
Optical Properties of Heavily Fluorinated Lanthanide Tris β-Diketonate Phosphine Oxide Adducts

Abstract
:
1. Introduction
2. Results and Discussion
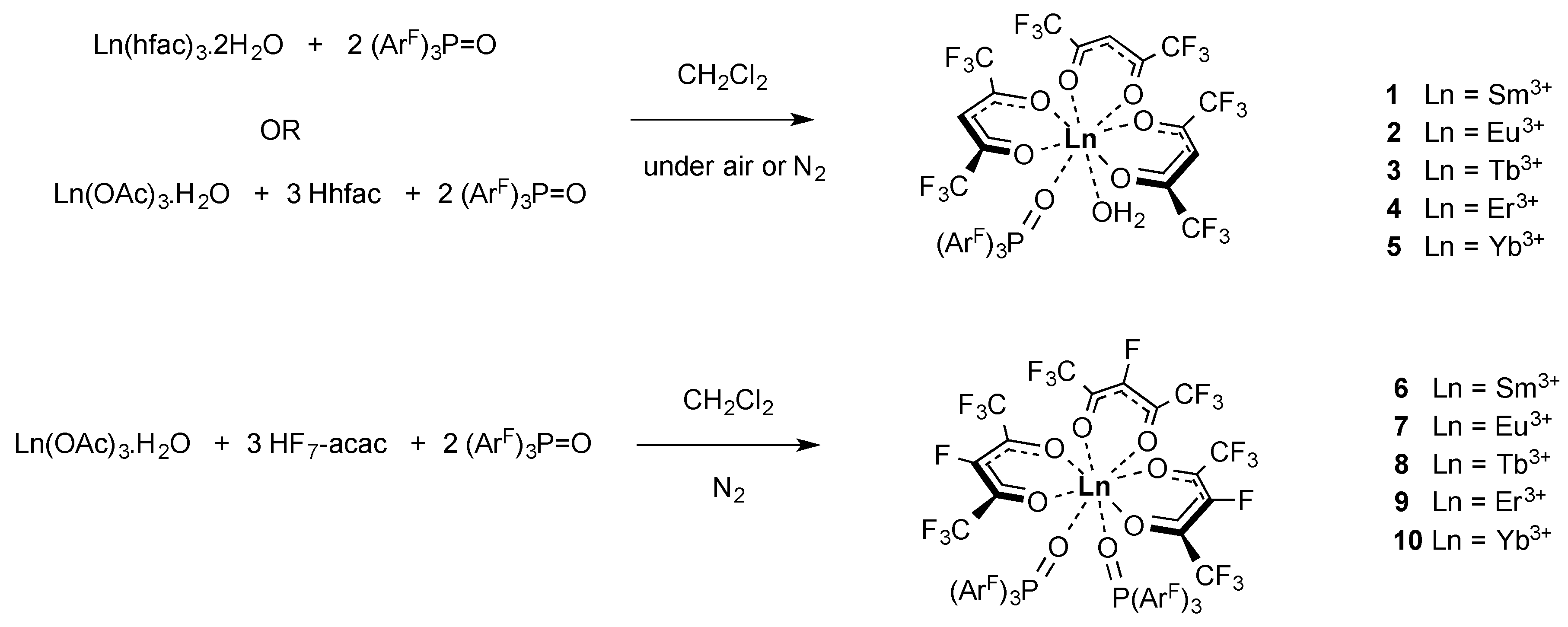
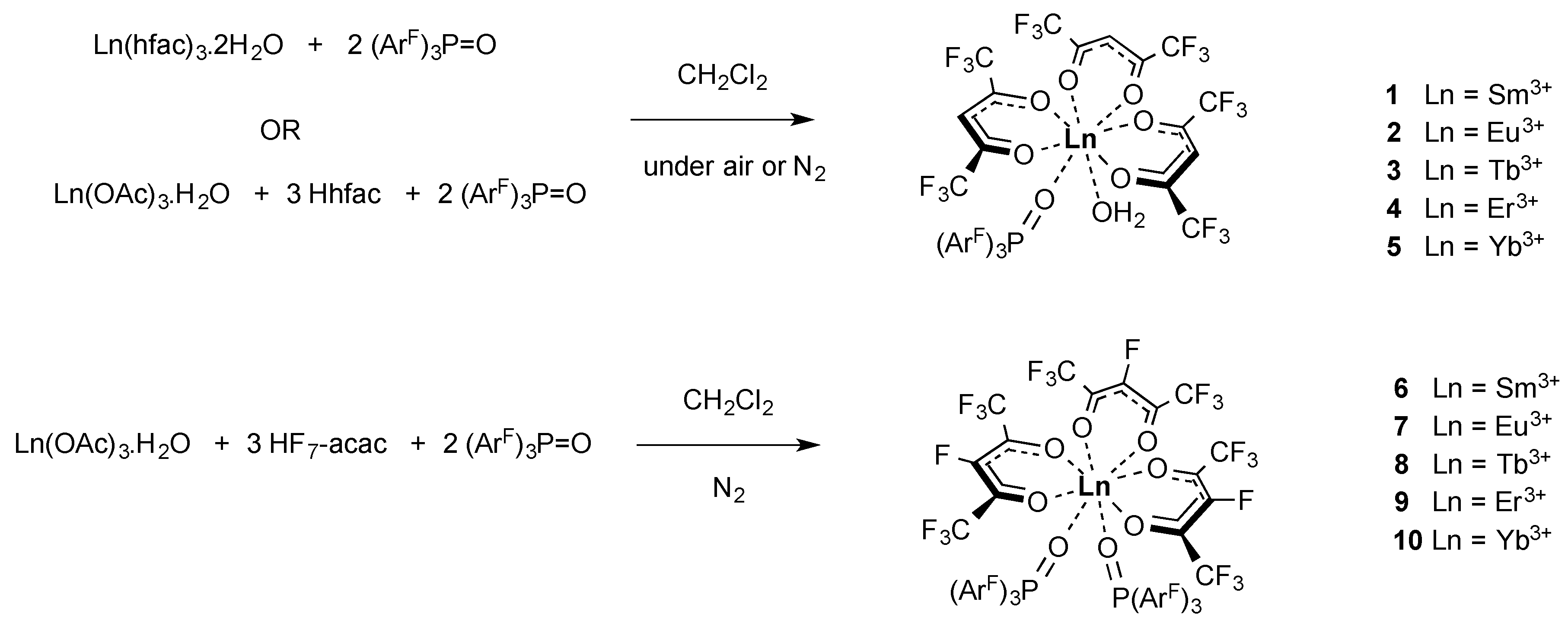
2.1. Synthesis of the Complexes
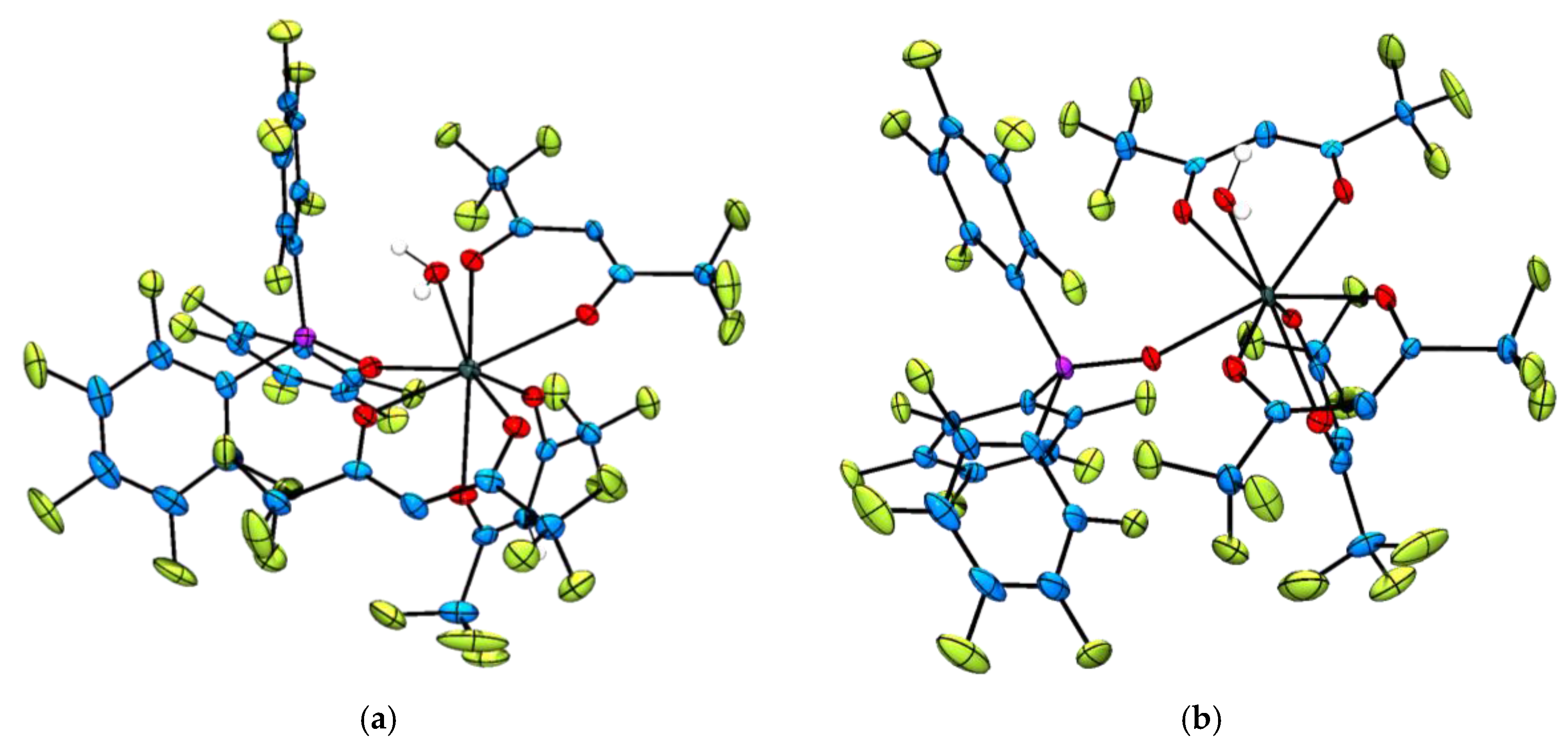
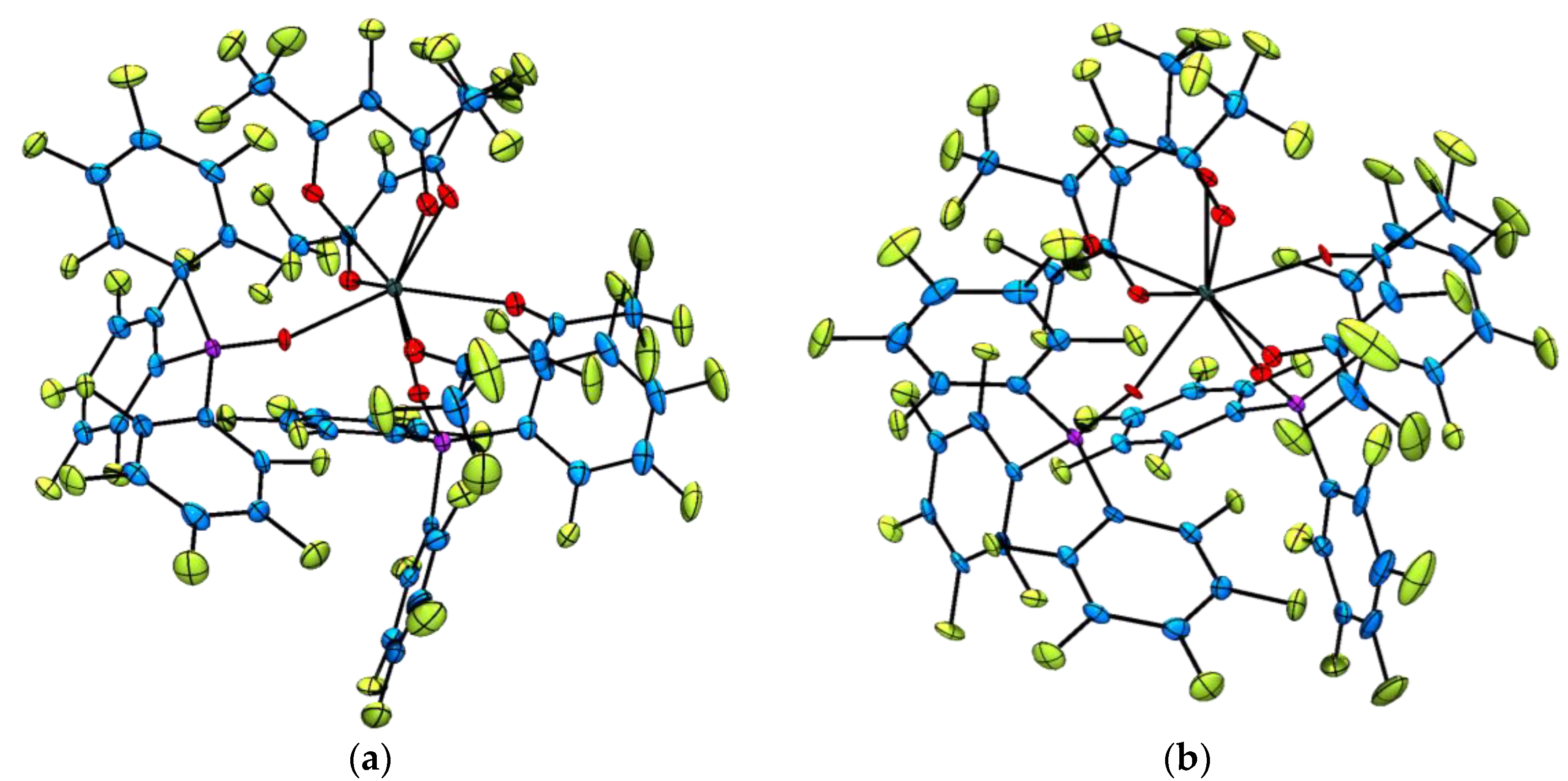
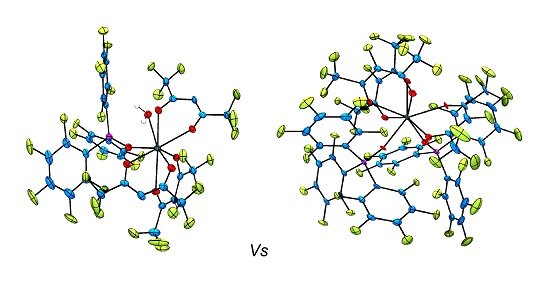
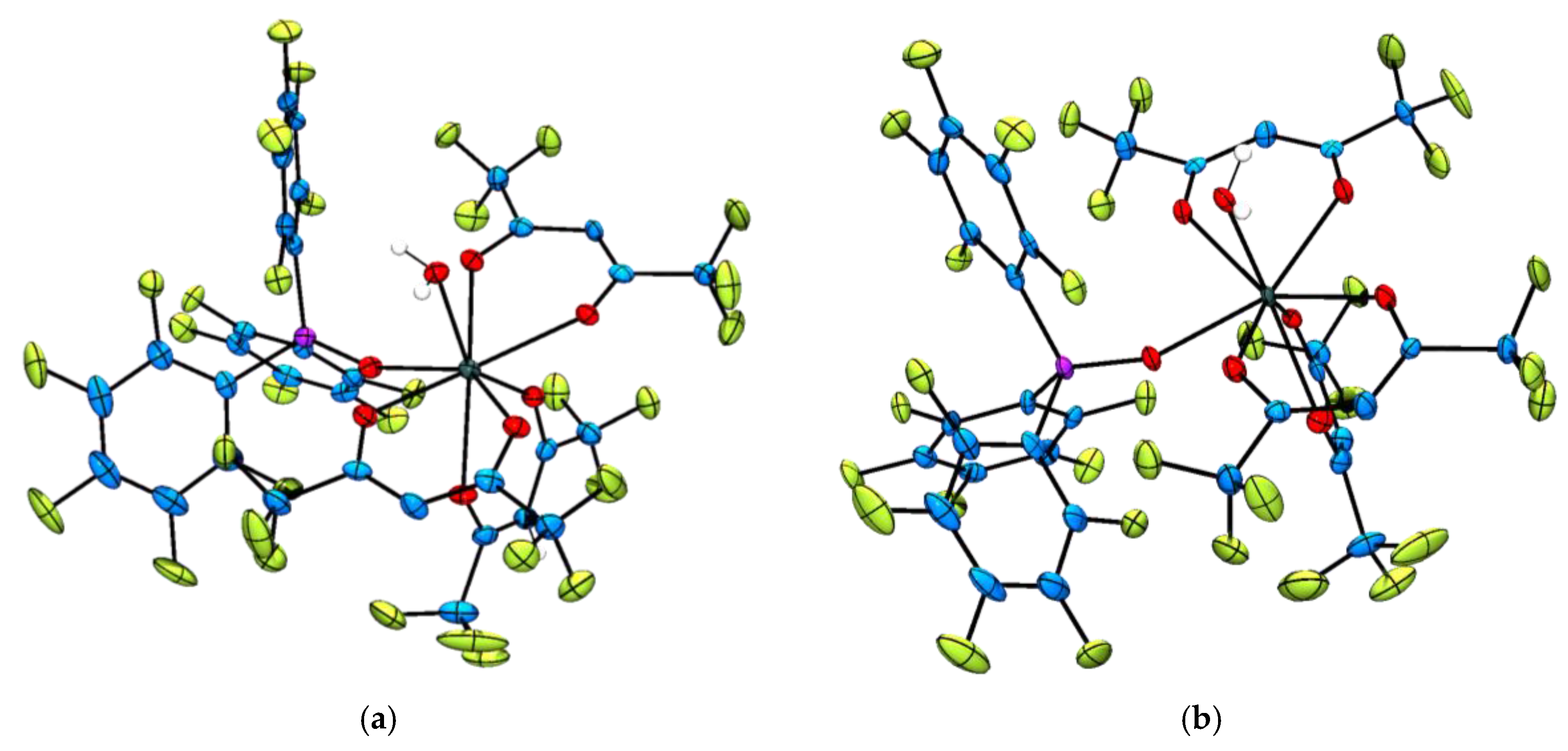
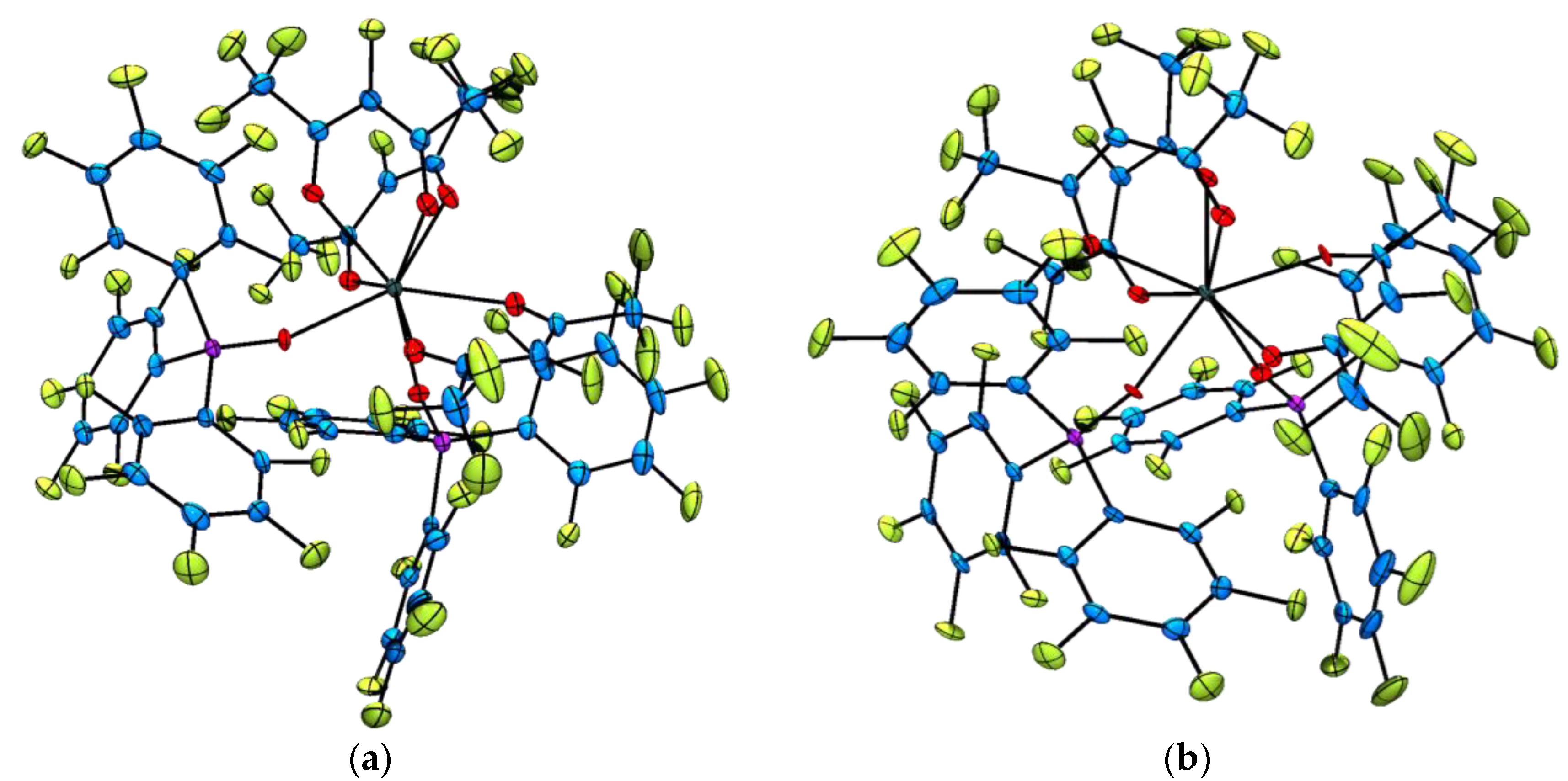
2.2. Single Crystal X-ray Diffraction Analysis
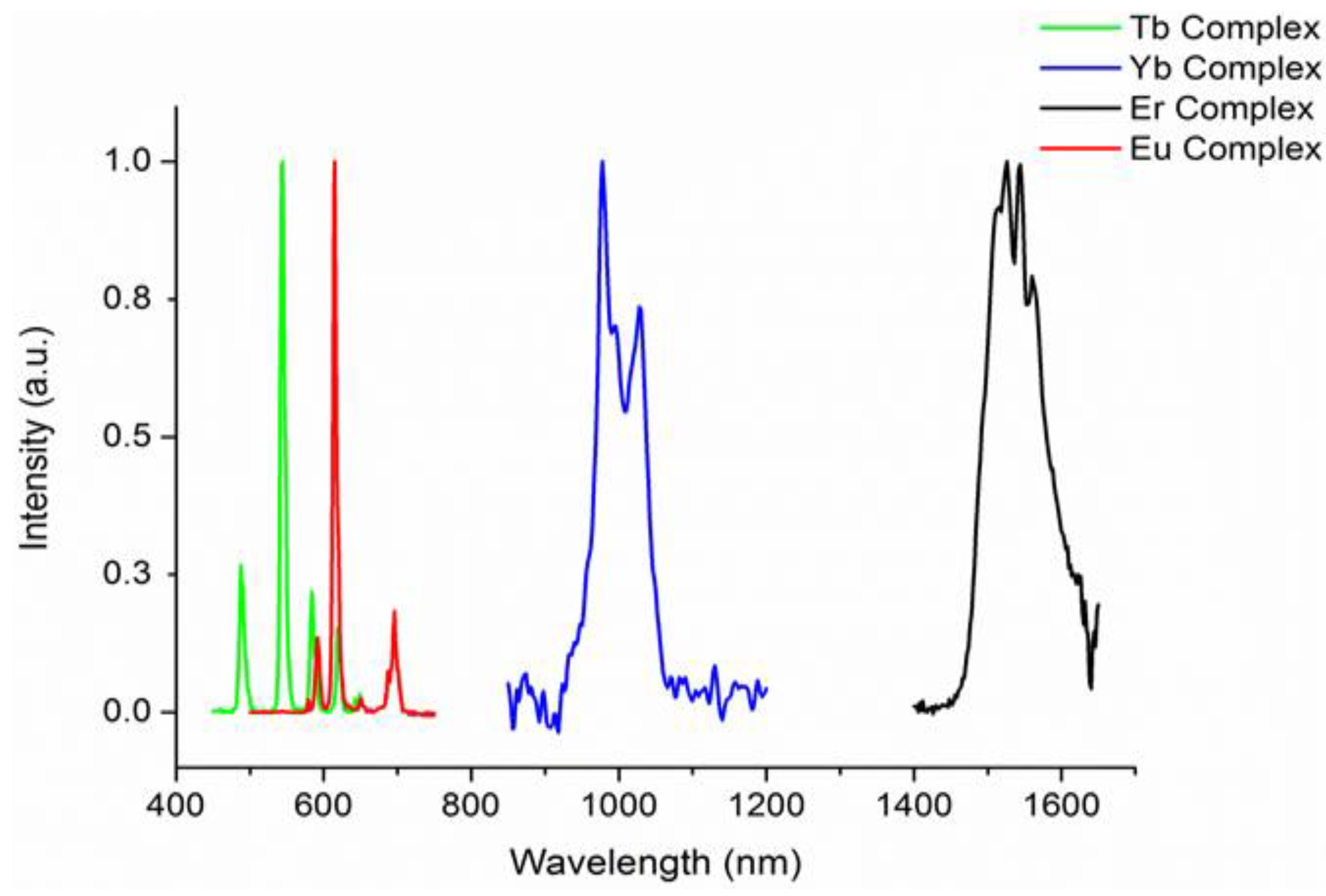
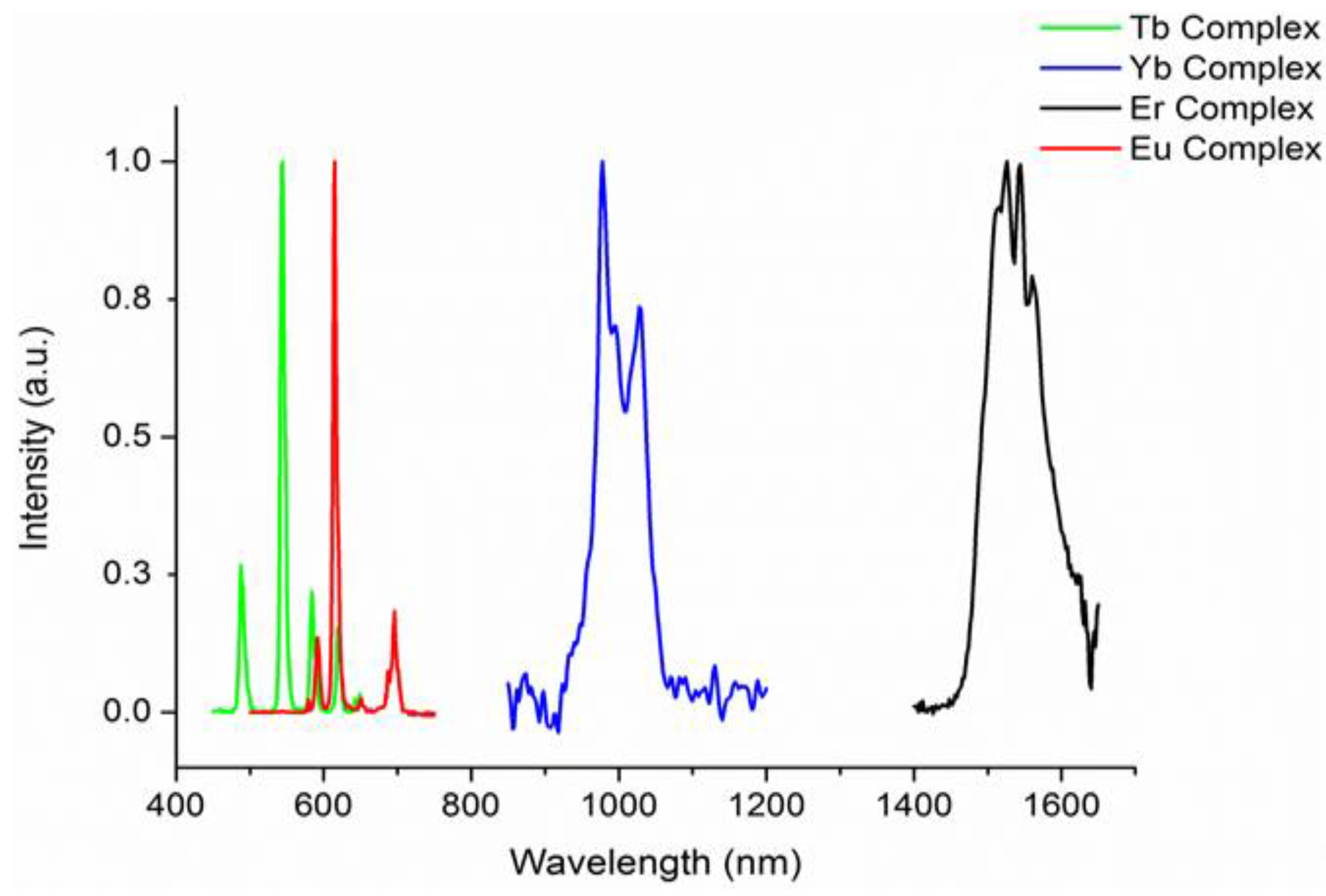
2.3. Photophysical Properties of the Complexes
3. Materials and Methods
3.1. General Details
3.2. Synthetic Procedures
3.2.1. Preparation of Tris(pentafluorophenyl)phosphine Oxide, (C6F5)3PO (ArF3PO)
3.2.2. Preparation of Ln(hfac)3·2H2O
3.2.3. Preparation of [Ln(hfac)3{(ArF)3PO}(H2O)], Ln = Sm3+, Eu3+, Tb3+, Er3+, Yb3+ (1–5) from Ln(hfac)3·2H2O
3.2.4. Preparation of [Ln(hfac)3{(ArF)3PO}(H2O)], Ln = Sm3+, Eu3+, Tb3+, Er3+, Yb3+ (1–5) from Ln(OAc)3·xH2O under N2
3.2.5. Preparation of [Ln(F7-acac)3{(ArF)3PO}2] Ln = Sm3+, Eu3+, Tb3+, Er3+, Yb3+ (6–10) from Ln(OAc)3·xH2O under N2
3.3. X-ray Crystallographic Data for Compounds 3, 4, 6, 7, 9 and 10
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| NMR | nuclear magnetic resonance |
| nIR | near Infra-Red |
| UV/vis | Ultra-violet/visible |
| hfac | 1,1,1,5,5,5-hexafluoro-2,4-pentanedione |
| F7-acac | 1,1,1,3,5,5,5-heptafluoroacetylacetone |
| (ArF3)PO | tris(pentafluorophenyl)phosphine oxide |
| TPIP | tetraphenylimidodiphosphinate |
| F-TPIP | perfluorotetraphenylimidodiphosphinate |
| OAc | acetate |
| Ln | lanthanide |
| DOTA | 1,4,7,10-tetraazacyclododecane- N′,N″,N′′′,N′′′′-tetraacetic acid |
References
- Bünzli, J.-C.G.; Eliseeva, S.V. Intriguing aspects of lanthanide luminescence. Chem. Sci. 2013, 4, 1939–1949. [Google Scholar] [CrossRef]
- Binnemans, K. Lanthanide-based luminescent hybrid materials. Chem. Rev. 2009, 109, 4283–4374. [Google Scholar] [CrossRef] [PubMed]
- De Bettencourt-Dias, A. Lanthanide-based emitting materials in light-emitting diodes. Dalton Trans. 2007, 2229–2241. [Google Scholar] [CrossRef] [PubMed]
