Abstract
The construction of lanthanide(III) chelates that exhibit superior photophysical properties holds great importance in biological and materials science. One strategy to increase the luminescence properties of lanthanide(III) chelates is to hinder competitive non-radiative decay processes through perfluorination of the chelating ligands. Here, the synthesis of two families of heavily fluorinated lanthanide(III) β-diketonate complexes bearing monodentate perfluorinated tris phenyl phosphine oxide ligands have been prepared through a facile one pot reaction [Ln(hfac)3{(ArF)3PO}(H2O)] and [Ln(F7-acac)3{(ArF)3PO}2] (where Ln = Sm3+, Eu3+, Tb3+, Er3+ and Yb3+). Single crystal X-ray diffraction analysis in combination with photophysical studies have been performed to investigate the factors responsible for the differences in the luminescence lifetimes and intrinsic quantum yields of the complexes. Replacement of both bound H2O and C–H oscillators in the ligand backbone has a dramatic effect on the photophysical properties of the complexes, particularly for the near infra-red emitting ion Yb3+, where a five fold increase in luminescence lifetime and quantum yield is observed. The complexes [Sm(hfac)3{(ArF)3PO}(H2O)] (1), [Yb(hfac)3{(ArF)3PO}(H2O)] (5), [Sm(F7-acac)3{(ArF)3PO}2] (6) and [Yb(F7-acac)3{(ArF)3PO}2] (10) exhibit unusually long luminescence lifetimes and attractive intrinsic quantum yields of emission in fluid solution (ΦLn = 3.4% (1); 1.4% (10)) and in the solid state (ΦLn = 8.5% (1); 2.0% (5); 26% (6); 11% (10)), which are amongst the largest values for this class of compounds to date.
1. Introduction
The unique optical properties of the trivalent lanthanides continue to garner appeal due to their numerous commercially exploitable applications in the biomedical and materials science fields [1,2]. Their inherent luminescent properties, which arise from parity forbidden intra f–f transitions, including long-lived emission (microsecond to millisecond range), high colour purity, insensitivity to dissolved molecular oxygen and resistance to photobleaching and blinking render these ions highly suitable as the emissive components in a number of growing technologies. These include, organic light emitting diodes [3], up and down-converting phosphors [4,5,6], luminescent sensors in biomedical diagnostics [7] and luminescent markers in cell microscopy [8,9,10]. More recently, the potential for near infra-red (nIR) emitting lanthanide chelates (Yb3+, Nd3+, Er3+, Pr3+) [11,12,13,14,15,16] to be exploited in mammalian cell imaging and in the telecommunications industry has been realized. Lanthanide ions that are emissive in the near infra-red region of the electromagnetic spectrum are particularly suited to biological imaging using confocal microscopy [17] and in optical amplifiers and waveguides [18,19,20] since their emission coincides with the more transparent wavelengths of biological tissue and silica. Above all however, the long-lived emission of the trivalent lanthanides is ideal for preventing spectral interference from scattered light and autofluorescence via time-gated luminescence detection [21] (pp. 6–8), which commonly occurs upon UV excitation in molecular complexes, particularly in biological media.
Lanthanide chelates that incorporate organic chromophores offer additional attractive properties for their use as optical materials, namely the use of low power excitation sources, a large Stokes’ shift between excitation and emission and theoretically very high quantum yields of emission. This is principally because the parity forbidden f–f transitions can be overcome by efficient energy transfer from the triplet state of the chromophore of the chelated organic ligand (antenna) to the lanthanide excited emissive state upon UV–visible excitation; this is termed the “antenna effect” or “sensitised emission” [22]. In this regard, β-diketonates and their substituted derivatives [23] have become synonymous with the development of highly luminescent lanthanide complexes used in optoelectronic devices and in fluoroimmunoassays such as DELFIA (dissociation-enhanced lanthanide fluoroimmunoassay) [21,24,25,26] (p. 5085). These monoanionic ligands form very kinetically and thermodynamically stable 1:3 charge neutral metal:ligand chelates and the appended chromophores can very efficiently sensitise both the visible and nIR f–f based emission from lanthanide ions [27].
However, the majority of these complexes only take advantage of the green and red emission of Tb3+ and Eu3+ respectively due to the fact that the excited states of these ions are only marginally quenched by frequency matched vibrational harmonics of proximate O–H and N–H oscillators and the luminescence lifetimes are of millisecond order. By contrast, the relative lower energy of the excited states of the nIR emitting lanthanides means that the emission from these ions is considerably quenched by lower energy vibrational overtones of X–H bond vibrations (particularly C–H oscillators) present within the ligand architecture and/or by closely diffusing solvent molecules [12]. Since the magnitude of vibrational quenching of the emissive excited state of a lanthanide ion is dictated by the energy gap law [28,29], X–H quenching is much more pronounced in nIR emitting lanthanides compared to those that emit in the visible region. In general, vibrational quenching is considered negligible if the energy gap between the emissive state and the next lowest lying energy state is greater than or equal to the sixth harmonic of the fundamental vibrational mode. However, in the case of the nIR emissive lanthanides, the emissive states often lie within the first and third vibrational overtones of X–H oscillators [30]. In this regard, the fact that O–D oscillators whose fundamental modes vibrate at lower frequency (e.g., 3450 cm−1 in an OH bond compared to 2500 cm−1 in an OD bond), can be exploited to increase the luminescence lifetime and quantum yield of Ln3+ based emission. Horrocks [31,32] and others [11,33] have exploited this effect to develop an empirical equation to determine the number of O–H oscillators and therefore water or methanol molecules bound to a Ln3+ ion in aqueous or methanolic solution to great effect (Ln3+ = Eu3+, Tb3+, Yb3+ and in a series of isostructural lanthanide(III) complexes, Nd3+). By extension, the luminescence lifetime of a given lanthanide complex can be increased by simply replacing the spectroscopic solvent to its deuterated counterpart. However, X–D vibrational quenching is often still operative in nIR emissive complexes; as an example, the third C–D overtone overlaps with the 4I13/2 excited state of Er3+ (in comparison to the first vibrational harmonic of C–H bonds).
To further overcome the limitations of vibrational quenching and thereby significantly increase the luminescence quantum yield of nIR (and visible) emitting lanthanide complexes, synthetic approaches based on partial or full deuteration [27,34,35,36,37] of the C–H bonds in organic ligands can be accomplished. For example, Seitz and Platas-Iglesias have synthesized several series of selectively deuterated Lehn cryptand complexes of Pr3+, Nd3+, Er3+ and Yb3+ and evaluated the spectral overlap integrals of the excited states with aromatic C–H and C–D overtones to develop a comprehensive picture of lanthanide C–H quenching combinations for luminescence enhancement in nIR emitting lanthanide complexes [30]. An alternative strategy is partial or perfluorination of C–H bonds of the supporting ligand [38,39,40,41]. This has also been utilized very effectively to prepare partially fluorinated complexes mainly based on diamine adducts of tris β-diketonate hexafluoroacetylacetonate (hfac) and heptafluoroacetylacetanoate (F7-acac) chelates that exhibit improved photophysical properties [23,42]. For example, the radiative lifetimes of the 4I13/2 → 4I13/2 transition at 1530 nm in crystalline samples of Er(hfac)3(H2O)2 and Cs[Er(hfac)4] have been determined to be ~9.5 and 23.4 ms respectively, reflecting the greater degree of fluorination in the latter [38].
Conversely, complexes of the lanthanides where all C–H bonds on all coordinated ligands have been fluorinated are rather rare [34,35,36,37]. Notable examples of sensitized nIR Ln3+ emission include the Er3+ complexes of tetrapentafluorophenylimidodiphosphinate (F-TPIP) [43,44], perfluorodiphenylphosphinic acid [41], perfluorinated nitrosopyrazolone [45] and perfluoro-benzophenone in Er3+ doped Zeolite L [46]. Remarkably, ligand sensitised nIR emission in the Er(F-TPIP)3 complex resulted in extremely long-lived Er3+ nIR emission with a reported lifetime of 200 μs, while the luminescence lifetime following direct intra 4f excitation at 978 nm in the perfluorodiphenylphosphinic acid complex reported by Song et al. was measured as 0.336 ms. In this contribution, we describe the synthesis and enhanced optical properties of two families of fluorinated tris β-diketonate lanthanide complexes of the visible and nIR emitting lanthanides Sm3+, Eu3+, Tb3+, Er3+ and Yb3+ where the eight coordinate coordination geometries are completed by the monodentate perfluorinated tris phenyl phosphine oxide (C6F5)3PO ((ArF)3PO) [41,45,47].
2. Results and Discussion
2.1. Synthesis of the Complexes
A series of lanthanide tris hexafluoroacetylacetonate (hfac) and heptafluoroacetylacetonate (F7-acac) bis perfluorinated tris aryl phosphine oxide complexes were originally targeted by two synthetic routes as outlined in Scheme 1. Either treatment of prepared Ln(hfac)3·2H2O [28] with two equivalents of (ArF)3PO (tris(pentafluorophenyl)phosphine oxide) in CH2Cl2 at room temperature sonicated for one hour or a one pot reaction of the corresponding lanthanide acetate, protonated β-diketonate and two equivalents of (ArF)3PO [41] at 2 × 10−3 M concentrations heated to reflux temperature for one hour yielded the complexes [Ln(hfac)3{(ArF)3PO}(H2O)] and (ArF)3PO (Ln = Sm3+, Eu3+, Tb3+, Er3+ and Yb3+, 1–5) and [Ln(F7-acac)3{(ArF)3PO}2] (Ln = Sm3+, Eu3+, Tb3+, Er3+ and Yb3+, 6–10) in moderate yields (26%–59%) after recrystallization from CH2Cl2 solutions (as verified by single crystal X-ray diffraction analysis). Interestingly, conducting the reactions in dry CH2Cl2 under air sensitive conditions at higher concentrations (1 × 10−2 M) in order to promote bis phosphine oxide substitution with the labile coordinated water molecules in the parent lanthanide acetate or β-diketonate complexes, resulted solely in the isolation of the mono phosphine oxide substituted complexes for the hexafluorinated β-diketonate hfac [47], whereas only the bis phosphine oxide substituted derivatives were isolated with the heptafluoro β-diketonate F7-acac using both standard and dry solvents in similar conditions (Scheme 1). This interesting divergence in reactivity can be attributed to the greater electron withdrawing effects of the F7-acac ligand relative to the hfac β-diketonate which promotes a higher affinity for the second phosphine oxide to bind to the electropositive lanthanide(III) metal centres. In the case of the hfac reactions, adjusting the stoichiometry of the phosphine oxide accordingly, resulted in slightly improved product yields in most cases apart from complex 4 (44%–53%).
