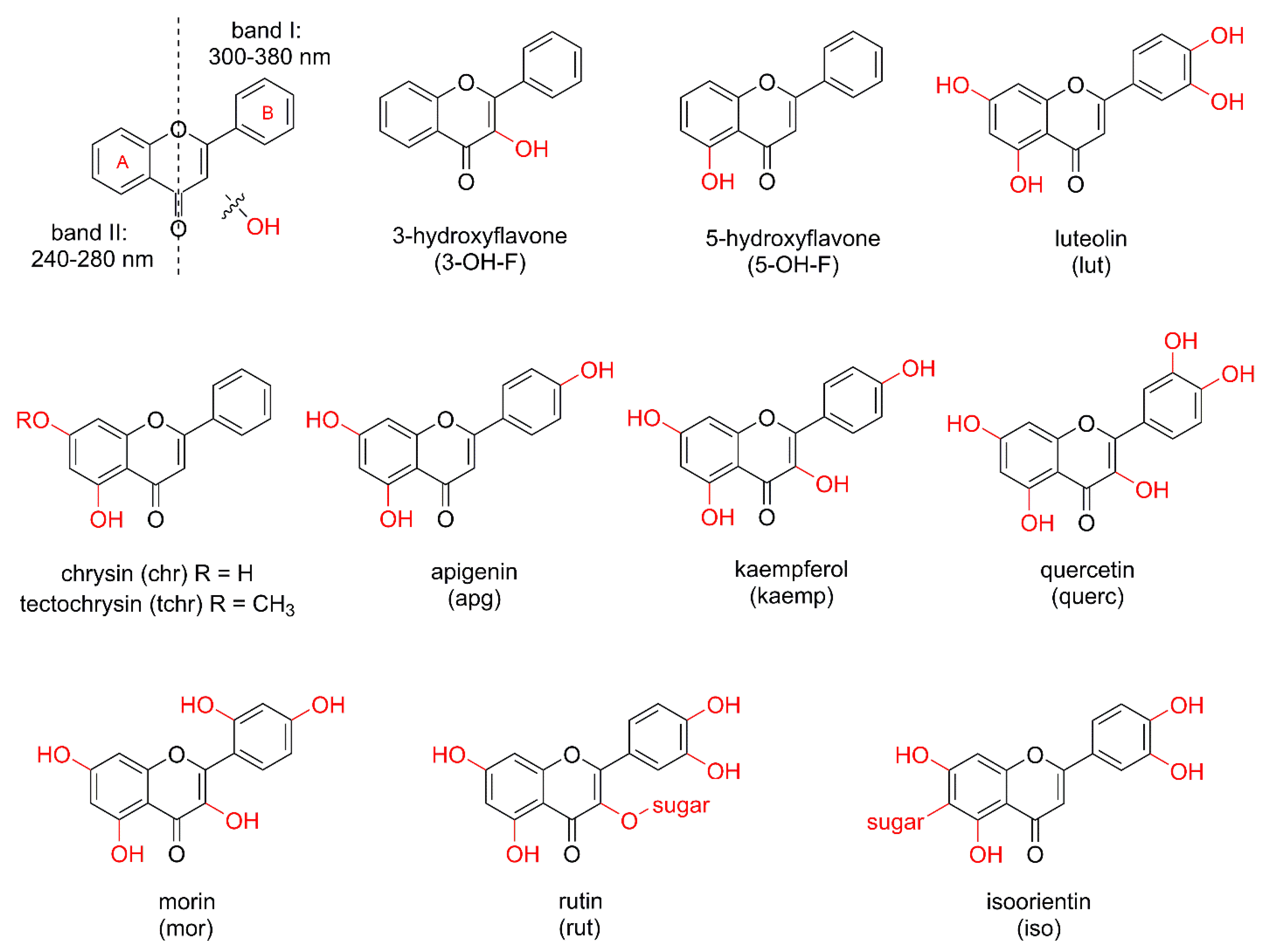