- Suyver, J.F.; Aebischer, A.; Biner, D.; Gerner, P.; Grimm, J.; Heer, S.; Krämer, K.E.; Reinhard, C.; Güdel, H.U. Novel materials doped with trivalent lanthanides and transition metal ions showing near-infrared to visible photon upconversion. Opt. Mater. 2005, 27, 1111–1130. [Google Scholar] [CrossRef]
- Meijerink, A.; Wegh, R.; Vergeer, P.; Vlugt, T. Photon management with lanthanides. Opt. Mater. 2006, 28, 575–581. [Google Scholar] [CrossRef]
- Harvey, P.; Oakland, C.; Driscoll, M.D.; Hay, S.; Natrajan, L.S. Ratiometric detection of enzyme turnover and flavin reduction using rare-earth upconverting phosphors. Dalton Trans. 2014, 43, 5265–5268. [Google Scholar] [CrossRef] [PubMed]
- Bünzli, J.-C.G. Lanthanide luminescence for biomedical analyses and imaging. Chem. Rev. 2010, 110, 2729–2755. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, C.P.; Murray, B.S.; New, E.J.; Pal, R.; Parker, D. Cell-penetrating metal complex optical probes: Targeted and responsive systems based on lanthanide luminescence. Acc. Chem. Res. 2009, 42, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Beeby, A.; Clarkson, I.M.; Faulkner, S.; Botchway, S.; Parker, D.; Williams, J.A.G.; Parker, A.W. Luminescence imaging microscopy and lifetime mapping using kinetically stable lanthanide (III) complexes. J. Photochem. Photobiol. B Biol. 2000, 57, 83–89. [Google Scholar] [CrossRef]
- Vuojola, J.; Soukka, T. Luminescent lanthanide reporters: New concepts for use in bioanalytical applications. Methods Appl. Fluoresc. 2014, 2, 1–28. [Google Scholar] [CrossRef]
- Beeby, A.; Burton-Pye, B.P.; Faulkner, S.; Motson, G.R.; Jeffery, J.C.; McCleverty, J.A.; Ward, M.D. Synthesis and near-IR luminescence properties of neodymium(III) and ytterbium(III) complexes with poly(pyrazolyl)borate ligands. J. Chem. Soc. Dalton Trans. 2002, 1923–1928. [Google Scholar] [CrossRef]
- Faulkner, S.; Carrie, M.-C.; Pope, S.J.A.; Squire, J.; Beeby, A.; Sammes, P.G. Pyrene-sensitised near-IR luminescence from ytterbium and neodymium complexes. Dalton Trans. 2004, 1405–1409. [Google Scholar] [CrossRef] [PubMed]
