intrinsic (mitochondrial pathway, caspase 9 dependent pathway) and external signals (extrinsic or death receptor pathway), while
26 induces apoptosis only via extrinsic receptor pathway [
40].
Further studies of the in vitro activity of
26 and
28 against 518A2 (melanoma), FaDu (head and neck carcinoma), HT-29 (colon cancer), MCF-7 (breast carcinoma), and SW1736 (thyroid cancer) cell lines showed the potent cytotoxic activity of
26 and
28 which induce apoptosis. These results were confirmed by the observation of membrane blebbing, translocation of phosphatidylserine, DNA fragmentation, and accumulation of cells in the Sub-G1 phase [
41].
Our group prepared two different 1D-polymeric triphenyltin(IV) carboxylate derivatives, based on the reaction of SnPh
3Cl with mesitylthioacetic acid and xylythioactic acid. The 1D-chains [{SnPh
3(O
2CCH
2SXyl)}
∞] (
29) (Xyl = 3,2-Me
2C
6H
3) and [{SnPh
3(O
2CCH
2SMes)}
∞] (
30) (Mes = 2,4,6-Me
3C
6H
2) were tested in vitro against 8505C, A253, A549, and DLD-1 cell lines observing that they present higher activity (from 8 to 85 times higher) than those of cisplatin (
Table 3) and between 285 and 2520 times higher than their gallium(III) and titanocene(IV) analogues, respectively [
42]. In addition, these studies showed that compounds
29 and
30 interacted with DNA by classical electrostatic interactions with intrinsic binding constants of 1.68 × 10
5 and 1.02 × 10
5 M
−1, respectively.
Thus, one can conclude from our results that tin compounds show potential due to the high cytotoxicity that they present in vitro, the possibility of overcoming multidrug resistance [
33], and the wide variety of cancer cells that they can treat [
43]. In addition, in the age of nanotechnology, their medicinal applications are being enhanced by simple conjugations with silica-based nanomaterials. For example, our group is now working on the support of novel organotin(IV) compounds onto nanostructured silica [
44]. Our latest results showed excellent in vitro [
45] and in vivo [
44] behavior of the new encapsulated systems which have the potential to be used in the future in phase I clinical trials.
4. Titanium-Based Metallodrugs
Although Ti
3+ can exist in aqueous media, the aqueous chemistry of titanium is dominated by oxidation state +4 and the tendency of free Ti
4+ to hydrolyze and precipitate, ultimately forming insoluble TiO