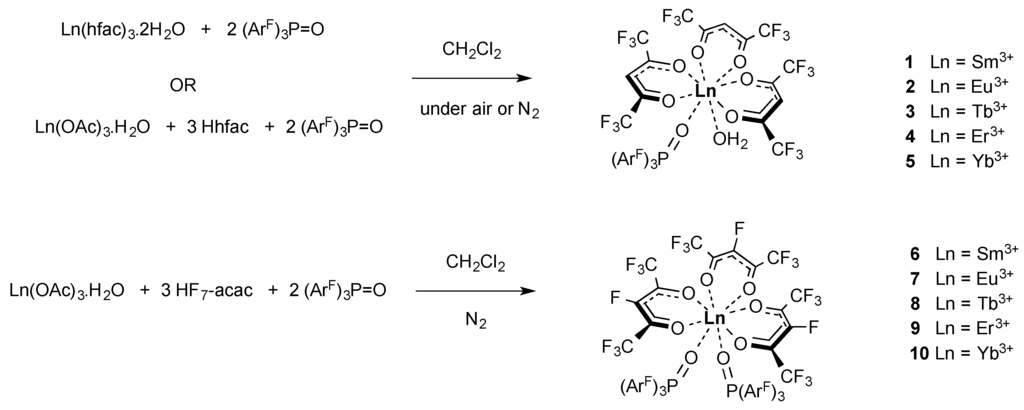
Scheme 1.
Synthesis of the heavily fluorinated lanthanide(III) complexes [Ln(hfac)3{(ArF)3PO}(H2O)] (1–5) and [Ln(F7-acac)3{(ArF)3PO}2] (6–10) described in this article.
The 31P and 19F NMR (nuclear magnetic resonance) spectra of all the complexes exhibited paramagnetically shifted phosphorous and fluorine resonances; the 19F resonances of the β-diketonate being significantly more shifted and broadened than the corresponding resonances in the coordinated (ArF)3PO ligands (see supplementary material). Notably, multinuclear NMR analysis of the crude powders from the hfac reactions taken after removal of all volatiles before recrystallization suggested no presence of a minor lanthanide species containing perfluorinated phosphine oxide ligands that could be formulated as [Ln(hfac)3{(ArF)3PO}2] as previously reported for Er3+ (vide infra) [47].
2.2. Single Crystal X-ray Diffraction Analysis
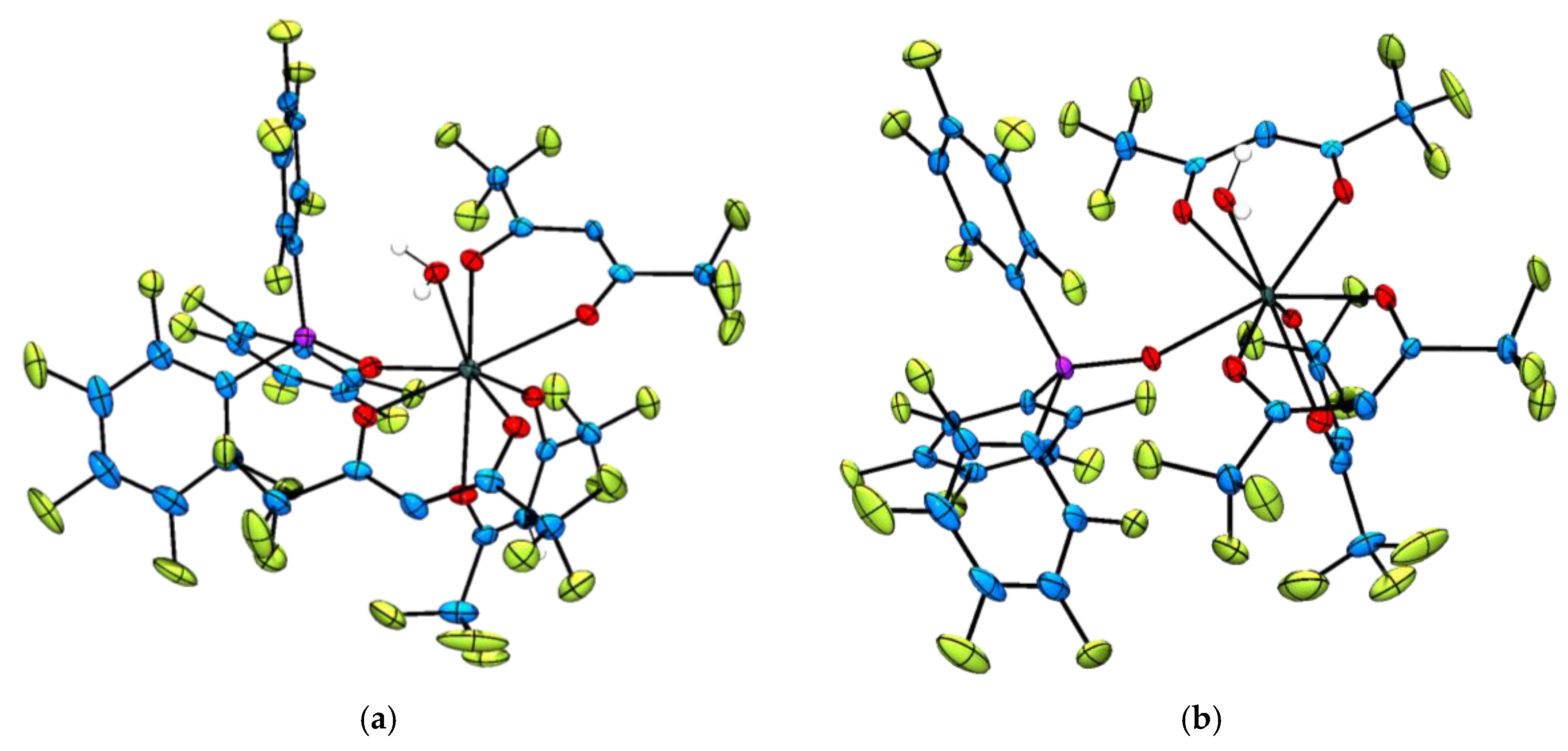
Representative single crystals suitable for X-ray diffraction analysis were grown for the complexes [Ln(hfac)3{(ArF)3PO}(H2O)] (Ln = Tb3+, 3 and Er3+, 5) and [Ln(F7-acac)3{(ArF)3PO}2] (Ln = Sm3+, 6, Eu3+, 7, Er3+, 9 and Yb3+, 10) by slow evaporation or slow cooling of saturated CH2Cl2 or CH2Cl2:pentane or hexane solutions to −18°C (Figure 1). A preliminary crystallographic data set was also collected for [Yb(hfac)3{(ArF)3PO}(H2O)], 5 (Table S1) and confirmed the same connectivity as the Tb3+ and Er3+ derivatives (complexes 3 and 4 respectively). Additionally, the single crystal X-ray structure of [Tm(F7-acac)3{(ArF)3PO}2] was also determined and found to be isostructural to its congeners 6, 7, 9 and 10.
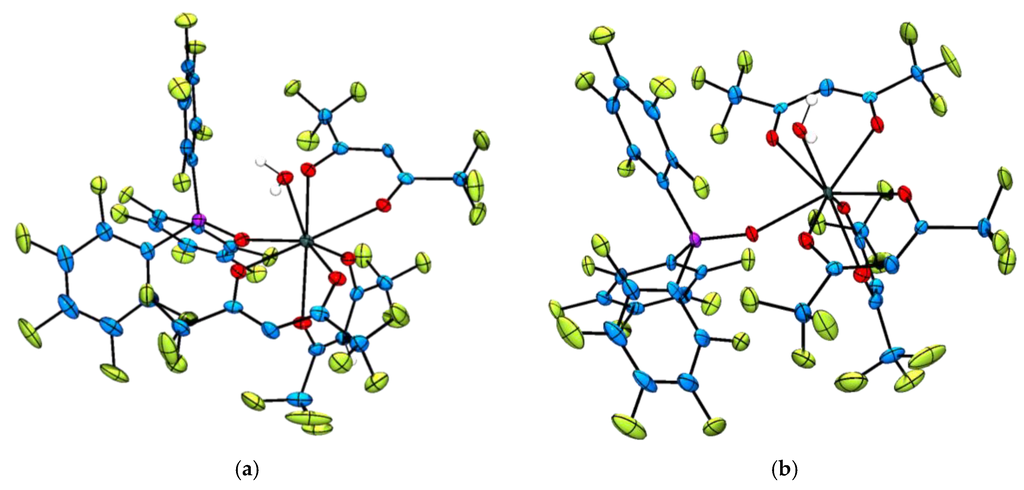
Figure 1.
Representative solid state molecular structures of the [Ln(hfac)3{(ArF)3PO}(H2O)] complexes with thermal ellipsoids set at the 50% probability level and C–H atoms omitted for clarity. Atom colour key: C, blue, Ln3+, dark green, O, red, H, white, P, pink, F, green. (a) Thermal ellipsoid plot of [Tb(hfac)3{(ArF)3PO}(H2O)], complex 3; (b) Thermal ellipsoid plot of [Er(hfac)3{(ArF)3PO}(H2O)], complex 4.
The molecular structures of the two hfac complexes [Ln(hfac)3{(ArF)3PO}(H2O)] (Ln = Tb3+, 3 and Er3+, 4) are isostructural and the lanthanide(III) ions are octacoordinated by three bidentate hfac ligands, one monodentate perfluorinated triphenylphosphine oxide and one water molecule (Figure 1). The coordination geometries at the lanthanide centres are best described as distorted square antiprismatic with two hfac ligands occupying one square plane and the third hfac, the phosphine oxide and coordinated water molecule describing the second square plane. The average hfac–oxygen–Tb3+ and hfac–oxygen–Er3+-bond distances are 2.383(3) and 2.357(5) Å respectively and lie in the range of bond distances previously reported [40,47]. Similarly, the (ArF)3PO–Tb3+ and (ArF)3PO–Er3+ bond distances of 2.314(3) and 2.284(5) Å compare well to those reported for the complex [Er(hfac)3{(ArF)3PO}2] (ave. = 2.323(4) Å) [34] and the Tb3+–OH2 and Er3+–OH2 bond distances are measured as 2.377(3) and 2.337(5) Å respectively, which are similar to the Ln–OH2 bond distances commonly measured in trivalent lanthanide complexes of DOTA; range = 2.590 Å ([CeDOTA]−) to 2.355 Å ([YbDOTA]−), (DOTA = 1,4,7,10-tetraazacyclododecane-N′,N″,N′′′,N′′′′-tetraacetic acid) [48].