- Shavaleev, N.M.; Scopelliti, R.; Gumy, F.; Bünzli, J.-C. Surprisingly bright near-infrared luminescence and short radiative lifetimes of ytterbium in hetero-binuclear Yb_Na chelates. Inorg. Chem. 2009, 48, 7937–7946. [Google Scholar] [CrossRef] [PubMed]
- Moudam, O.; Rowan, B.C.; Alamiry, M.; Richardson, P.; Richards, B.S.; Jones, A.C.; Robertson, N. Europium complexes with high total photoluminescence quantum yields in solution and in PMMA. Chem. Commun. 2009, 6649–6651. [Google Scholar] [CrossRef] [PubMed]
- Magennis, S.W.; Ferguson, A.J.; Bryden, T.; Jones, T.S.; Beeby, A.; Samuel, I.D.W. Time-dependence of erbium(III) tris(8-hydroxyquinolate) near-infrared photoluminescence: Implications for organic light-emitting diode efficiency. Synth. Metals 2003, 138, 463–469. [Google Scholar] [CrossRef]
- Lunstroot, K.; Nockemann, P.; Van Hecke, K.; Van Meervelt, L.; Görller-Walrand, C.; Binnemans, K.; Driesen, K. Visible and near-infrared emission by Samarium(III)-containing ionic liquid mixtures. Inorg. Chem. 2009, 48, 3018–3026. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Tropiano, T.; Mantulnikovs, K.; Faulkner, S.; Vosch, T.; Sørensen, T.J. Spectrally resolved confocal microscopy using lanthanide centred near-IR emission. Chem. Commun. 2015, 51, 2372–2375. [Google Scholar] [CrossRef] [PubMed]
- Digonnet, M. (Ed.) Rare Earth Doped Fiber Lasers and Amplifiers; Marcel Dekker: New York, NY, USA, 1993.
- Desurvire, E. Erbium Doped Fiber Amplifiers; Wiley: New York, NY, USA, 1994. [Google Scholar]
- Desurvire, E. The golden age of optical fiber amplifiers. Phys. Today 1994, 47, 20–27. [Google Scholar] [CrossRef]
- Faulkner, S.; Burton-Pye, B.P.; Pope, S.J.A. Lanthanide complexes for luminescence imaging applications. Appl. Spectrosc. Rev. 2005, 40, 1–31. [Google Scholar] [CrossRef]
- Faulkner, S.; Natrajan, L.S.; Perry, W.S.; Sykes, D. Sensitised luminescence in lanthanide containing arrays and d–f hybrids. Dalton Trans. 2009, 3890–3899. [Google Scholar] [CrossRef] [PubMed]
- Binnemans, K. Rare-earth β-diketonates. In Handbook on the Physics and Chemistry of Rare Earths; Gschneidner, K.A., Jr., Bünzli, J.-C.G., Pecharsky, V.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; Volume 35, pp. 107–271. [Google Scholar]