2, is very high. However, hydrolytic reactions can be minimized by surrounding the metal with the appropriate ligands which decreases the rate of the hydrolysis reactions. Thus, the titanium β-diketonate complex, budotitane, was the first non-platinum metal complex to enter clinical trials for treatment of cancer. In this context, cyclopentadienyl ligands are also ideal candidates for improving the hydrolytic stability of titanium(IV) with potential anticancer properties of titanocene dihalide derivatives being observed by Köpf-Maier and Köpf in the 1980s [
46].
The preclinical trials of titanium compounds indicated their potential as therapeutic metallodrugs against different tumors [
47]. The main biological target of titanium-based metallodrugs is the inhibition of DNA synthesis, triggering apoptosis [
48]. Some additional recent studies have reported the inhibition of the enzyme topoisomerase II by titanocene dichloride and this, therefore, may be an alternative cell death induction pathway [
49].
Titanocene dichloride was also studied in phase I clinical trials in 1993 [
50] and later in phase II clinical trials [
51,
52] and became very important in the field of antitumor metallodrugs. Although the results of phase II clinical trials were not satisfactory because of the lack of activity against the studied tumors, the excellent research on titanium compounds published by Tacke, Meléndez, McGowan, Baird, Valentine, and Tshuva, reignited the interest in novel titanium compounds with anticancer properties [
53,
54,
55,
56,
57,
58,
59,
60].
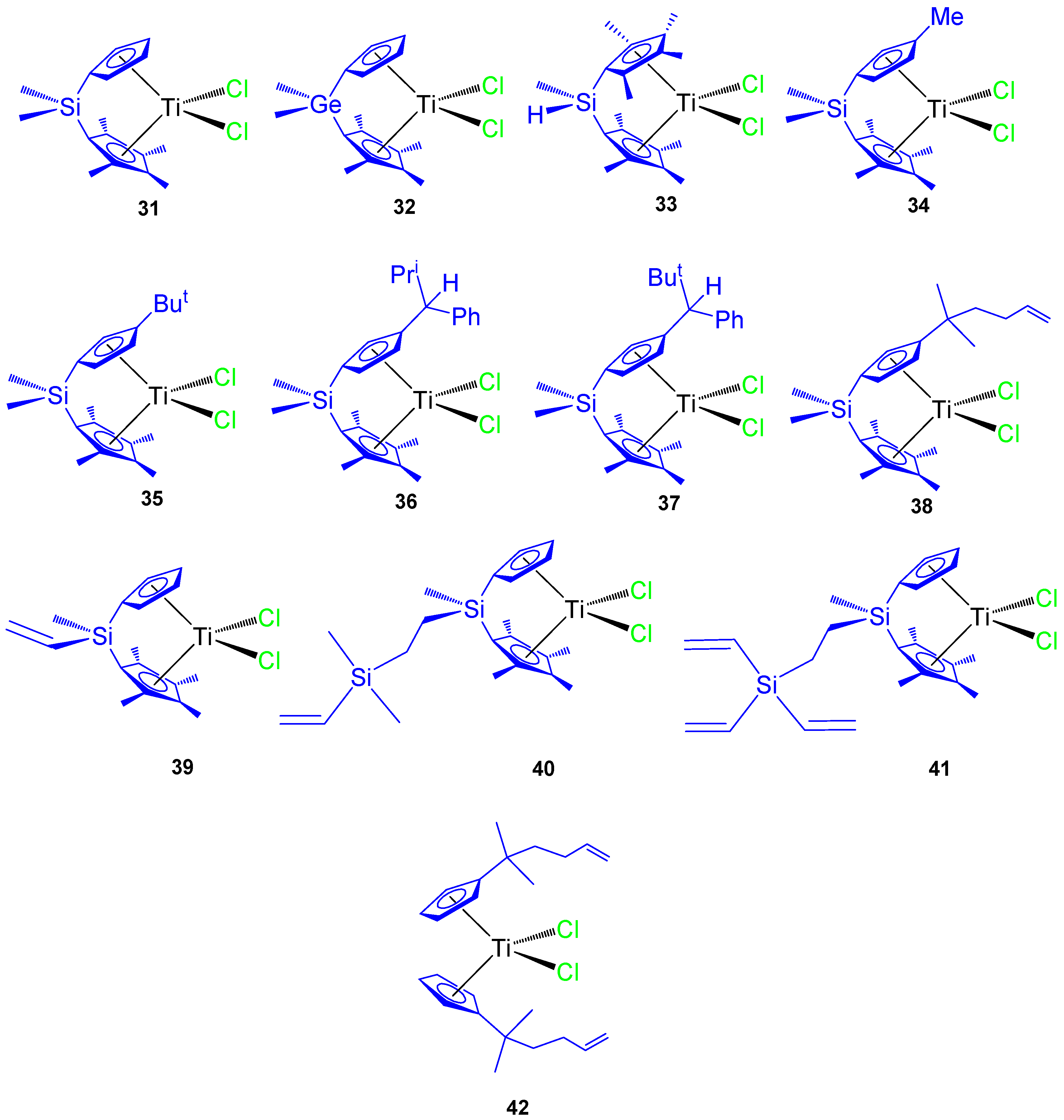
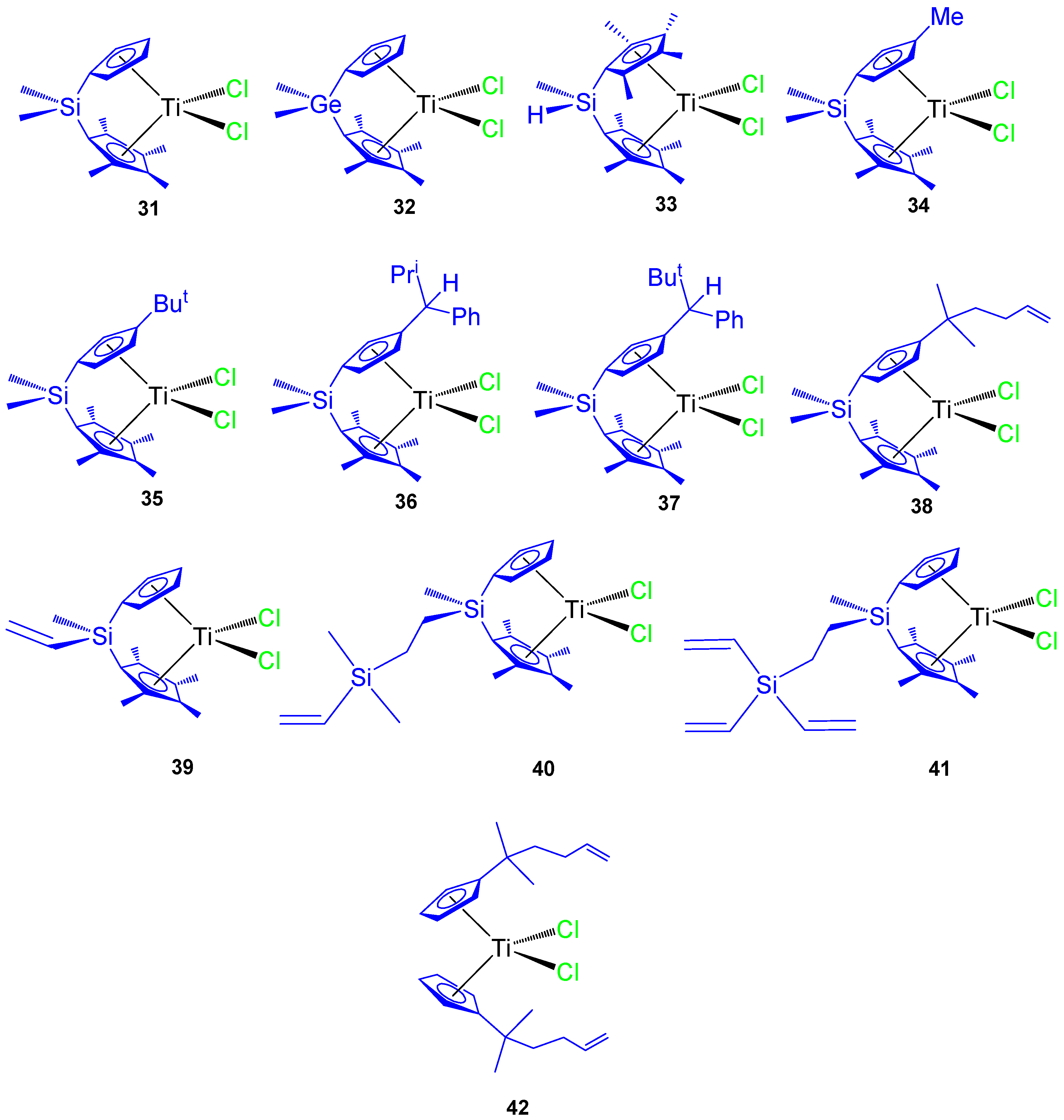
Since 2007 our research group has synthesized different titanocene derivatives which have demonstrated high activity against a series of cancer cell lines. In our first study, titanocene and
ansa-titanocene compounds with different alkyl and alkenyl ligands (
Figure 9 compounds
31–
42) were prepared and characterized. Most of the compounds were active against all the studied cancer cell lines and the activity was dependent on the substituent at the Cp ring or at the
ansa-bridge [
61,
62]. Of special interest were the alkenyl-substituted compounds
38,
39, and
42 which showed improved cytotoxic activity against the studied cell lines HeLa, K562, and Fem-x (
Table 4).
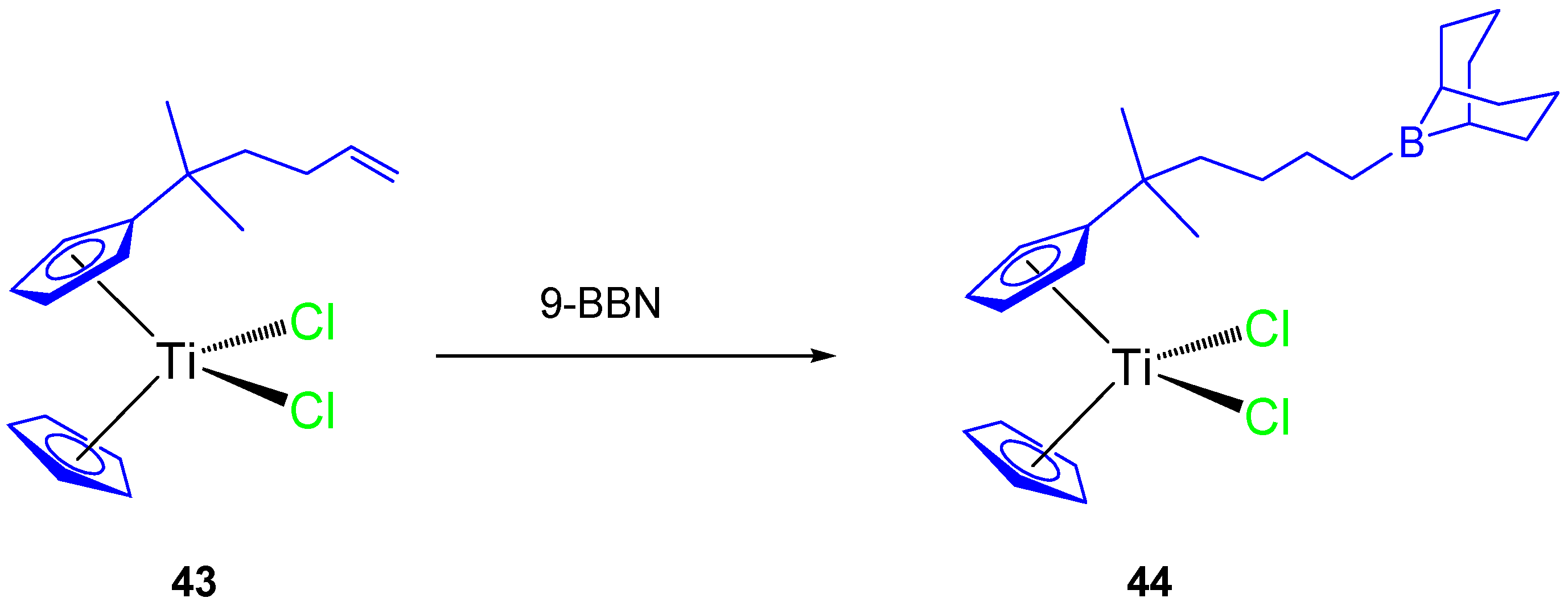
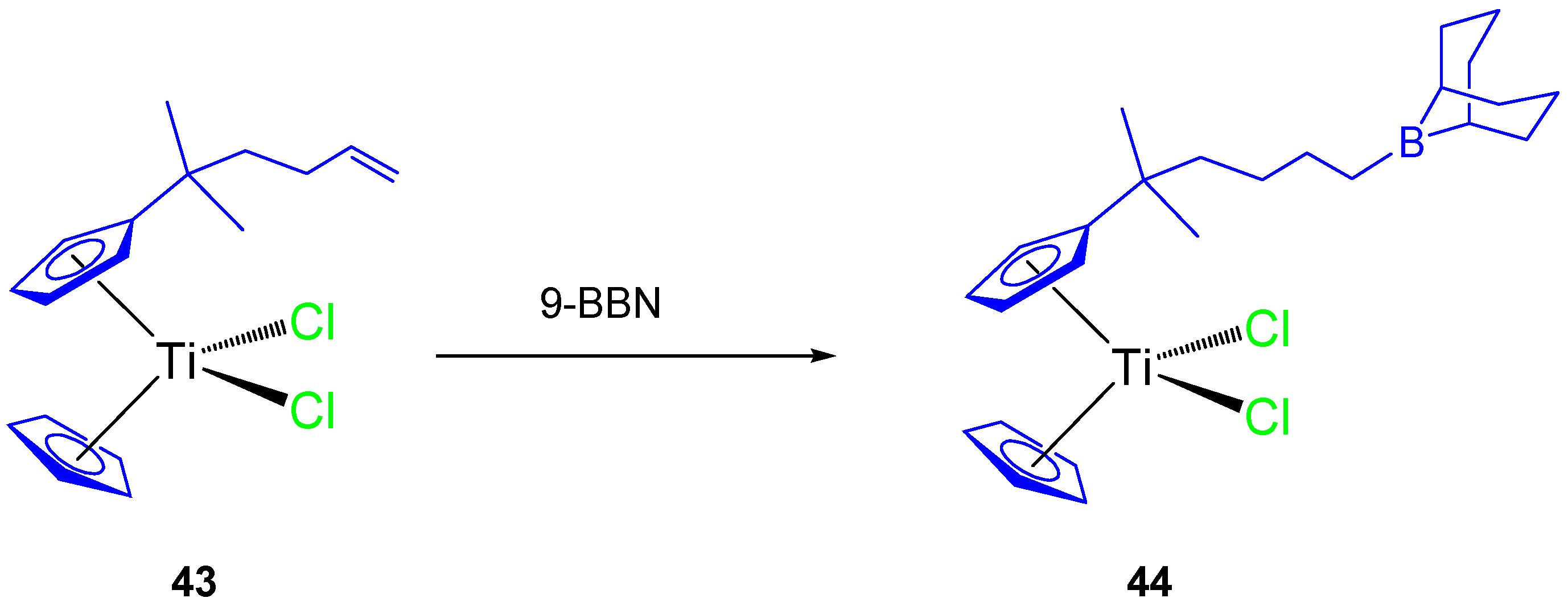
Thus, a subsequent study with an alkenyl monosubstituted titanocene complex (
43) and its 9-BBN hydroboration product (
44,
Scheme 4) were synthesized and characterized. Both compounds were tested against HeLa, K562, and MBA-MB-361 cell lines [
63] and showed a dose-dependent antiproliferative effect towards all cell lines and on human PBMC and stimulated PBMC (
Table 4).