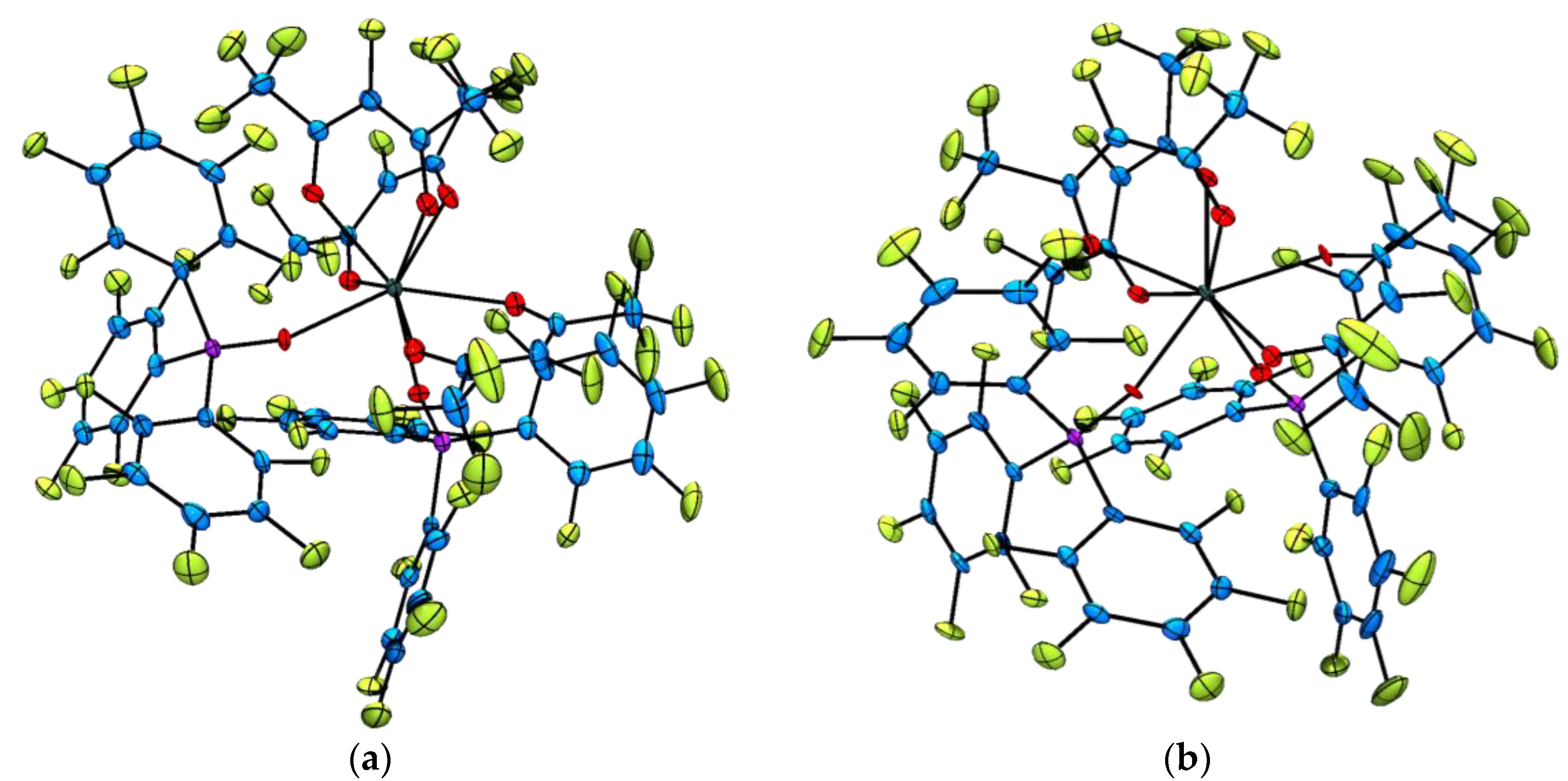
Single X-ray diffraction analysis of the fully perfluorinated lanthanide(III) complexes [Ln(F7-acac)3{(ArF)3PO}2] (where Ln = Sm3+, 6, Eu3+, 7, Er3+, 9, and Yb3+, 10) confirmed the expected connectivity (Figure 2). Again, this series of complexes are isostructural and all crystallise with a single CH2Cl2 solvent molecule in the asymmetric unit cell. In each structure, the lanthanide(III) ion is eight coordinate and the coordination geometry best described as distorted square antiprismatic, where the perfluorinated phosphine oxide ligands lie in a mutually cis arrangement, but are located in different square planes, presumably to minimize steric interactions. Average F7-acac oxygen–lanthanide(III) bond distances are 2.409(5) Å (6), 2.394(4) Å (7), 2.338(4) Å (9) and 2.317(4) Å (10) and follow the 8-coordinate lanthanide contraction as the series is progressed. Similarly, the average (ArF)3PO–Ln3+ distances become smaller traversing the 4f series (2.368(5) Å for 6 to 2.269(4) Å for 10) and lie in the range of those reported previously for the complex [Er(F7-acac)3{(ArF)3PO}2] which is a polymorph of 9 [47] and for the related complex [Eu(F7-acac)3{Ph3PO}2] [49].
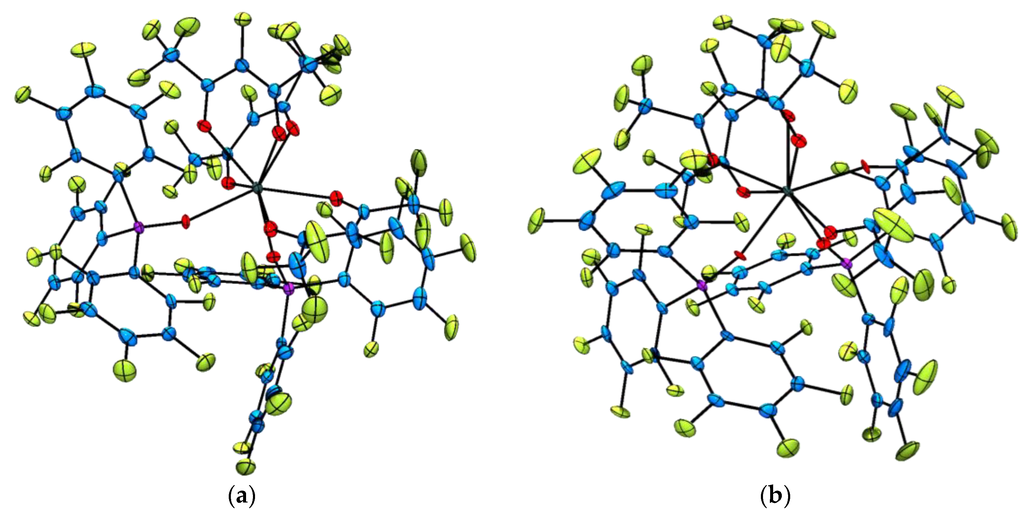
Figure 2.
Representative solid state molecular structures of the [Ln(F7-acac)3{(ArF)3PO}2] complexes with thermal ellipsoids set at the 50% probability level and lattice CH2Cl2 molecules omitted for clarity. Atom colour key: C, blue, Ln3+, dark green, O, red, P, pink, F, green. (a) Thermal ellipsoid plot of [Sm(F7-acac)3{(ArF)3PO}2], complex 6; (b) Thermal ellipsoid plot of [Yb(F7-acac)3{(ArF)3PO}2], complex 10.
2.3. Photophysical Properties of the Complexes
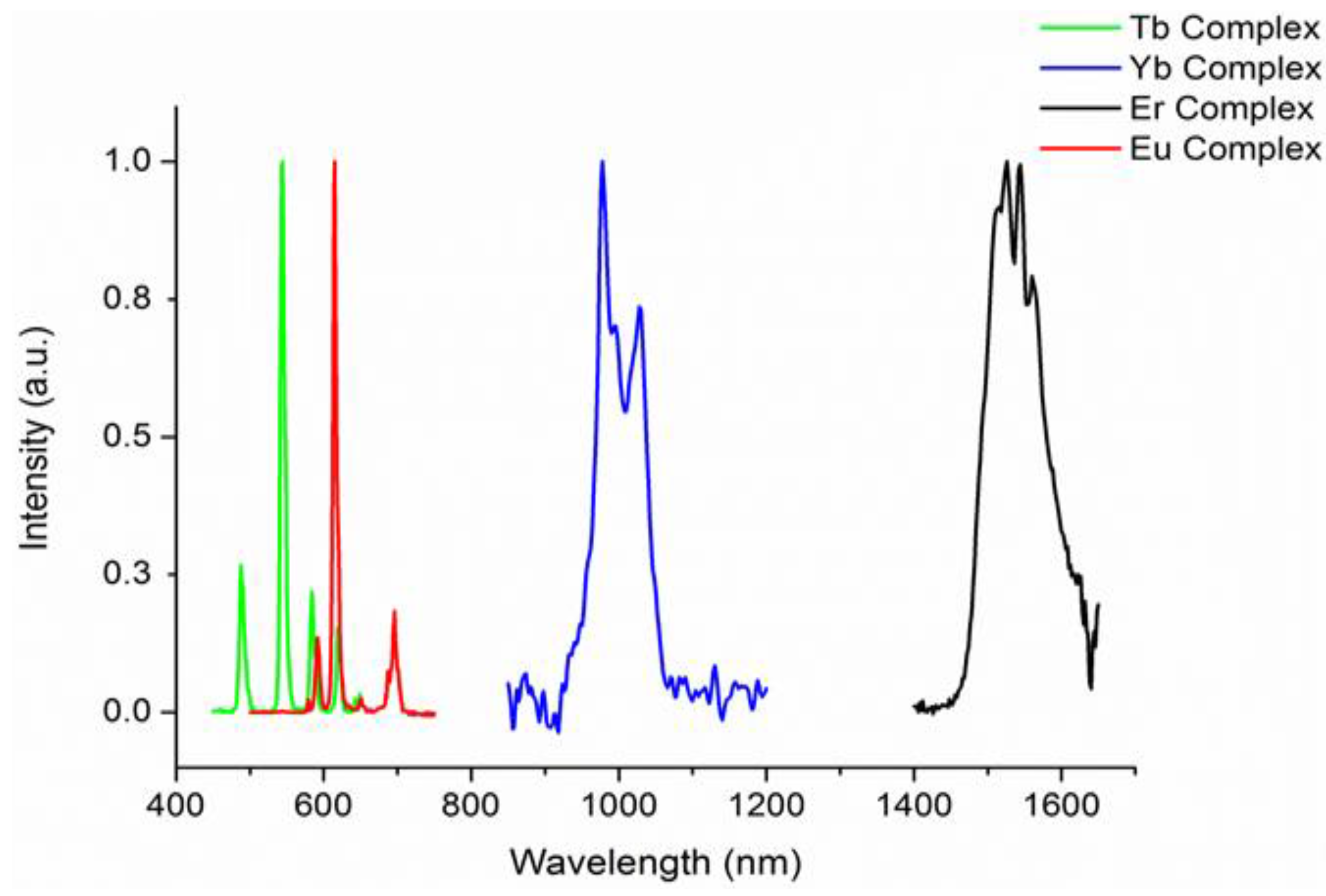
With two families of complexes incorporating different degrees of fluorination in hand, we next resolved to investigate the effects of perfluorination on the optical properties of the complexes. The UV–visible absorption spectra of all the complexes 1–10 exhibit intense absorptions in the UV region of the electromagnetic spectrum with absorption maxima at ca. 230 and 300 nm (complexes 1–5) attributable to the π–π* transitions of the perfluorinated-phenyl and β-diketonate chromophores respectively. In the absorption spectra of complexes 6–10, an additional maximum centered at ca. 325 nm is also observed. Following UV excitation into all ligand absorption bands at 230, 280 or 355 nm, all the complexes exhibited lanthanide f-centered emission in the visible and near infra-red region at typical wavelengths for a given lanthanide(III) ion (Figure 3). In all cases, ligand sensitised emission was confirmed by recording the excitation spectrum at the emission maximum, which matched well to the absorption spectra indicating that both chromophores are involved in the sensitization process. Representative emission spectra for the [Ln(F7-acac)3{(ArF)3PO}2] family of complexes are illustrated in Figure 3. Interestingly, the relative intensities of the visible and near infra-red emission, in particular of the Er3+ and Yb3+ complexes (9 and 10), are much greater than those recorded for the Er3+ and Yb3+ nIR analogues of [Ln(hfac)3{(ArF)3PO}(H2O)] (4 and 5) reflecting the greater degree of vibrational quenching from the coordinated water molecule in the latter.