- Hemilla, A. Applications of Fluorescence in Immunoassays; Wiley Interscience: New York, NY, USA, 1991. [Google Scholar]
- Yuan, J.; Wang, G. Lanthanide complex-based fluorescence label for time-resolved fluorescence bioassay. J. Fluoresc. 2005, 15, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Sy, M.; Nonat, A.; Hildebrandt, N.; Charbonnière, L.J. Lanthanide-based luminescence biolabelling. Chem. Commun. 2016, 52, 5080–5095. [Google Scholar] [CrossRef] [PubMed]
- Browne, W.R.; Vos, J.G. The effect of deuteriation on the emission lifetime of inorganic compounds. Coord. Chem. Rev. 2001, 219–221, 761–787. [Google Scholar] [CrossRef]
- Stein, G.; Wurzberg, E. Energy gap law in the solvent isotope effect on radiationless transitions of rare earth Ions. J. Chem. Phys. 1975, 62, 208–213. [Google Scholar] [CrossRef]
- Heller, A. Formation of hot OH Bonds in the radiationless relaxations of excited rare earth ions in aqueous solutions. J. Am. Chem. Soc. 1966, 88, 2058–2059. [Google Scholar] [CrossRef]
- Doffek, C.; Alzakhem, N.; Bischof, C.; Wahsner, J.; Güden-Silber, T.; Lugger, J.; Platas-Iglesias, C.; Seitz, M. Understanding the quenching effects of aromatic C–H– and C–D– oscillators in Near-IR Lanthanoid Luminescence. J. Am. Chem. Soc. 2012, 134, 16413–16423. [Google Scholar] [CrossRef] [PubMed]
- Horrocks, W.D.; Sudnick, D.R. Lanthanide ion luminescence probes of the structure of biological macromolecules. Acc. Chem. Res. 1981, 14, 384–392. [Google Scholar] [CrossRef]
- Supkowski, R.M.; Horrocks, W.D. On the determination of the number of water molecules, q, coordinated to europium(III) ions in solution from luminescence decay lifetimes. Inorg. Chim. Acta 2002, 340, 44–48. [Google Scholar] [CrossRef]
- Beeby, A.; Clarkson, I.M.; Dickins, R.S.; Faulkner, S.; Parker, D.; Royle, L.; de Sousa, A.S.; Williams, J.A.G.; Woods, M. Non-radiative deactivation of the excited states of europium, terbium and ytterbium complexes by proximate energy-matched OH, NH and CH oscillators: An improved luminescence method for establishing solution hydration states. J. Chem. Soc. Perkin Trans. 2 1999, 493–503. [Google Scholar] [CrossRef]