The alkenyl-substituted complex
44, presented good activity against K562 (IC
50 96.6 ± 3.4 µM) and moderate activity on HeLa (IC
50 149.2 ± 2.9 µM) and Fem-x (IC
50 133.6 ± 9.4 µM), while complex
43 presented only moderate activity on K562, HeLa ,and Fem-x (
Table 4).
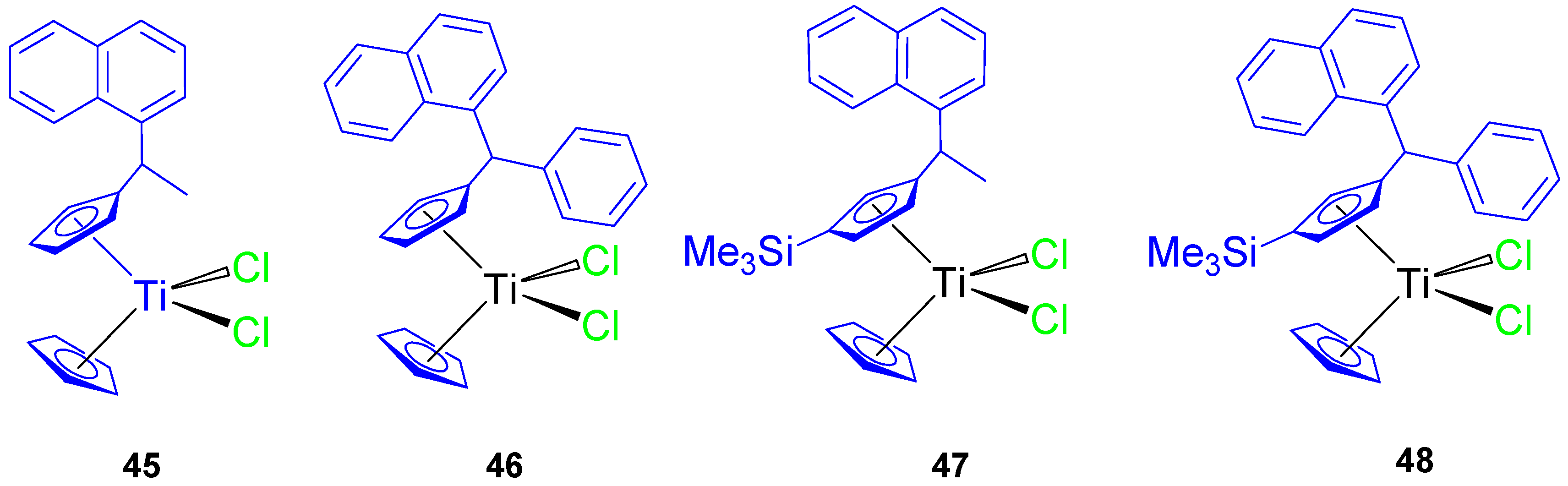
Subsequently, a series of naphthyl-substituted titanocene compounds (
45–
48) were also synthesized and characterized by our group (
Figure 10) [
64]. The molecular structure of
46 was established by single-crystal X-ray diffraction studies.
In anticancer tests against 8505C, A549, A2780, DLD-1, and FaDu, the titanocene(IV) complexes (
45–
48) showed a significant cytotoxic activity with IC
50 values (
Table 5) lower than titanocene dichloride. Compound
48 was the most active of all the tested compounds, with IC
50 values between 35.65 ± 4.95 and 69.02 ± 1.67 µM. The improvements in cytotoxic activity of
48 were due to the presence of the trimethylsilyl group.
In addition, several carbon and silicon-bridged
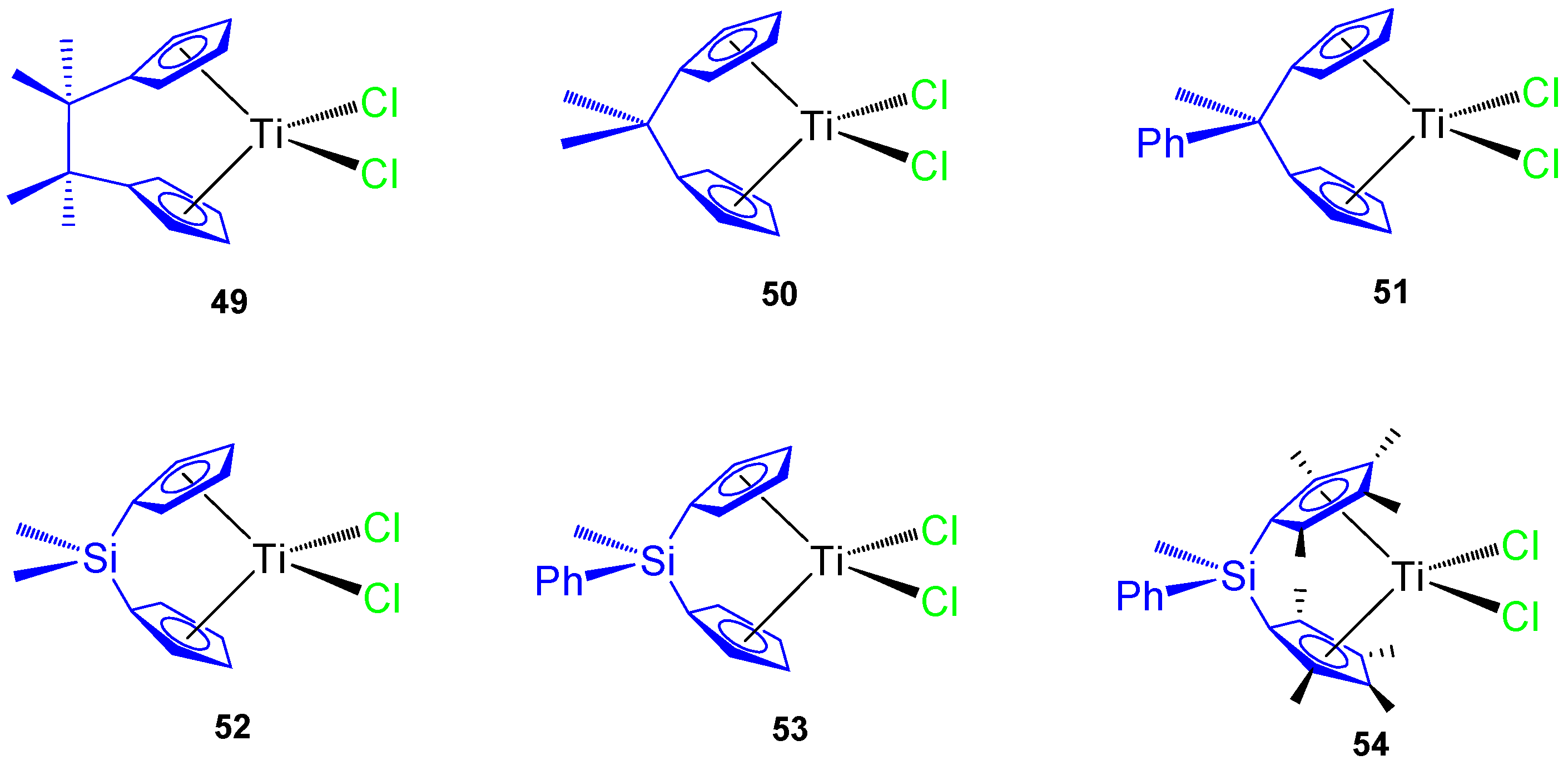
ansa-titanocene(IV) derivatives were synthesized (