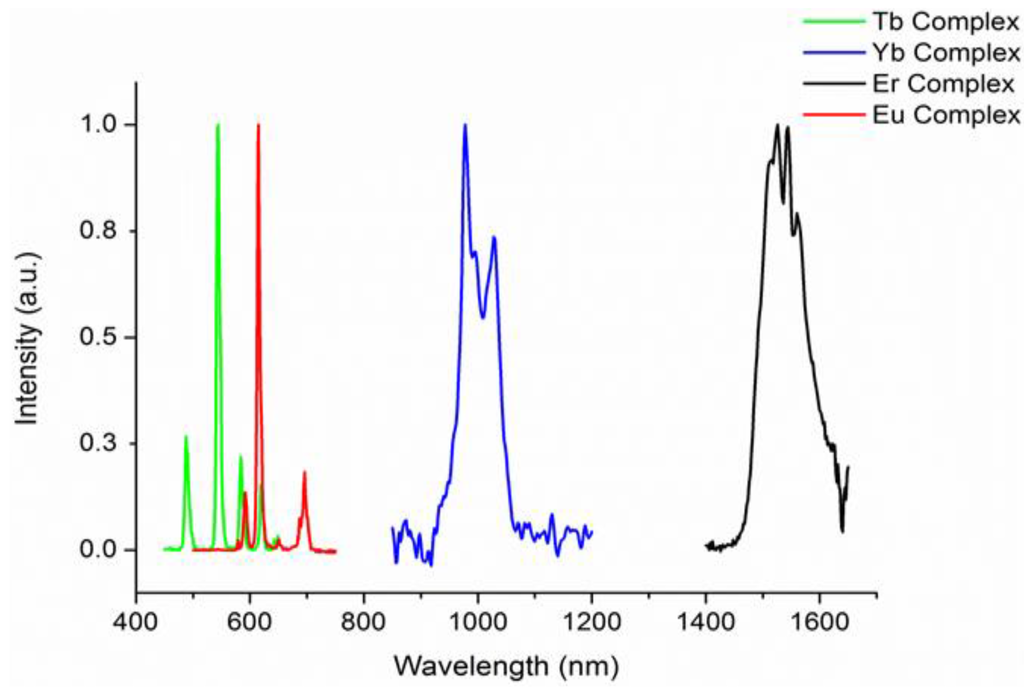
Figure 3.
Corrected and normalized steady state emission spectra of [Ln(F7-acac)3{(ArF)3PO}2] complexes following 280 nm excitation in CH2Cl2 at 298 K (Ln3+ = Eu, 7, Tb, 8, Er, 9, and Yb, 10).
The luminescence lifetimes of the [Ln(hfac)3{(ArF)3PO}(H2O)] complexes recorded at the emission maxima are typical for both the visible and nIR emitting Ln3+ complexes, being in the microsecond range (Table 1). Interestingly, for the shorter lived Er3+ and Yb3+ complexes 4 and 5, the kinetic traces following 337 nm excitation were best fitted to a biexponential decay, suggestive of two non-interconverting emissive species in solution on the timescale of the experiment. Given that the longer lived component of the kinetic trace of 5 (τ = 1.94 μs) can be compared to the related complex Er(TPIP)3 (TPIP = tetraphenylimidodiphosphinate) [43], where τCDCl3 = 5.0 μs, it seems reasonable to assume that the shorter lived component is due to the aqua species [Ln(hfac)3{(ArF)3PO}(H2O)], whereas the longer lived species is devoid of a coordinated solvent molecule. In the case of the longer lived visible emitting lanthanide ions in this series of hfac complexes, dynamic exchange of the labile water molecule is faster than the timescale of the experiment leading to the observation of an averaged solution lifetime [48]. It is worth noting here, that the luminescence lifetimes of all the complexes measured in solution are exceptionally sensitive to small amounts of water present in the solvent, therefore the solvent was thoroughly dried prior to use and reported values are reproducible in at least three independent measurements. Additionally, initial discrepancies in kinetic measurements led us to additionally record the photophysical properties of the complexes in the solid state (Table 1).

Table 1.
Photophysical properties of the complexes recorded in dry CH2Cl2 and in the solid state at 298 K upon excitation at 280 nm.
ΦLn = τobs/τrad
Since we were unable to reliably determine the total quantum yield of emission for all the complexes, (due in part to the small f–f molar absorption extinction coefficients) we have instead estimated the intrinsic quantum yield of emission (ΦLn) based on calculated and published values of the Ln3+ radiative lifetime of a given emissive excited state in the absence of any non-radiative deactivation processes, τrad (Equation (1)) [2,37,40,47]. The quantum efficiency of the Ln3+ based emission can be determined by measuring the quantum yield based on emission following excitation into the f–f absorption bands. Alternatively, and in the absence of sufficiently intense emission upon direct excitation, the intrinsic quantum yield can be estimated from the ratio of the observed luminescence lifetime (τobs) with the radiative lifetime (τrad). However, since τrad values depend heavily on the coordination environment and refractive index of the medium of a given lanthanide ion, we have here chosen larger τrad values reported for structurally similar complexes as far as possible to give more reasonable estimations of ΦLn (Table 1) [14,15,39,50,51,52,53,54]. Nevertheless, caution must be exercised when interpreting these values since very small changes in the ligand structure, solvent and coordination geometry of the complex can lead to large discrepancies in τrad values. As a result, the values reported here therefore only serve as a guide. For the series of the [Ln(hfac)3{(ArF)3PO}(H2O)] complexes (1–5), generally the intrinsic quantum yields in fluid solution are typical for partially fluorinated β-diketonate complexes, but notably, the quantum yield values for the Sm3+ and Yb3+ derivatives 1 and 5 both in solution and in the solid state are relatively large (3.4%, 8.5%, 0.3% and 2% respectively). However, concentration quenching effects or local heating in the solid state samples cannot be ruled out and these values may in fact be considerably larger.
The effect of complete fluorination on the photophysical properties of the complexes is further evidenced by the significant increase in the luminescence lifetimes of the [Ln(F7-acac)3{(ArF)3PO}2] series of complexes 6–10 (Table 1) of approximately one order of magnitude. This effect is particularly pronounced for the nIR emitting lanthanide ions Er3+ and Yb3+, where in the case of the Er3+ derivative, 9, the lifetimes measured in solution and the solid state are very similar (15.3 and 16.8 μs respectively). This observation is in agreement with those of Monguzzi and co-workers who recorded identical lifetimes for this complex in CDCl3 solution and in the solid state. For complex 9, the similar solution state and solid state lifetimes suggest that the Er3+ ions may be shielded to the same degree in their immediate coordination environment. Again, the detrimental effect of intermolecular quenching between neighbouring Er3+ ions and/or heating of the samples may result in an apparent lowering of the actual lifetime and quantum yield values. Here, the intrinsic quantum yield of 0.4% is considerably lower than the theoretical value where the radiative lifetime is equal to 4 ms [47], which suggests that residual CH2Cl2 solvent (as observed by X-ray crystallography) may play a role in lowering the overall quantum yield of emission. This observation is borne out to a certain degree by examination of the kinetic data for the Yb3+ complex 10, where the Yb3+ excited state is less susceptible to vibrational quenching by C–H oscillators [42]. This complex exhibits a marked enhancement (by a factor of >5) in both solution and solid state luminescence lifetimes and quantum yields when compared to the hexafluorinated aqua complex [Yb(hfac)3{(ArF)3PO}(H2O)], 5. Indeed, the fully perfluorinated derivative 10 exhibits a remarkably long luminescence lifetime of 139 μs in the solid phase (18 μs in CH2Cl2 solution) and an intrinsic quantum yield of Yb3+ based emission of 11%. This compares well to the Yb3+ bis-bipyridine N-oxide derivative of the Lehn cryptand described by Seitz that exhibits a room temperature solution luminescence lifetime of 26.1 μs in deuterated methanol [31]. In this system, perdeuteration of the ligand results in an extraordinarily long solution lifetime of 172 μs and the highest reported intrinsic quantum yield of Yb3+ emission in solution to date (26%). Together, these observations further highlight the fact that purposeful reduction in competitive vibrational quenching thereby substantially increasing the lifetime of the lanthanide based emission is key to achieving large nIR quantum yields of emission.
Interestingly, in the case of the Sm3+ complex 6, no f-centered emission was observed in solution at room temperature; the emission spectrum being dominated by residual ligand centered transitions. This clearly indicates that the F7-acac ligand is a poor sensitizer for Sm3+ and that the C–F bond in the β-diketonate unit quenches the Sm3+ based emission considerably [44]. In the solid state however, complex 6 exhibits a typical Sm3+ based emission profile with a long luminescence lifetime of 506 μs and a large calculated intrinsic quantum yield of 26% [16,55].