- Yasuchika Hasegawa, H.; Kimura, Y.; Murakoshi, K.; Wada, Y.; Kim, J.-H.; Nakashima, N.; Yamanaka, T.; Yanagida, S. Enhanced emission of deuterated tris(hexafluoroacetylacetonato)neodymium(III) complex in solution by suppression of radiationless transition via vibrational excitation. J. Phys. Chem. 1996, 100, 10201–10205. [Google Scholar] [CrossRef]
- Wahsner, J.; Seitz, M. Perdeuterated 2,2′-Bipyridine-6,6′-dicarboxylate: An extremely efficient sensitizer for thulium luminescence in solution. Inorg. Chem. 2013, 52, 13301–13303. [Google Scholar] [CrossRef] [PubMed]
- Bischof, C.; Wahsner, J.; Scholten, J.; Troslen, S.; Seitz, M. Quantification of C–H quenching in near-IR luminescent ytterbium and neodymium cryptates. J. Am. Chem. Soc. 2010, 132, 14334–14335. [Google Scholar] [CrossRef] [PubMed]
- Doffeck, C.; Seitz, M. The radiative lifetime in near-IR-luminescent ytterbium cryptates: The key to extremely high quantum yields. Angew. Chem. Int. Ed. 2015, 54, 9719–9721. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.-Q.; Peng, Y.; Li, Z.; Wang, C.-C.; Zheng, Y.-X.; Motevalli, M.; Wyatt, P.B.; Gillin, W.P.; Hernańdez, I. Effect of fluorination on the radiative properties of Er3+ organic complexes: An opto-structural correlation study. J. Phys. Chem. C 2013, 117, 23970–23975. [Google Scholar] [CrossRef]
- Quochi, F.; Orrú, R.; Cordella, F.; Mura, A.; Bongiovanni, G.; Artizzu, F.; Deplano, P.; Mercuri, M.L.; Pilia, L.; Serpe, A. Near infrared light emission quenching in organolanthanide complexes. J. Appl. Phys. 2006, 99, 053520. [Google Scholar] [CrossRef]
- Congiu, M.; Alamiry, M.; Moudam, O.; Ciorba, S.; Richardson, P.R.; Maron, L.; Jones, A.C.; Bryce, S.; Richards, B.S.; Robertson, N. Preparation and photophysical studies of [Ln(hfac)3DPEPO], Ln = Eu, Tb, Yb, Nd, Gd; of total photoluminescence quantum yields. Dalton Trans. 2013, 42, 13537–13545. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Hu, J.; Wang, J.; Liu, X.; Zhen, Z. Novel perfluorodiphenylphosphinic acid lanthanide (Er or Er–Yb) complex with high NIR photoluminescence quantum yield. Photochem. Photobiol. Sci. 2008, 7, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.H.C.; Motevalli, M.; Abrahams, I.; Wyatt, P.B.; Gillin, W.P. IR luminescence of Er, Nd and Yb β-diketonates. J. Phys. Chem. B 2006, 110, 24476–24479. [Google Scholar] [CrossRef] [PubMed]