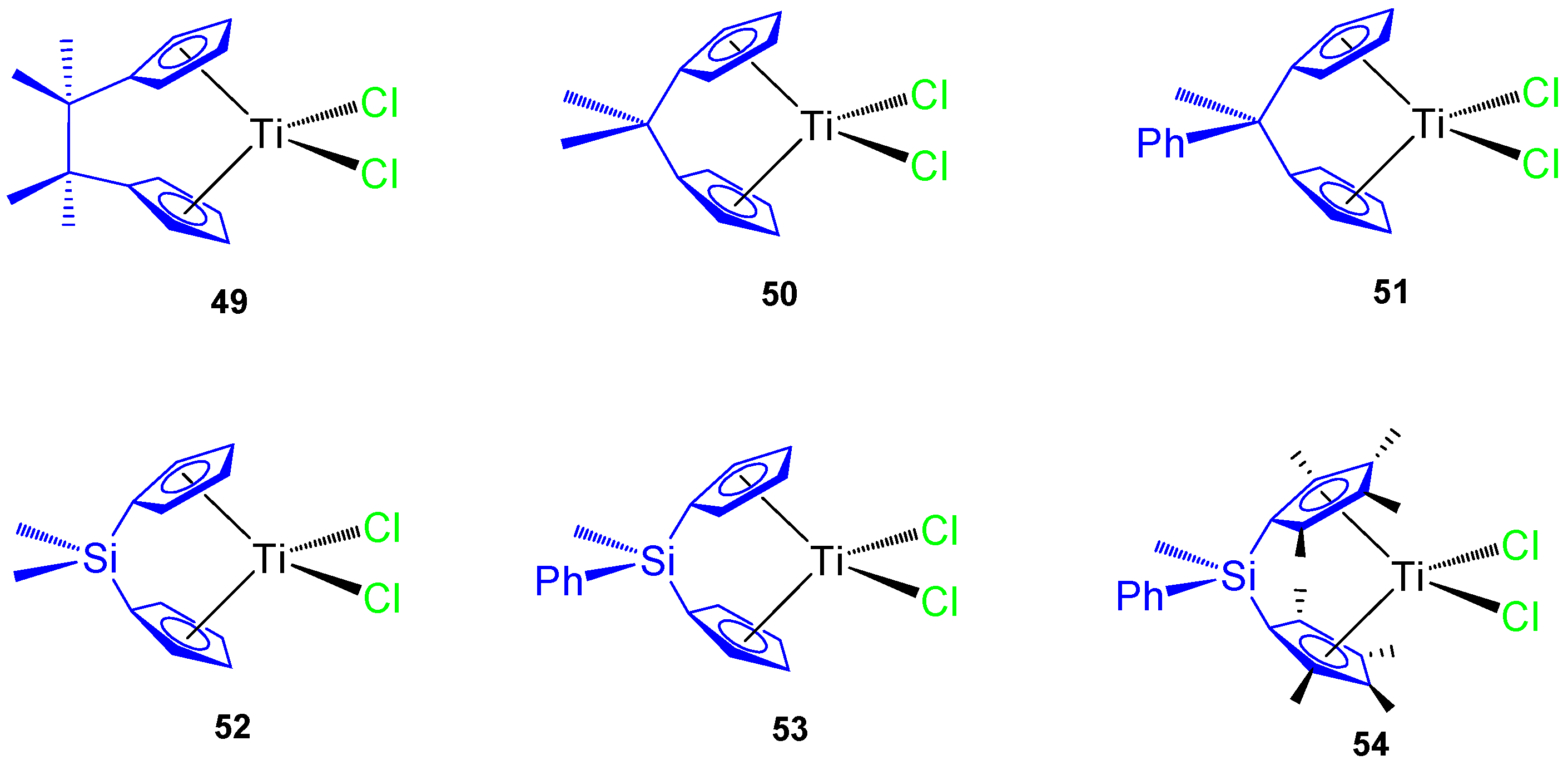
Figure 11) and tested against different tumor cell lines, namely murine melanoma B16, human melanoma A375, colon cancer HCT116 and SW620, prostate cancer LNCaP and DU145, and mouse colon cancer CT26CL25.
The C-bridged
ansa-titanocene derivatives showed an increase in cytotoxic activity with an ethylene bridge (complex
49) and poor activity with methylene (complexes
50 and
51) while the incorporation of a phenyl ring attached directly to the bridging atom decreases the viability of the cancer cells in carbon-bridged compounds (
51), but increases the viability of cancer cells in silicon-bridged systems (
53 and
54) (
Table 6). The most cytotoxic titanocene complexes
49 and
54 were also tested against primary mouse keratinocynates and lung fibroblasts while observing a large viability of both primary cells and that
54 was nontoxic to primary cells [
65]. In addition,
49 and
54 showed accumulation of hypodiploid cells in subG compartment in cisplatin resistant HCT116 and SW620.
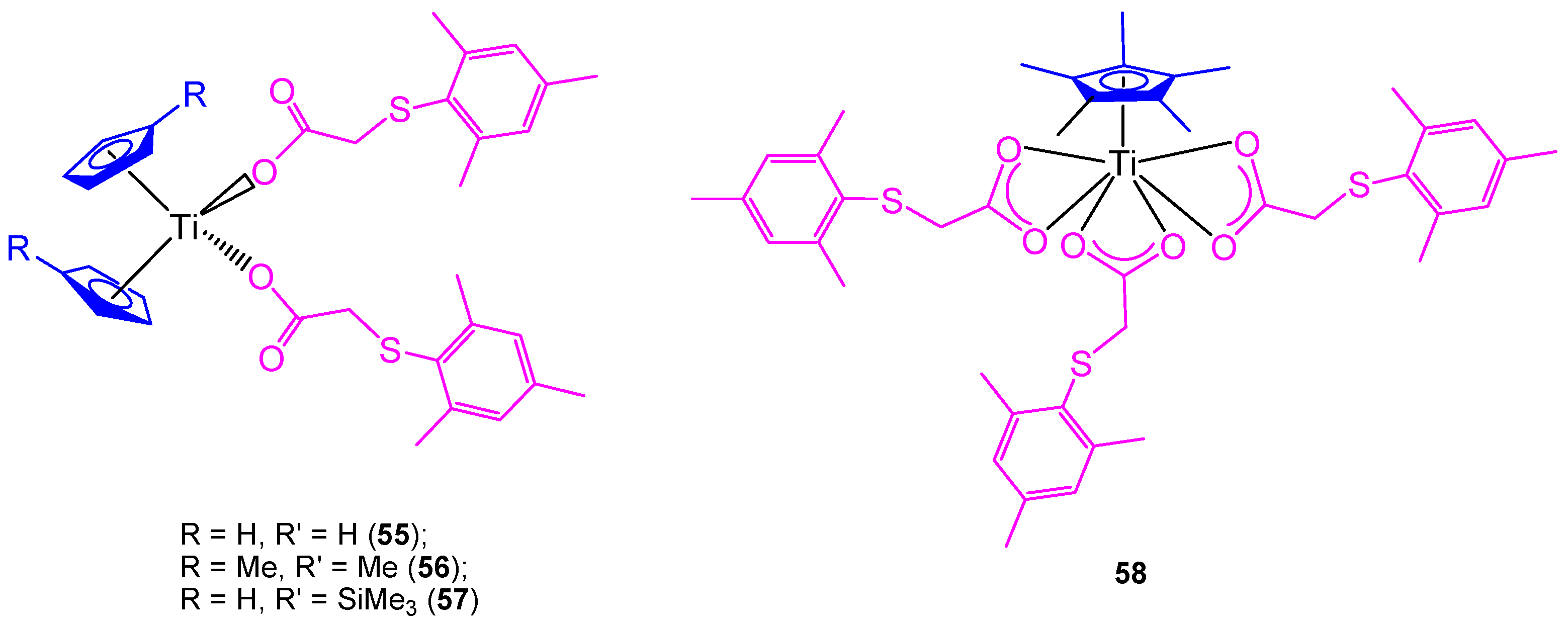
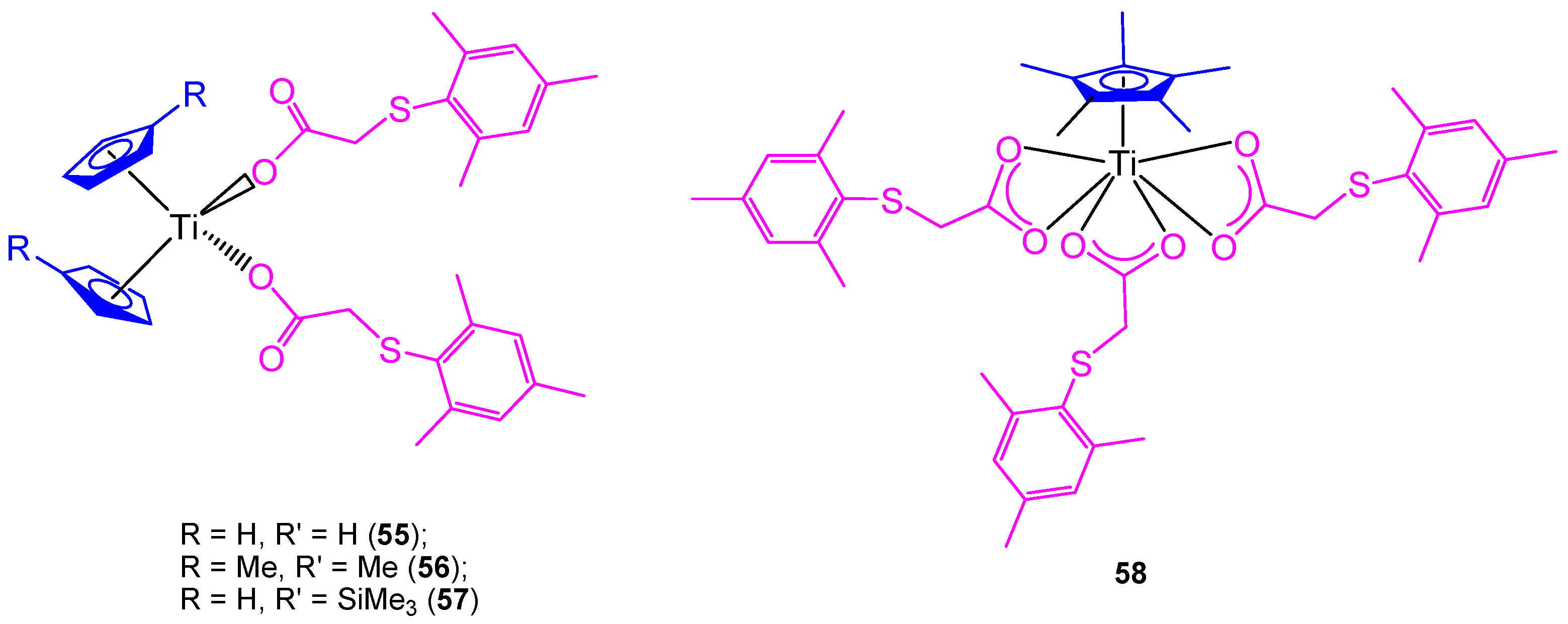
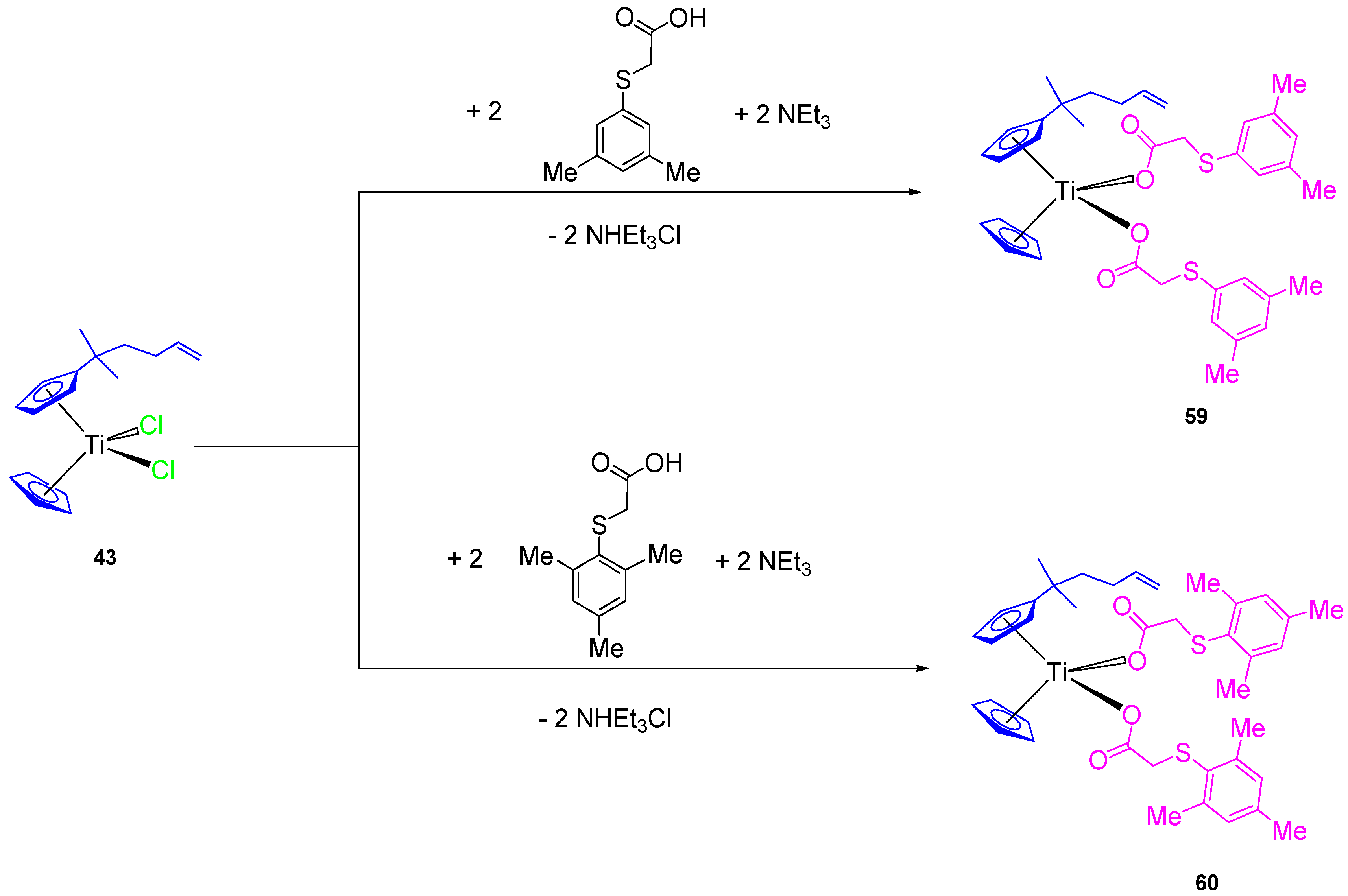