3. Materials and Methods
3.1. General Details
All chemical reagents were obtained from Sigma-Aldrich chemical company (Dorset, England) apart from the lanthanide acetate hydrate salts (Ln(OAc)3·xH2O), which were obtained from Alfa Aesar (Heysham, UK) and 1,1,1,3,5,5,5-heptafluoroacetylacetone (F7-acac) from Apollo Scientific (Manchester, UK) and were used as supplied or recrystallised before use. The compounds tris(pentafluorophenyl)phosphine oxide (ArF3PO) [41] and lanthanide(III) tris(1,1,1,5,5,5-hexafluoro-2,4-pentanedionate) dihydrates (Ln(hfac)3·2H2O, Ln = Sm3+, Eu3+, Tb3+, Tm3+, Er3+ and Yb3+) [34] were prepared according to literature and modified literature procedures. Reagent grade anhydrous solvents were either obtained by passing through activated alumina columns (Innovative Technologies) or dried over potassium (hexane and pentane) or over CaH2 (CH2Cl2) and were distilled and degassed prior to use. Deuterated solvents for multinuclear NMR studies were dried over 4 Å molecular sieves before use. All air sensitive experiments were performed using standard Schlenk line techniques.
NMR spectra were recorded on either a Bruker Avance 400 spectrometer (Bruker UK, Coventry, UK), operating frequency 400 MHz (1H), 101 MHz (13C), 162 MHz (31P) and 376 MHz (19F) variable temperature unit set at 295 K, or on a Varian Mecury-200 spectrometer (Varian Associates, Palo Alto, CA, USA) at 298 K (1H at 200 MHz, 13C at 50 MHz, 31P at 81 MHz, 19F at 188 MHz). Chemical shifts are reported in parts per million (ppm) relative to TMS (1H), 85% H3PO4 (ortho-phosphoric acid) (31P{1H}) and CFCl3 (19F). All 1H NMR spectra were internally referenced to residual proton resonances in CDCl3 or d6-acetone.
Mass spectra were obtained using either MALDI from CH2Cl2 solutions with a dithrinol matrix on a Shimadzu Axima confidence spectrometer (Shimazdu, Kratos site, Manchester, UK, for all complexes) or electrospray mass spectrometry, performed on a Micromass Platform II system (ligands and precursors). For all of the complexes 1–10, no identifiable molecular ion peaks or fragmentation products were observable.
Elemental analyses on the compounds were performed by M. Jennings and colleagues in the microanalytical laboratory in the School of Chemistry at the University of Manchester; a Carlo ERBA Instruments CHNS–O EA1108 elemental analyzer (Carlo ERBA Instruments, Milan, Italy) was used for C, H and N analysis and a Fisons Horizon elemental analysis ICP-OED spectrometer (VG Elemental, Winsford, UK) for metals.
X-ray diffraction data for compounds 3, 6, 7, and [Tm(F7-acac)3{(ArF)3PO}2]) were collected using an Rigaku Oxford Diffraction Xcalibur, Sapphire2 diffractometer (Rigaku, Tokyo, Japan), utilising graphite-monochromated Mo Kα X-ray radiation (λ = 0.71073 Å); compounds 9 and 10, or on a Bruker X8 Prospector diffractometer (Bruker, Billerica, MA, USA) utilising graphite-monochromated Cu Kα X-ray radiation (λ = 1.54178 Å) and compound 4 on a Bruker AXS SMART diffractometer (Bruker, Billerica, MA, USA) using graphite-monochromated Mo Kα X-ray radiation (λ = 0.71073 Å). Data were corrected for Lorentz and polarisation factors and empirical absorption corrections applied [56,57]. Crystal data, data collection and structural refinement parameters are given in Table S1. The structures were solved by direct methods using the program SHELXT [58] or OLEX-2 [59]. Full matrix refinement on F2 and all further calculations were performed using OLEX-2 or ShelXL [58]. The non-H atoms were refined anisotropically and hydrogen atoms were positioned in idealised sites and were allowed to ride on their parent C or N atoms.
Absorption spectra were recorded in dry CH2Cl2 on a T60U spectrometer (PG Instruments Ltd., Lutterworth, UK) using fused quartz cells with a path length of 1 cm or on a double-beam Cary Varian 500 scan UV–vis–nIR spectrophotometer over the range 300–1300 nm.
All solution luminescence measurements were recorded on compounds dissolved in dry CH2Cl2 solutions using screw or Teflon™ (Hellma UK Ltd., Southend on Sea, UK) capped fused quartz cuvettes with a 1 cm or 0.1 cm path length. All measurements were recorded within 30 min of sample preparation to avoid ingress of oxygen and moisture. Luminescence measurements of solid samples were recorded using finely divided powdered samples held in between two 10 cm2 quartz plates. All steady state emission and excitation spectra were recorded on an Edinburgh Instrument FP920 Phosphorescence Lifetime Spectrometer (Edinburgh Instruments, Livingston, Scotland) equipped with a 450 watt steady state xenon lamp, a 5 watt microsecond pulsed xenon flashlamp, (with single 300 mm focal length excitation and emission monochromators in Czerny Turner configuration), a red sensitive photomultiplier in peltier (air cooled) housing (Hamamatsu R928P), and a liquid nitrogen cooled nIR photomultiplier (Hamamatsu, Hamamatsu City, Shizuoka Prefecture, Japan). Lifetime data were recorded following excitation with the microsecond flashlamp using time correlated single photon counting (PCS900 plug-in PC card for fast photon counting). Lifetimes were obtained by tail fit on the data obtained and quality of fit judged by minimization of reduced chi-squared and residuals squared. For the Er3+ and Yb3+ complexes 4 and 5, the sample was excited using a pulsed nitrogen laser (337 nm) operating at 10 Hz. Light emitted at right angles to the excitation beam was focused onto the slits of a monochromator, which was used to select the appropriate wavelength. The growth and decay of the luminescence at selected wavelengths was detected using a germanium photodiode (Edinburgh Instruments, EI-P, Edinburgh Instruments, Livingstone, Scotland) and recorded using a digital oscilloscope (Tektronix TDS220, Tektronix Inc., Beaverton, OR, USA) before being transferred to a PC for analysis. Luminescence lifetimes were obtained by iterative reconvolution of the detector response (obtained by using a scatterer) with exponential components for growth and decay of the metal centred luminescence, using a spreadsheet running in Microsoft Excel. The details of this approach have been discussed elsewhere [60,61]. Unless otherwise stated, fitting to a double exponential decay yielded no improvement in fit as judged by minimisation of residual squared and reduced chi squared. Note that the Tm3+ complexes [Tm(hfac)3{(ArF)3PO}(H2O)] and [Tm(F7-acac)3{(ArF)3PO}2] were found to be non-emissive in CH2Cl2 solutions at room temperature upon UV excitation (230–360 nm).
3.2. Synthetic Procedures
3.2.1. Preparation of Tris(pentafluorophenyl)phosphine Oxide, (C6F5)3PO (ArF3PO)
According to a modification of a literature procedure [41], tris(pentafluorophenyl)phosphine (1.031 g, 1.9 mmol) was dissolved in chloroform (40 mL), cooled to 0 °C in an ice bath and stirred for 30 min. 3-Chloroperoxybenzoic acid (0.3013 g, 2.2 mmol) was dissolved in chloroform (10 mL) and then added dropwise to the reaction mixture over 10 min. The reaction mixture was again cooled to 0 °C in an ice bath and left to warm to room temperature and stirred for 48 h. After this time, saturated sodium hydrogen carbonate solution (50 mL) was added and the reaction mixture stirred for 30 min. The organic soluble products were extracted with chloroform (3 × 50 mL) and dried over MgSO4. The organic layer was removed by rotary evaporation and the white powder dried in vacuo to give 0.89 g of tris(pentafluorophenyl)phosphine oxide (81% yield). All data were consistent with those documented in the literature. ES+ MS (MeCN) m/z 549 [M + H]+ (44%), 571 [M + Na]+ (100%), 1119 [2M + Na]+ (62%), 1667 (3M + Na]+ (67%).
NMR/CDCl3 (400 MHz) δF: 131.30 (d, 2F, 3JFF = 22.4 Hz, ortho-CF), −141.32 (t, 1F, 3JFF = 18.6, 22.6 Hz, para-CF), −157.27 (tt, 2F, 3JFF = 22.6, 18.8 Hz, 4JFF = 7.5, 3.8 Hz, meta-CF); δP: −8.26 (s, ArF3PO). UV–vis (CH2Cl2) λ = 230, 277 nm.
3.2.2. Preparation of Ln(hfac)3·2H2O
According to a literature procedure reported for Nd3+ [34], The corresponding lanthanide acetate hydrate (Ln(OAc)3·xH2O) (15 mmol) was dissolved in deionised water (20 mL) with stirring in an ice bath. 1,1,1,5,5,5-hexafluoro-2,4-pentanedione (hfac) (5.0 g, 24 mmol) was dissolved in methanol and was added dropwise to the lanthanide acetate solution. The reaction mixture was stirred for 3 h in an ice bath and a further 65 h at room temperature. The solvent was then removed using a rotary evaporator and the resulting product was recrystallised from MeOH.
Yields: Tb(hfac)3·2H2O = 93% (pale green-blue powder), Sm(hfac)3·2H2O (pale yellow powder) = 93%, Er(hfac)3·2H2O (pink powder) = 86%, Yb(hfac)3·2H2O (cream powder) = 56%, Tm(hfac)3·2H2O (white powder) = 43%, Eu(hfac)3·2H2O (cream powder) = 69%. All spectroscopic data were consistent with the formulation of the compounds as Ln(hfac)3·2H2O as previously reported.
3.2.3. Preparation of [Ln(hfac)3{(ArF)3PO}(H2O)], Ln = Sm3+, Eu3+, Tb3+, Er3+, Yb3+ (1–5) from Ln(hfac)3·2H2O
In air, Ln(hfac)3·2H2O (0.31 mmol) and FPh3PO (0.31 mmol) were added to a round bottomed flask and dissolved in CH2Cl2 (150 mL). The reaction mixture was sonicated for 1 h. The reaction mixture was then filtered under gravity. All volatiles were removed by rotary evaporation and the resultant crude powders recrystallised from CH2Cl2 by slow evaporation and the crystalline products filtered under gravity, washed with CH2Cl2 and dried in vacuo. Once isolated, all the complexes are air stable for extended time periods (>4 years) are moderately soluble in acetone and sparingly soluble in CH2Cl2 and CHCl3 precluding acquisition of NMR data in some cases.
[Sm(hfac)3{(ArF)3PO}(H2O)] (1): Isolated in 41% yield as pale yellow crystals (170 mg). NMR/CDCl3 (400 MHz) δH: 7.51 (s, CH-hfac); δF: −77.79 (s, 18F, CF3-hfac), −131.37 (s, 6F) ortho-CF), −140.37 (br, 3F, para-CF), −157.05 (m, 6F, meta-CF) δP {1H}: −7.44 (s, (ArF)3PO). UV–vis (CH2Cl2) λ = 234 nm ((ArF)3PO), 306 nm ((ArF)3PO and hfac). Anal. Calcd. For C33H5O8F33PSm: C 29.63, H 0.37, N 0.0, P 2.31, Sm 11.24. Found: C 29.52, H 0.0, N 0.0, P 2.06, Sm 11.63.