- Mancino, G.; Ferguson, A.J.; Beeby, A.; Long, N.J.; Jones, T.S. Dramatic increases in the lifetime of the Er3+ ion in a molecular complex using a perfluorinated imidodiphosphinate sensitizing ligand. J. Am. Chem. Soc. 2005, 127, 524–525. [Google Scholar] [CrossRef] [PubMed]
- Glover, P.B.; Bassett, A.P.; Nockemann, P.; Benson, M.; Kariuki, M.; Van Deun, R.; Pikramenou, Z. Fully fluorinated imidodiphosphinate shells for visible- and NIR-emitting lanthanides: Hitherto unexpected effects of sensitizer fluorination on lanthanide emission properties. Chem. Eur. J. 2007, 13, 6308–6320. [Google Scholar] [CrossRef] [PubMed]
- Beverina, L.; Crippa, M.; Sassi, M.; Monguzzi, A.; Meinardi, F.; Tubino, R.; Pagani, G.A. Perfluorinated nitrosopyrazolone-based erbium chelates: A new efficient solution processable NIR emitter. Chem. Commun. 2009, 34, 5103–5105. [Google Scholar] [CrossRef] [PubMed]
- Mech, A.; Monguzzi, A.; Meinardi, F.; Mezyk, J.; Macchi, G.; Tubino, R. Sensitized NIR Erbium(III) emission in confined geometries: A new strategy for light emitters in telecom applications. J. Am. Chem. Soc. 2010, 132, 4574–4576. [Google Scholar] [CrossRef] [PubMed]
- Monguzzi, A.; Tubino, R.; Meinardi, F.; Biroli, A.O.; Maddalena Pizzotti, M.; Demartin, F.; Quochi, F.; Cordella, F.; Loi, M.A. Novel Er3+ perfluorinated complexes for broadband sensitized near infrared emission. Chem. Mater. 2009, 21, 128–135. [Google Scholar] [CrossRef]
- Natrajan, L.S.; Khoabane, N.M.; Dadds, B.L.; Muryn, C.A.; Pritchard, R.G.; Heath, S.L.; Alan, M.; Kenwright, A.M.; Ilya Kuprov, I.; Faulkner, S. Probing the structure, conformation, and stereochemical Exchange in a family of lanthanide complexes derived from tetrapyridyl-appended cyclen. Inorg. Chem. 2010, 49, 7700–7710. [Google Scholar] [CrossRef] [PubMed]
- Petrov, V.A.; Marshall, W.J.; Grushin, V.V. The first perfluoroacetylacetonate metal complexes: As unexpectedly robust as tricky to make. Chem. Commun. 2002, 520–521. [Google Scholar] [CrossRef]