All the previously described titanocene and
ansa-titanocene compounds were chloride derivatives, however, a complete study of the substitution of the chlorido by carboxylato ligands was carried out using mesitylthioacetic acid and different cyclopentadienyl ligands (
Figure 12) [
66].
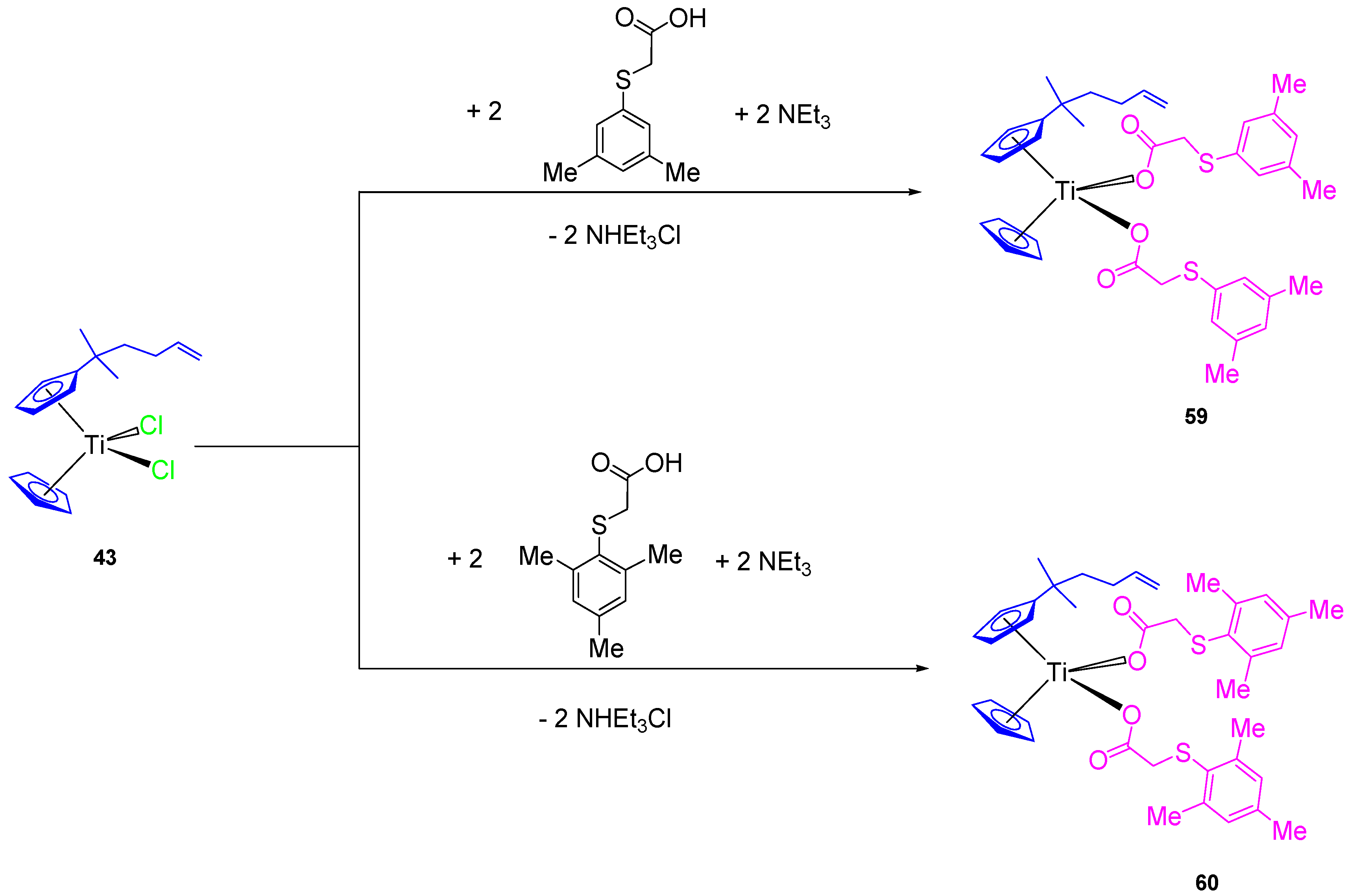
In addition, two new alkenyl-substituted titanocene(IV) carboxylate complexes containing the mesitylthioacetato and the xilylthioacetato ligands (
59 and
60, respectively,
Scheme 5) were synthesized and characterized. The comparison of cytotoxic activities of the titanocene(IV) carboxylate and titanocene(IV) dichloride against 8505C, A253, A549, and DLD-1, showed that titanocene(IV) carboxylates (
59 and
60) are less active against all the studied cells than their corresponding dichloride counterpart (
43) (
Table 5), indicating that the effect of the carboxylato ligands on the cytotoxicity is not synergistic but negative in the case of alkenyl-substituted titanocene compounds [
67]. This was confirmed in the DNA interaction tests, where complex
43 showed a higher intrinsic binding constant than
59 and
60.