[Eu(hfac)3{(ArF)3PO}(H2O)] (2): Isolated in 38% yield as colourless crystals (158 mg). NMR/CDCl3 (400 MHz) δH, δF, δP {1H}: no signals observed. UV–vis (CH2Cl2) λ = 232 nm ((ArF)3PO), 301 nm ((ArF)3PO and hfac). Anal. Calcd. For C33H5O8F33PEu: C 29.59, H 0.38, N 0.0. Found: C 28.64, H 0.14, N 0.0.
[Tb(hfac)3{(ArF)3PO}(H2O)] (3): Isolated in 49% yield as pale green crystals (205 mg). NMR/CDCl3 (400 MHz) δH: −0.19 (br, CH-hfac); δF: −45.54 (br, CH-hfac), −131.88 (br, ortho-CF), −141.34 (br, para-CF), −157.33 (br, meta–CF). δP {1H}: −10.06 (br). UV–vis (CH2Cl2) λ = 231 nm ((ArF)3PO), 303 nm ((ArF)3PO and hfac). Anal. Calcd. For C33H5O8F33PTb: C 29.44, H 0.37, N 0.0, P 2.30, Tb 11.80. Found: C 29.32, H 0.0, N 0.0, P 1.99, Tb 10.80.
[Er(hfac)3{(ArF)3PO}(H2O)] (4): Isolated in 49% yield as pink crystals (206 mg). NMR/CDCl3 (400 MHz) δH: no signal observed; δF: −94.62 (br, CF3-hfac), −137.20 (br, ortho/meta/para-CF), −153.32 (br, 6F, ortho/meta/para-CF); δP {1H}: no signal observed. UV–vis (CH2Cl2) λ = 233 nm ((ArF)3PO), 302 nm ((ArF)3PO and hfac). Anal. Calcd. For C33H5O8F33PEr: C 29.25, H 0.37, N 0.0, P 2.29, Er 12.38. Found: C 29.28, H 0.0, N 0.0, P 1.97, Er 12.10.
[Yb(hfac)3{(ArF)3PO}(H2O)] (5): Isolated in 59% yield as colourless crystals (249 mg). NMR/CDCl3 (400 MHz) δH: 8.07 (br, CH-hfac); δF: −87.83 (s, 2F, ortho/meta CF), −123.75 (br, CF3-hfac), −138.98 (s, 1F, para-CF), −155.68 (s, 2F, ortho/meta CF); δP {1H}: no signal observed. UV–vis (CH2Cl2) λ = 232 ((ArF)3PO), 302 nm ((ArF)3PO and hfac). Anal. Calcd. For C33H5O8F33PYb: C 29.14, H 0.37, N 0.0, P 2.27. Found: C 28.82, H 0.0, N 0.0, P 1.73.
[Tm(hfac)3{(ArF)3PO}(H2O)]: Isolated in 26% yield as colourless crystals (109 mg). Anal. Calcd. For C33H5O8F33PTm: C 29.22, H 0.37, N 0.0, P 2.28, Tm 12.45. Found: C 29.18, H 0.0, N 0.0, P 2.05, Tm 12.36.
3.2.4. Preparation of [Ln(hfac)3{(ArF)3PO}(H2O)], Ln = Sm3+, Eu3+, Tb3+, Er3+, Yb3+ (1–5) from Ln(OAc)3·xH2O under N2
Under N2, an oven dried Schlenk was charged with 0.26 mmol of the appropriate lanthanide acetate hydrate (Ln(OAc)3·xH2O) and 0.26 mmol (142 mg) (ArF)3PO and 25 mL of dry CH2Cl2 added by cannula or syringe. A solution of Hfac (0.78 mmol, 154 mg) in minimal CH2Cl2 (~2 mL) was then added dropwise and the reaction mixture heated at reflux temperature for 1 hour under a flow of N2 and then cooled to room temperature. After this time, the solution was filtered and the filtrate layered with hexane or pentane and placed in the freezer at −18 °C. After several days, the crystalline products were isolated by filtration, washed with cold CH2Cl2 and dried in vacuo to yield the title product.
[Sm(hfac)3{(ArF)3PO}(H2O)] (1): Isolated in 53% yield (184 mg). All characterization data are consistent with the proposed structure.
[Tb(hfac)3{(ArF)3PO}(H2O)] (3): Isolated in 44% yield (155 mg). All characterization data are consistent with the proposed structure.
[Er(hfac)3{(ArF)3PO}(H2O)] (4): Isolated in 16% yield (55 mg). All characterization data are consistent with the proposed structure.
[Yb(hfac)3{(ArF)3PO}(H2O)] (5): Isolated in 48% yield (170 mg). All characterization data are consistent with the proposed structure.
3.2.5. Preparation of [Ln(F7-acac)3{(ArF)3PO}2] Ln = Sm3+, Eu3+, Tb3+, Er3+, Yb3+ (6–10) from Ln(OAc)3·xH2O under N2
Under N2, an oven dried Schlenk was charged with 0.33 mmol of the appropriate lanthanide acetate hydrate (Ln(OAc)3·xH2O) and 0.66 mmol (142 mg) (ArF)3PO. 25 mL of dry CH2Cl2 was then added by cannula or syringe. 1,1,1,3,5,5,5-heptfluoro-2,4-pentanedione (0.94 mmol) was subsequently rapidly added and the reaction mixture heated at reflux temperature for 1 hour under a flow of N2, then cooled to room temperature. After this time, dry pentane (40 mL) was added and the Schlenk flask placed in the freezer at −18 °C. After one week, the crystalline products were isolated by filtration, washed with cold CH2Cl2 and dried under vacuum suction to yield the title compounds. The products are slightly hygroscopic when stored under ambient conditions for extended periods of time (>1 year). All complexes are soluble in acetone and sparingly soluble in CH2Cl2 and CHCl3.
[Sm(F7-acac)3{(ArF)3PO}2] (6): Isolated in 42% yield as a pale yellow crystalline solid (289 mg). NMR/d6-acetone (200 MHz) δF: −74.67 (d, 18F, 4JFF = 18.3 Hz, CF3-hfac), −134.15 (d, 12F, 3JFF = 10.9 Hz, 4JFF = 8.1 Hz, ortho-CF), −145.68 (m, 6F, para-CF), −161.28 (m, 12F, meta-CF), −192.30 (br, 3H, CF F7-acac); δP {1H}: −9.20 (s, (ArF)3PO). UV–vis (CH2Cl2) λ = 230, 272 nm ((ArF)3PO), 328, 356 (sh) nm (F7-acac). Anal. Calcd. For C51O8F51P2Sm·2CH2Cl2: C 30.43, H 0.19, N 0.0, Sm 7.20. Found: C 30.25, H 0.0, N 0.0, Sm 6.85.
[Eu(F7-acac)3{(ArF)3PO}2] (7): Isolated in 37% yield as a pale yellow crystalline solid (242 mg). NMR/d6-acetone (200 MHz) δF: −78.14 (d, 18F, 4JFF = 17.4 Hz, CF3 F7-acac), −134.11 (dd, 12F, 3JFF = 22.0 Hz, 4JFF = 1.9 Hz, ortho-CF), −145.68 (m, 6F, para-CF), −161.29 (m, 12F, meta-CF), −170.01 (m, 3F, CF F7-acac); δP {1H}: −7.80 (s, (ArF)3PO). UV–vis (CH2Cl2) λ = 232, 274 nm ((ArF)3PO), 324, 358 (sh) nm (F7-acac). For C51O8F51P2Eu·CH2Cl2: C 31.10, H 0.1, N 0.0, Eu 7.57. Found: C 30.73, H 0.0, N 0.0, Eu 8.57.
[Tb(F7-acac)3{(ArF)3PO}2] (8): Isolated in 47% yield as a pale yellow crystalline solid (312 mg). NMR/d6-acetone (200 MHz) δF: −58.84 (br, CF3 F7-acac), −134.20 (d, 6F, 4JFF = 20.7 Hz, ortho-CF), −145.65 (m, 3F, para-CF), −161.28 (m, 6F, meta-CF), −108.00 (br, CF F7-acac); δP{1H}: −11.61 (br, (ArF)3PO). UV–vis (CH2Cl2) λ = 230, 276 nm ((ArF)3PO), 330 nm (F7-acac). Anal. Calcd. For C51O8F51P2Tb·CH2Cl2: C 30.99, H 0.1, N 0.0, Tb 7.89. Found: C 30.92, H 0.0, N 0.0, Tb 7.20.
[Er(F7-acac)3{(ArF)3PO}2] (9): Isolated in 40% yield as a pale yellow crystalline solid (289 mg). NMR/d6-acetone (200 MHz) δF: −87.28 (br, CF3 F7-acac), −133.90 (d, 6H, 4JFF = 21.8 Hz, ortho-CF), −145.47 (m, 3H, para-CF), −161.27 (m, 6H, meta-CF), −213.41 (br, CF F7-acac); δP{1H}: −10.63 (s, (ArF)3PO). UV–vis (CH2Cl2) λ = 232, 274 nm ((ArF)3PO), 322, 354 (sh) nm (F7-acac). Anal. Calcd. For C51O8F51P2Er·3CH2Cl2: C 29.57, H 0.28, N 0.0, Er 7.63. Found: C 29.10, H 0.0, N 0.0, Er 7.22.
[Yb(F7-acac)3{(ArF)3PO}2] (10): Isolated in 27% yield as a pale yellow crystalline solid (213 mg). NMR/d6-acetone (200 MHz) δF: −87.37 (br, CF3 F7-acac), −133.99 (d, 6H, 4JFF = 20.0 Hz, ortho-CF), −145.66 (m, para-CF), −161.28 (m, 6H, meta-CF), −208.20 (br, CF F7-acac); δP {1H}: −8.93 (br, (ArF)3PO). UV–vis (CH2Cl2) λ = 230, 274 nm ((ArF)3PO), 322, 354 (sh) nm (F7-acac). Anal. Calcd. For C51O8F51P2Yb·5CH2Cl2: C 28.39, H 0.43, N 0.0, Yb 7.30. Found: C 27.82, H 0.0, N 0.0, Yb 6.98.