- Chen, X.Y.; Jensen, M.P.; Liu, G.K. Analysis of energy level structure and excited-state dynamics in a Sm3+ complex with soft-donor ligands: Sm(Et2Dtc)3(bipy). J. Phys. Chem. B 2005, 109, 13991–13999. [Google Scholar] [CrossRef] [PubMed]
- Aebischer, A.; Gumy, F.; Bünzli, J.-C.G. Intrinsic quantum yields and radiative lifetimes of lanthanide tris(dipicolinates). Phys. Chem. Chem. Phys. 2009, 11, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Werts, M.H.V.; Jukes, R.T.F.; Verhoeven, J.W. The emission spectrum and the radiative lifetime of Eu3+ in luminescent lanthanide complexes. Phys. Chem. Chem. Phys. 2002, 4, 1542–1548. [Google Scholar] [CrossRef]
- He, H.; Sykes, A.G.; May, P.S.; He, G. Structure and photophysics of near-infrared emissive ytterbium(III) monoporphyrinate acetate complexes having neutral bidentate ligands. Dalton Trans. 2009, 36, 7454–7461. [Google Scholar] [CrossRef] [PubMed]
- Duhamel-Henry, N.; Adam, J.L.; Jacquier, B.; Linarès, C. Photoluminescence of new fluorophosphate glasses containing a high concentration of terbium (III) ions. Opt. Mater. 1996, 5, 197–207. [Google Scholar] [CrossRef]
- Natrajan, L.S.; Weinstein, J.A.; Wilson, C.; Arnold, P.L. Synthesis and luminescence studies of mono- and C3-symmetric, tris(ligand) complexes of Sm(III), Y(III) and Eu(III) with sulfur-bridged binaphtholate ligands. Dalton Trans. 2004, 3748–3755. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SADABS, Empirical Absorption Correction Program Based upon the Method of Blessing; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Blessing, R.H. An empirical correction for absorption anisotropy. Acta Crystallogr. 1995, 51, 33–38. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Beeby, A.; Faulkner, S. Luminescence from neodymium(III) in solution. Chem. Phys. Lett. 1997, 266, 116–122. [Google Scholar] [CrossRef]
- Beeby, A.; Faulkner, S.; Parker, D.; Williams, J.A.G. Sensitised luminescence from phenanthridine appended lanthanide complexes: Analysis of triplet mediated energy transfer processes in terbium, europium and neodymium complexes. J. Chem. Soc. Perkin Trans. 2 2001, 1268–1273. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Complex | λem (nm) | τCH2Cl2 (μs) 1 | ΦCH2Cl2(%) 2 | τsolid (μs) 1 | Φsolid (%) 2 |