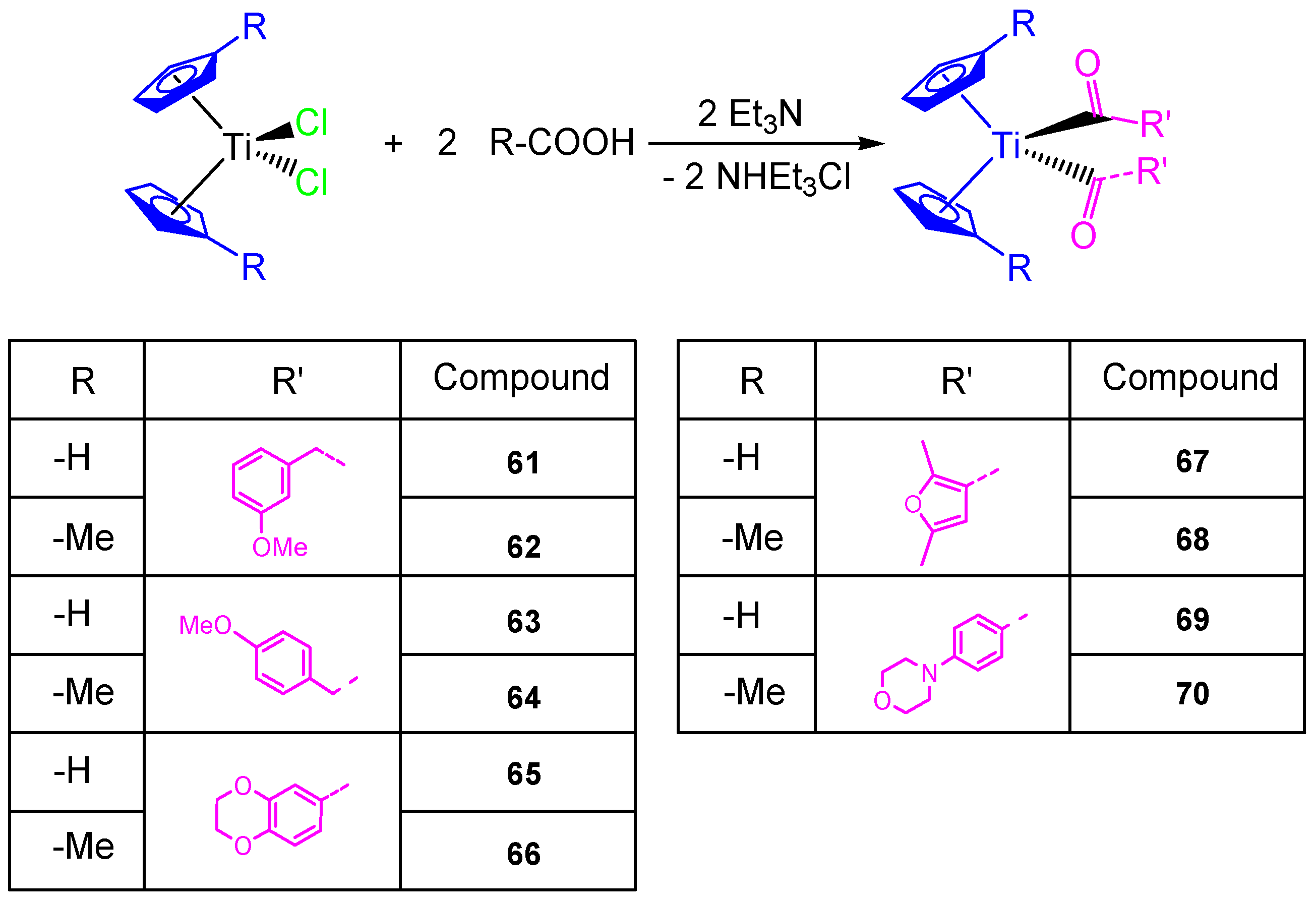
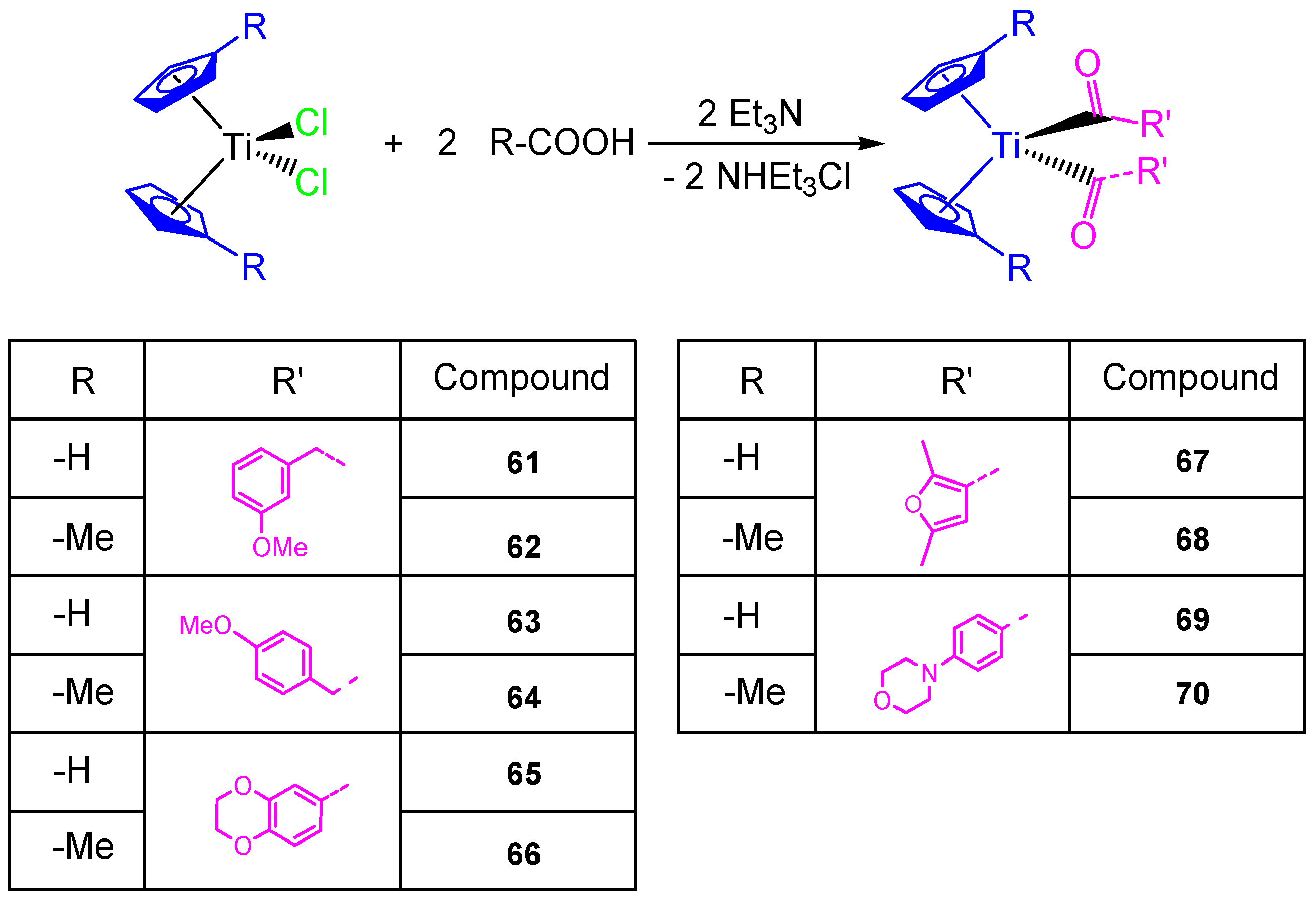
This study on the influence of carboxylato ligands on the cytotoxic activity of titanocene complexes was completed by the preparation of a wide variety of titanocene carboxylate derivatives of the type [Ti(η
5-C
5H
5)
2(OOC-L)
2] and [Ti(η
5-C
5H
4Me)
2(OOC-L)
2] with different carboxylato ligands such as 3-methoxyphenylacetato, 4-methoxyphenylacetato, 1,4-benzodioxane-6-carboxylato, 2,5-dimethyl-3-furoato, and 4-(4-morpholinyl)benzoato (
Scheme 6). All the carboxylate compounds
61–
70 showed a higher cytotoxic activity than [Ti(η
5-C
5H
5)
2Cl
2] or [Ti(η
5-C
5H
4Me)
2Cl
2] against ovarian cell line (A2780), with IC
50 values from 40.54 ± 4.39 to 82.14 ± 8.95 µM (
Table 5). In addition, the DNA binding studies carried out in simulated body fluid showed the weak interaction of the titanocene compounds with DNA [
68].