[Tm(F7-acac)3{(ArF)3PO}2]: Isolated in 48% yield as a white crystalline solid (321 mg). NMR/d6-acetone (200 MHz) δF: −98.08 (br, CF3 F7-acac), −133.88 (s, 6H, ortho-CF), −145.47 (m, para-CF), −161.27 (m, 6H, meta-CF), −245.04 (br, CF F7-acac); δP: −9.10 (br, (ArF)3PO). Anal. Calcd. For C51O8F51P2Tm·CH2Cl2: C 30.84, H 0.0, N 0.0, Tm 8.34. Found: C 30.47, H 0.0, N 0.0, Tm 8.74.
3.3. X-ray Crystallographic Data for Compounds 3, 4, 6, 7, 9 and 10
The X-ray crystallographic information files (cif) for all the single crystal X-ray structures reported herein have been deposited with the Cambridge Crystallographic Database (CCDC), numbers: 1472376–1472382. These data are available free of charge at www.ccdc.cam.ac.uk. For data collection and structural refinement details, see Table S1.
4. Conclusions
In summary, two families of emissive heavily fluorinated lanthanide(III) β-diketonate complexes bearing monodentate perfluorinated tris phenyl phosphine oxide ligands, [Ln(hfac)3{(ArF)3PO}(H2O)] and [Ln(F7-acac)3{(ArF)3PO}2] (where Ln = Sm3+, Eu3+, Tb3+, Er3+ and Yb3+) have been prepared and characterized by NMR spectroscopy, single crystal X-ray diffraction and luminescence spectroscopy. In depth photophysical studies on the complexes in CH2Cl2 solution and in the solid state have shown that replacing both the bound inner sphere H2O molecule and C–H oscillator in the β-diketonate ligand backbone in [Ln(hfac)3{(ArF)3PO}(H2O)] by a second (ArF)3PO ligand and C–F bond respectively in [Ln(F7-acac)3{(ArF)3PO}2] has a dramatic effect on the photophysical properties of the complexes. This effect is particularly notable for the near infra-red emitting ion Yb3+, where a five fold increase in luminescence lifetime and quantum yield is observed in [Yb(F7-acac)3{(ArF)3PO}2] (10) compared to [Ln(hfac)3{(ArF)3PO}(H2O)] (5). The presented data herein conclude that replacing all C–H oscillators in the immediate coordination environment of the Ln3+ ions with C–F bonds substantially reduces the degree of competitive vibrational quenching, particularly for the nIR emitting trivalent lanthanides in solution which in turn leads to more intense and longer lived f-centered emission. Here, this is particularly evident for the Sm3+ and Yb3+ complexes 1, 5 and 10, which exhibit both long luminescence lifetimes and relatively large intrinsic quantum yields of emission. Such an approach combined with intentional reduction of the radiative lifetime of the lanthanide based emission as demonstrated by Seitz [37] may find increasing use in the development of optical imaging probes and highly emissive materials for optoelectronics amongst other applications in the near future.
Supplementary Materials
The following are available online at www.mdpi.com/2304-6740/4/3/27/s1, Table S1: Single Crystal X-ray Data Collection and Structural Refinement for the Complexes and unit cell parameters for 5; Figures S1–S7: 19F and 31P{1H} NMR spectra for selected complexes (1, 3, 4, 5, 8, 9 and 10).
Acknowledgments
We thank the EPSRC for funding a Career Acceleration Fellowship (Louise S. Natrajan), postdoctoral funding (Adam N. Swinburne) and a studentship (Simon Randall) (grant number EP/G004846/1). We also thank the Leverhulme Trust for additional postdoctoral funding (Adam N. Swinburne, Fabrizio Ortu) (RL-2012-072) and a research Leadership award (Louise S. Natrajan) and the University of Manchester for project student support. This work was also funded by the EPSRC (grant number EP/K039547/1). We are additionally grateful to Stephen Faulkner for the loan of his facilities for the nIR measurements of complexes 4 and 5.
Author Contributions
Louise S. Natrajan conceived and designed the experiments; Adam N. Swinburne, Tsz Ling Chan, Madeleine H. Langford Paden and Louise S. Natrajan performed the experiments; Adam N. Swinburne, Louise S. Natrajan, Simon Randall, Fabrizio Ortu and Alan M. Kenwright analyzed the data; Alan M. Kenwright contributed analysis tools; Louise S. Natrajan wrote the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| NMR | nuclear magnetic resonance |
| nIR | near Infra-Red |
| UV/vis | Ultra-violet/visible |
| hfac | 1,1,1,5,5,5-hexafluoro-2,4-pentanedione |
| F7-acac | 1,1,1,3,5,5,5-heptafluoroacetylacetone |
| (ArF3)PO | tris(pentafluorophenyl)phosphine oxide |
| TPIP | tetraphenylimidodiphosphinate |
| F-TPIP | perfluorotetraphenylimidodiphosphinate |
| OAc | acetate |
| Ln | lanthanide |
| DOTA | 1,4,7,10-tetraazacyclododecane- N′,N″,N′′′,N′′′′-tetraacetic acid |
References
- Bünzli, J.-C.G.; Eliseeva, S.V. Intriguing aspects of lanthanide luminescence. Chem. Sci. 2013, 4, 1939–1949. [Google Scholar] [CrossRef]
- Binnemans, K. Lanthanide-based luminescent hybrid materials. Chem. Rev. 2009, 109, 4283–4374. [Google Scholar] [CrossRef] [PubMed]
- De Bettencourt-Dias, A. Lanthanide-based emitting materials in light-emitting diodes. Dalton Trans. 2007, 2229–2241. [Google Scholar] [CrossRef] [PubMed]
- Suyver, J.F.; Aebischer, A.; Biner, D.; Gerner, P.; Grimm, J.; Heer, S.; Krämer, K.E.; Reinhard, C.; Güdel, H.U. Novel materials doped with trivalent lanthanides and transition metal ions showing near-infrared to visible photon upconversion. Opt. Mater. 2005, 27, 1111–1130. [Google Scholar] [CrossRef]
- Meijerink, A.; Wegh, R.; Vergeer, P.; Vlugt, T. Photon management with lanthanides. Opt. Mater. 2006, 28, 575–581. [Google Scholar] [CrossRef]
- Harvey, P.; Oakland, C.; Driscoll, M.D.; Hay, S.; Natrajan, L.S. Ratiometric detection of enzyme turnover and flavin reduction using rare-earth upconverting phosphors. Dalton Trans. 2014, 43, 5265–5268. [Google Scholar] [CrossRef] [PubMed]
- Bünzli, J.-C.G. Lanthanide luminescence for biomedical analyses and imaging. Chem. Rev. 2010, 110, 2729–2755. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, C.P.; Murray, B.S.; New, E.J.; Pal, R.; Parker, D. Cell-penetrating metal complex optical probes: Targeted and responsive systems based on lanthanide luminescence. Acc. Chem. Res. 2009, 42, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Beeby, A.; Clarkson, I.M.; Faulkner, S.; Botchway, S.; Parker, D.; Williams, J.A.G.; Parker, A.W. Luminescence imaging microscopy and lifetime mapping using kinetically stable lanthanide (III) complexes. J. Photochem. Photobiol. B Biol. 2000, 57, 83–89. [Google Scholar] [CrossRef]
- Vuojola, J.; Soukka, T. Luminescent lanthanide reporters: New concepts for use in bioanalytical applications. Methods Appl. Fluoresc. 2014, 2, 1–28. [Google Scholar] [CrossRef]
- Beeby, A.; Burton-Pye, B.P.; Faulkner, S.; Motson, G.R.; Jeffery, J.C.; McCleverty, J.A.; Ward, M.D. Synthesis and near-IR luminescence properties of neodymium(III) and ytterbium(III) complexes with poly(pyrazolyl)borate ligands. J. Chem. Soc. Dalton Trans. 2002, 1923–1928. [Google Scholar] [CrossRef]
- Faulkner, S.; Carrie, M.-C.; Pope, S.J.A.; Squire, J.; Beeby, A.; Sammes, P.G. Pyrene-sensitised near-IR luminescence from ytterbium and neodymium complexes. Dalton Trans. 2004, 1405–1409. [Google Scholar] [CrossRef] [PubMed]
- Shavaleev, N.M.; Scopelliti, R.; Gumy, F.; Bünzli, J.-C. Surprisingly bright near-infrared luminescence and short radiative lifetimes of ytterbium in hetero-binuclear Yb_Na chelates. Inorg. Chem. 2009, 48, 7937–7946. [Google Scholar] [CrossRef] [PubMed]
- Moudam, O.; Rowan, B.C.; Alamiry, M.; Richardson, P.; Richards, B.S.; Jones, A.C.; Robertson, N. Europium complexes with high total photoluminescence quantum yields in solution and in PMMA. Chem. Commun. 2009, 6649–6651. [Google Scholar] [CrossRef] [PubMed]
- Magennis, S.W.; Ferguson, A.J.; Bryden, T.; Jones, T.S.; Beeby, A.; Samuel, I.D.W. Time-dependence of erbium(III) tris(8-hydroxyquinolate) near-infrared photoluminescence: Implications for organic light-emitting diode efficiency. Synth. Metals 2003, 138, 463–469. [Google Scholar] [CrossRef]
- Lunstroot, K.; Nockemann, P.; Van Hecke, K.; Van Meervelt, L.; Görller-Walrand, C.; Binnemans, K.; Driesen, K. Visible and near-infrared emission by Samarium(III)-containing ionic liquid mixtures. Inorg. Chem. 2009, 48, 3018–3026. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Tropiano, T.; Mantulnikovs, K.; Faulkner, S.; Vosch, T.; Sørensen, T.J. Spectrally resolved confocal microscopy using lanthanide centred near-IR emission. Chem. Commun. 2015, 51, 2372–2375. [Google Scholar] [CrossRef] [PubMed]
- Digonnet, M. (Ed.) Rare Earth Doped Fiber Lasers and Amplifiers; Marcel Dekker: New York, NY, USA, 1993.