|---|---|---|---|---|---|---|
| [Sm(hfac)3{(ArF)3PO}(H2O)] | 1 | 650 | 67.1 | 3.4 | 169 | 8.5 |
| [Eu(hfac)3{(ArF)3PO}(H2O)] | 2 | 617 | 5 | 5 | 116 (30%), 799 (70%) | 53 |
| [Tb(hfac)3{(ArF)3PO}(H2O)] | 3 | 545 | 79.3 | 1.6 | 638 | 13 |
| [Er(hfac)3{(ArF)3PO}(H2O)] | 4 | 1530 | 1.94 (14%), 0.394 (86%) 4 | 0.05 3 | 3.60 | 0.1 |
| [Yb(hfac)3{(ArF)3PO}(H2O)] | 5 | 980 | 3.31 (65%), 0.742 (35%) 4 | 0.25 3 | 26.1 | 2.0 |
| [Sm(F7-acac)3{(ArF)3PO}2] | 6 | 650 | 5 | 5 | 506 | 26 |
| [Eu(F7-acac)3{(ArF)3PO}2] | 7 | 617 | 638 | 58 | 220 (14%), 741 (86%) | 67 3 |
| [Tb(F7-acac)3{(ArF)3PO}2] | 8 | 545 | 1520 | 30 | 421 (33%), 1400 (67%) | 27 3 |
| [Er(F7-acac)3{(ArF)3PO}2] | 9 | 1530 | 15.3 6 | 0.4 | 16.8 | 0.4 |
| [Yb(F7-acac)3{(ArF)3PO}2] | 10 | 980 | 18.2 6 | 1.4 | 139 | 11 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swinburne, A.N.; Langford Paden, M.H.; Chan, T.L.; Randall, S.; Ortu, F.; Kenwright, A.M.; Natrajan, L.S. Optical Properties of Heavily Fluorinated Lanthanide Tris β-Diketonate Phosphine Oxide Adducts. Inorganics 2016, 4, 27. https://doi.org/10.3390/inorganics4030027
Swinburne AN, Langford Paden MH, Chan TL, Randall S, Ortu F, Kenwright AM, Natrajan LS. Optical Properties of Heavily Fluorinated Lanthanide Tris β-Diketonate Phosphine Oxide Adducts. Inorganics. 2016; 4(3):27. https://doi.org/10.3390/inorganics4030027
Chicago/Turabian StyleSwinburne, Adam N., Madeleine H. Langford Paden, Tsz Ling Chan, Simon Randall, Fabrizio Ortu, Alan M. Kenwright, and Louise S. Natrajan. 2016. "Optical Properties of Heavily Fluorinated Lanthanide Tris β-Diketonate Phosphine Oxide Adducts" Inorganics 4, no. 3: 27. https://doi.org/10.3390/inorganics4030027
APA StyleSwinburne, A. N., Langford Paden, M. H., Chan, T. L., Randall, S., Ortu, F., Kenwright, A. M., & Natrajan, L. S. (2016). Optical Properties of Heavily Fluorinated Lanthanide Tris β-Diketonate Phosphine Oxide Adducts. Inorganics, 4(3), 27. https://doi.org/10.3390/inorganics4030027