Our group also synthesized titanocene compounds containing the α,α’-dimercapto-
o-xylene as a thiolato ligand (
71 and
72,
Figure 13) and tested their efficacy against human tumor cell lines HeLa, Fem-x, K562 (
Table 4) [
36]. When the biological activity was analyzed, an improvement of the cytotoxic activity was observed compared with [Ti(η
5-C
5H
5)
2Cl
2] and [Ti(η
5-C
5H
4Me)
2Cl
2] against the cell lines tested K562, HeLa, and Fem-x with a higher cytotoxicity of
72 and a slight preference against K562 (
Table 4) [
36].
Our group also synthesized two titanium(IV) complexes anchored by a tripodal diamine bis(phenolate) ligands (
73 and
74) which showed hydrolytic stability and a high cytotoxic activity against HeLa, K562, Fem-x and MDA-MB-453 with IC
50 values between 22.4 ± 1.2 and 48.9 ± 0.8 µM [
69].
After the extensive study of our group on the biological applications of titanocene derivatives with different substituents either at the Cp ring or directly bound to titanium (
31–
72), we observed that most of the synthesized compounds (especially those containing thiolato or carboxylato ligands) show a low hydrolytic stability. Therefore, the anticancer action of these compounds is usually due to decomposition products which are soluble in water and/or DMSO and that are formed after the elimination of one or more ligands [
70]. Thus, we focused on the formulation of these titanium derivatives via functionalization of mesoporous silica-based nanostructured materials such as MCM-41, SBA-15, MSU-2, alumina or hydroxyapatite [
71,
72,
73,
74] in order to overcome the problems associated with the low hydrolytic stability of titanocene compounds.
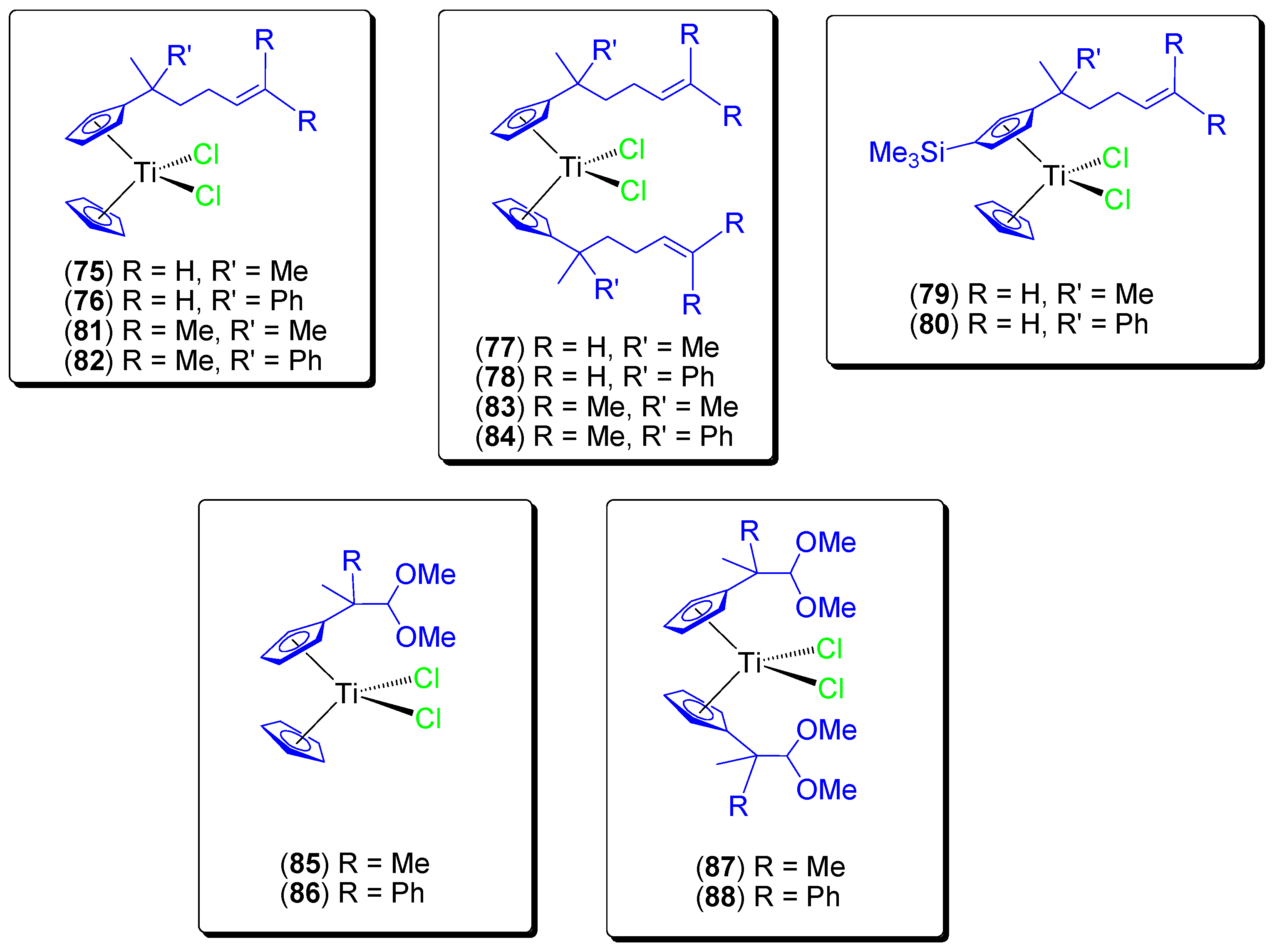
During these studies and in an effort to encapsulate titanocene compounds on KIT-6, additional alkenyl substituted (
75–
80) [
75] and ether-substituted (
81–
84) [
76] titanocene(IV) dichloride compounds were synthesized (
Figure 14), characterized and tested in vitro against a wide variety of cancer cell lines (
Table 7). We observed a very high cytotoxicity (IC
50 values in the range of those described in the literature for the most active cytotoxic titanocene compounds such as titanocene-Y synthesized by Tacke) with high selectivity towards cancer cell lines.
Finally, after the incorporation of the compounds in KIT-6, a higher Ti-uptake by the treated cancer cells (from 4% to 23% of the initial amount of Ti) was observed when compared with the “free” titanocene compounds giving clear insights on the positive effect of the encapsulation with nanostructured silica.