- Desurvire, E. Erbium Doped Fiber Amplifiers; Wiley: New York, NY, USA, 1994. [Google Scholar]
- Desurvire, E. The golden age of optical fiber amplifiers. Phys. Today 1994, 47, 20–27. [Google Scholar] [CrossRef]
- Faulkner, S.; Burton-Pye, B.P.; Pope, S.J.A. Lanthanide complexes for luminescence imaging applications. Appl. Spectrosc. Rev. 2005, 40, 1–31. [Google Scholar] [CrossRef]
- Faulkner, S.; Natrajan, L.S.; Perry, W.S.; Sykes, D. Sensitised luminescence in lanthanide containing arrays and d–f hybrids. Dalton Trans. 2009, 3890–3899. [Google Scholar] [CrossRef] [PubMed]
- Binnemans, K. Rare-earth β-diketonates. In Handbook on the Physics and Chemistry of Rare Earths; Gschneidner, K.A., Jr., Bünzli, J.-C.G., Pecharsky, V.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; Volume 35, pp. 107–271. [Google Scholar]
- Hemilla, A. Applications of Fluorescence in Immunoassays; Wiley Interscience: New York, NY, USA, 1991. [Google Scholar]
- Yuan, J.; Wang, G. Lanthanide complex-based fluorescence label for time-resolved fluorescence bioassay. J. Fluoresc. 2005, 15, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Sy, M.; Nonat, A.; Hildebrandt, N.; Charbonnière, L.J. Lanthanide-based luminescence biolabelling. Chem. Commun. 2016, 52, 5080–5095. [Google Scholar] [CrossRef] [PubMed]
- Browne, W.R.; Vos, J.G. The effect of deuteriation on the emission lifetime of inorganic compounds. Coord. Chem. Rev. 2001, 219–221, 761–787. [Google Scholar] [CrossRef]
- Stein, G.; Wurzberg, E. Energy gap law in the solvent isotope effect on radiationless transitions of rare earth Ions. J. Chem. Phys. 1975, 62, 208–213. [Google Scholar] [CrossRef]
- Heller, A. Formation of hot OH Bonds in the radiationless relaxations of excited rare earth ions in aqueous solutions. J. Am. Chem. Soc. 1966, 88, 2058–2059. [Google Scholar] [CrossRef]
- Doffek, C.; Alzakhem, N.; Bischof, C.; Wahsner, J.; Güden-Silber, T.; Lugger, J.; Platas-Iglesias, C.; Seitz, M. Understanding the quenching effects of aromatic C–H– and C–D– oscillators in Near-IR Lanthanoid Luminescence. J. Am. Chem. Soc. 2012, 134, 16413–16423. [Google Scholar] [CrossRef] [PubMed]
- Horrocks, W.D.; Sudnick, D.R. Lanthanide ion luminescence probes of the structure of biological macromolecules. Acc. Chem. Res. 1981, 14, 384–392. [Google Scholar] [CrossRef]
- Supkowski, R.M.; Horrocks, W.D. On the determination of the number of water molecules, q, coordinated to europium(III) ions in solution from luminescence decay lifetimes. Inorg. Chim. Acta 2002, 340, 44–48. [Google Scholar] [CrossRef]
- Beeby, A.; Clarkson, I.M.; Dickins, R.S.; Faulkner, S.; Parker, D.; Royle, L.; de Sousa, A.S.; Williams, J.A.G.; Woods, M. Non-radiative deactivation of the excited states of europium, terbium and ytterbium complexes by proximate energy-matched OH, NH and CH oscillators: An improved luminescence method for establishing solution hydration states. J. Chem. Soc. Perkin Trans. 2 1999, 493–503. [Google Scholar] [CrossRef]
- Yasuchika Hasegawa, H.; Kimura, Y.; Murakoshi, K.; Wada, Y.; Kim, J.-H.; Nakashima, N.; Yamanaka, T.; Yanagida, S. Enhanced emission of deuterated tris(hexafluoroacetylacetonato)neodymium(III) complex in solution by suppression of radiationless transition via vibrational excitation. J. Phys. Chem. 1996, 100, 10201–10205. [Google Scholar] [CrossRef]
- Wahsner, J.; Seitz, M. Perdeuterated 2,2′-Bipyridine-6,6′-dicarboxylate: An extremely efficient sensitizer for thulium luminescence in solution. Inorg. Chem. 2013, 52, 13301–13303. [Google Scholar] [CrossRef] [PubMed]
- Bischof, C.; Wahsner, J.; Scholten, J.; Troslen, S.; Seitz, M. Quantification of C–H quenching in near-IR luminescent ytterbium and neodymium cryptates. J. Am. Chem. Soc. 2010, 132, 14334–14335. [Google Scholar] [CrossRef] [PubMed]
- Doffeck, C.; Seitz, M. The radiative lifetime in near-IR-luminescent ytterbium cryptates: The key to extremely high quantum yields. Angew. Chem. Int. Ed. 2015, 54, 9719–9721. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.-Q.; Peng, Y.; Li, Z.; Wang, C.-C.; Zheng, Y.-X.; Motevalli, M.; Wyatt, P.B.; Gillin, W.P.; Hernańdez, I. Effect of fluorination on the radiative properties of Er3+ organic complexes: An opto-structural correlation study. J. Phys. Chem. C 2013, 117, 23970–23975. [Google Scholar] [CrossRef]
- Quochi, F.; Orrú, R.; Cordella, F.; Mura, A.; Bongiovanni, G.; Artizzu, F.; Deplano, P.; Mercuri, M.L.; Pilia, L.; Serpe, A. Near infrared light emission quenching in organolanthanide complexes. J. Appl. Phys. 2006, 99, 053520. [Google Scholar] [CrossRef]
- Congiu, M.; Alamiry, M.; Moudam, O.; Ciorba, S.; Richardson, P.R.; Maron, L.; Jones, A.C.; Bryce, S.; Richards, B.S.; Robertson, N. Preparation and photophysical studies of [Ln(hfac)3DPEPO], Ln = Eu, Tb, Yb, Nd, Gd; of total photoluminescence quantum yields. Dalton Trans. 2013, 42, 13537–13545. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Hu, J.; Wang, J.; Liu, X.; Zhen, Z. Novel perfluorodiphenylphosphinic acid lanthanide (Er or Er–Yb) complex with high NIR photoluminescence quantum yield. Photochem. Photobiol. Sci. 2008, 7, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.H.C.; Motevalli, M.; Abrahams, I.; Wyatt, P.B.; Gillin, W.P. IR luminescence of Er, Nd and Yb β-diketonates. J. Phys. Chem. B 2006, 110, 24476–24479. [Google Scholar] [CrossRef] [PubMed]
- Mancino, G.; Ferguson, A.J.; Beeby, A.; Long, N.J.; Jones, T.S. Dramatic increases in the lifetime of the Er3+ ion in a molecular complex using a perfluorinated imidodiphosphinate sensitizing ligand. J. Am. Chem. Soc. 2005, 127, 524–525. [Google Scholar] [CrossRef] [PubMed]
- Glover, P.B.; Bassett, A.P.; Nockemann, P.; Benson, M.; Kariuki, M.; Van Deun, R.; Pikramenou, Z. Fully fluorinated imidodiphosphinate shells for visible- and NIR-emitting lanthanides: Hitherto unexpected effects of sensitizer fluorination on lanthanide emission properties. Chem. Eur. J. 2007, 13, 6308–6320. [Google Scholar] [CrossRef] [PubMed]
- Beverina, L.; Crippa, M.; Sassi, M.; Monguzzi, A.; Meinardi, F.; Tubino, R.; Pagani, G.A. Perfluorinated nitrosopyrazolone-based erbium chelates: A new efficient solution processable NIR emitter. Chem. Commun. 2009, 34, 5103–5105. [Google Scholar] [CrossRef] [PubMed]
- Mech, A.; Monguzzi, A.; Meinardi, F.; Mezyk, J.; Macchi, G.; Tubino, R. Sensitized NIR Erbium(III) emission in confined geometries: A new strategy for light emitters in telecom applications. J. Am. Chem. Soc. 2010, 132, 4574–4576. [Google Scholar] [CrossRef] [PubMed]
- Monguzzi, A.; Tubino, R.; Meinardi, F.; Biroli, A.O.; Maddalena Pizzotti, M.; Demartin, F.; Quochi, F.; Cordella, F.; Loi, M.A. Novel Er3+ perfluorinated complexes for broadband sensitized near infrared emission. Chem. Mater. 2009, 21, 128–135. [Google Scholar] [CrossRef]
- Natrajan, L.S.; Khoabane, N.M.; Dadds, B.L.; Muryn, C.A.; Pritchard, R.G.; Heath, S.L.; Alan, M.; Kenwright, A.M.; Ilya Kuprov, I.; Faulkner, S. Probing the structure, conformation, and stereochemical Exchange in a family of lanthanide complexes derived from tetrapyridyl-appended cyclen. Inorg. Chem. 2010, 49, 7700–7710. [Google Scholar] [CrossRef] [PubMed]
- Petrov, V.A.; Marshall, W.J.; Grushin, V.V. The first perfluoroacetylacetonate metal complexes: As unexpectedly robust as tricky to make. Chem. Commun. 2002, 520–521. [Google Scholar] [CrossRef]
- Chen, X.Y.; Jensen, M.P.; Liu, G.K. Analysis of energy level structure and excited-state dynamics in a Sm3+ complex with soft-donor ligands: Sm(Et2Dtc)3(bipy). J. Phys. Chem. B 2005, 109, 13991–13999. [Google Scholar] [CrossRef] [PubMed]
- Aebischer, A.; Gumy, F.; Bünzli, J.-C.G. Intrinsic quantum yields and radiative lifetimes of lanthanide tris(dipicolinates). Phys. Chem. Chem. Phys. 2009, 11, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Werts, M.H.V.; Jukes, R.T.F.; Verhoeven, J.W. The emission spectrum and the radiative lifetime of Eu3+ in luminescent lanthanide complexes. Phys. Chem. Chem. Phys. 2002, 4, 1542–1548. [Google Scholar] [CrossRef]
- He, H.; Sykes, A.G.; May, P.S.; He, G. Structure and photophysics of near-infrared emissive ytterbium(III) monoporphyrinate acetate complexes having neutral bidentate ligands. Dalton Trans. 2009, 36, 7454–7461. [Google Scholar] [CrossRef] [PubMed]
- Duhamel-Henry, N.; Adam, J.L.; Jacquier, B.; Linarès, C. Photoluminescence of new fluorophosphate glasses containing a high concentration of terbium (III) ions. Opt. Mater. 1996, 5, 197–207. [Google Scholar] [CrossRef]
- Natrajan, L.S.; Weinstein, J.A.; Wilson, C.; Arnold, P.L. Synthesis and luminescence studies of mono- and C3-symmetric, tris(ligand) complexes of Sm(III), Y(III) and Eu(III) with sulfur-bridged binaphtholate ligands. Dalton Trans. 2004, 3748–3755. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SADABS, Empirical Absorption Correction Program Based upon the Method of Blessing; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Blessing, R.H. An empirical correction for absorption anisotropy. Acta Crystallogr. 1995, 51, 33–38. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Beeby, A.; Faulkner, S. Luminescence from neodymium(III) in solution. Chem. Phys. Lett. 1997, 266, 116–122. [Google Scholar] [CrossRef]
- Beeby, A.; Faulkner, S.; Parker, D.; Williams, J.A.G. Sensitised luminescence from phenanthridine appended lanthanide complexes: Analysis of triplet mediated energy transfer processes in terbium, europium and neodymium complexes. J. Chem. Soc. Perkin Trans. 2 2001, 1268–1273. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).