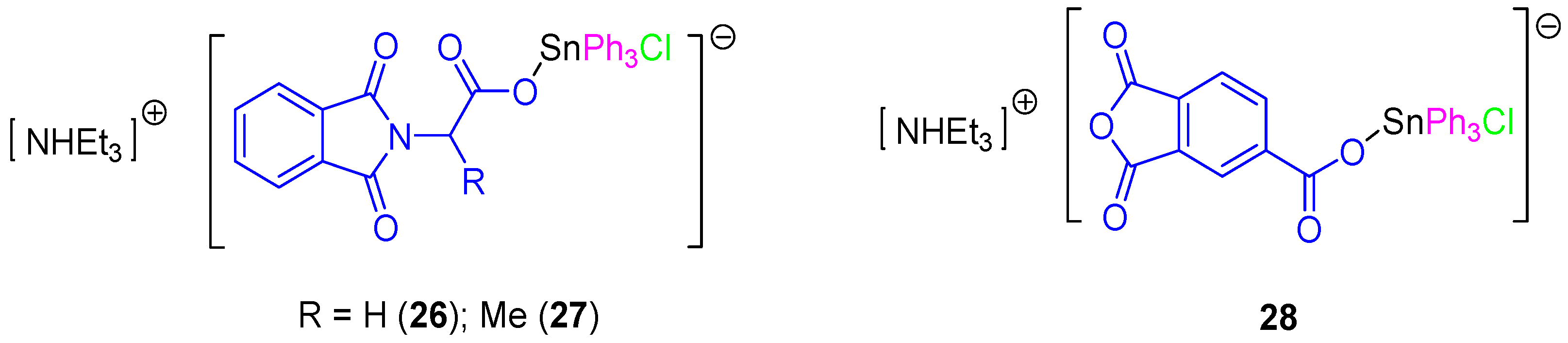
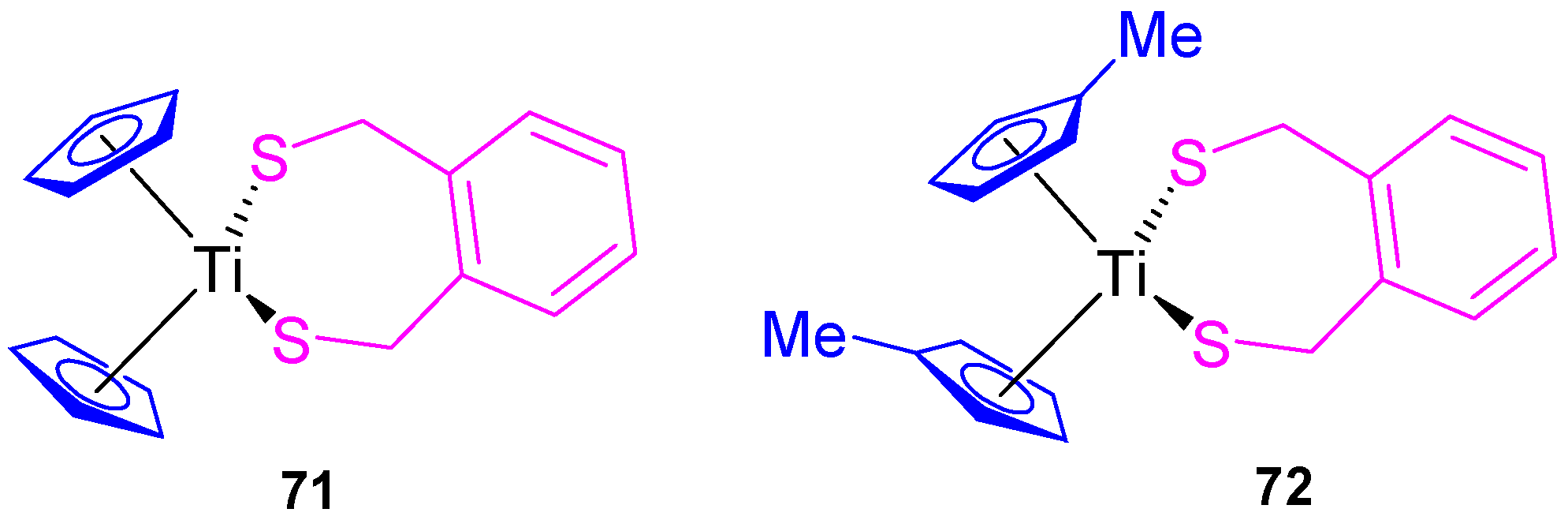


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
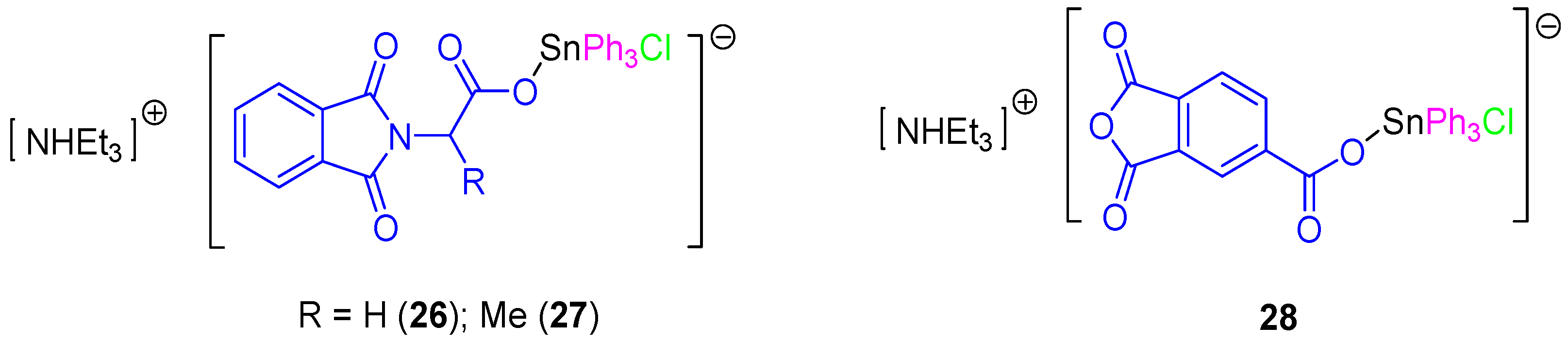{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
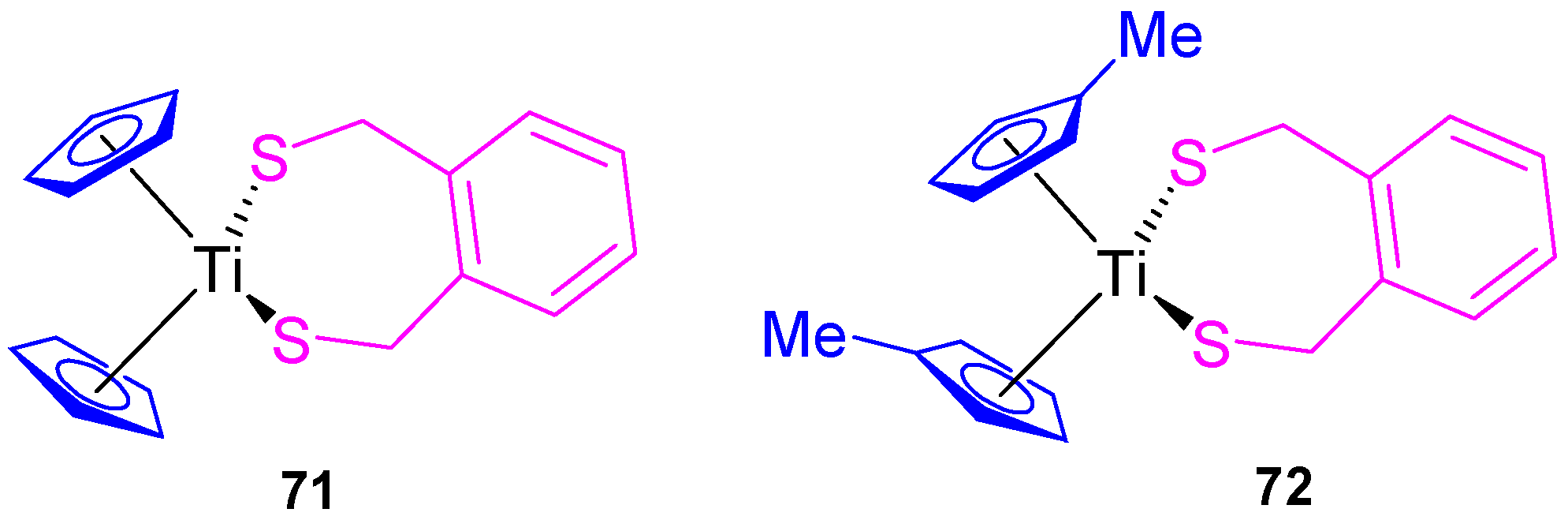{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}