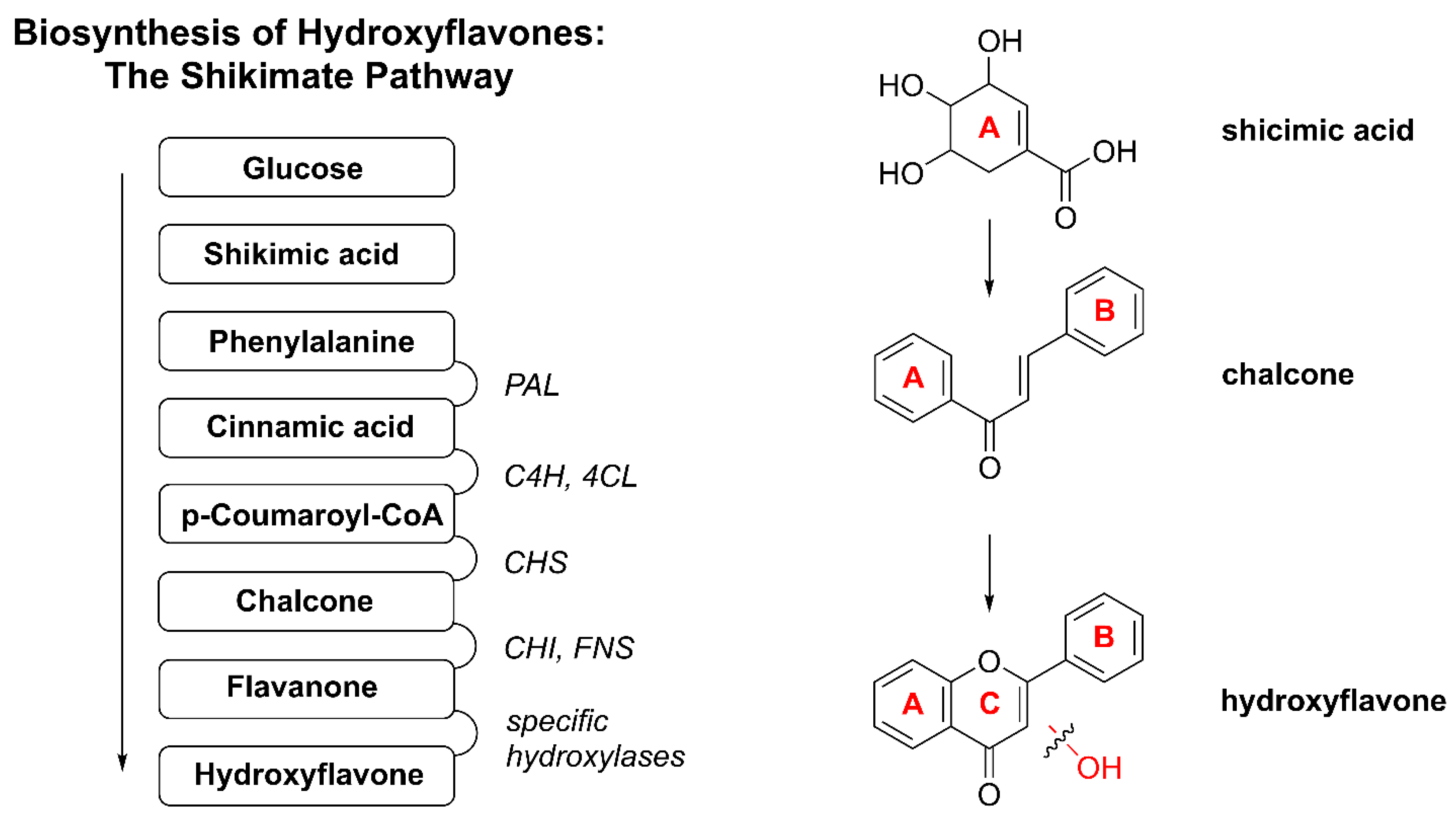
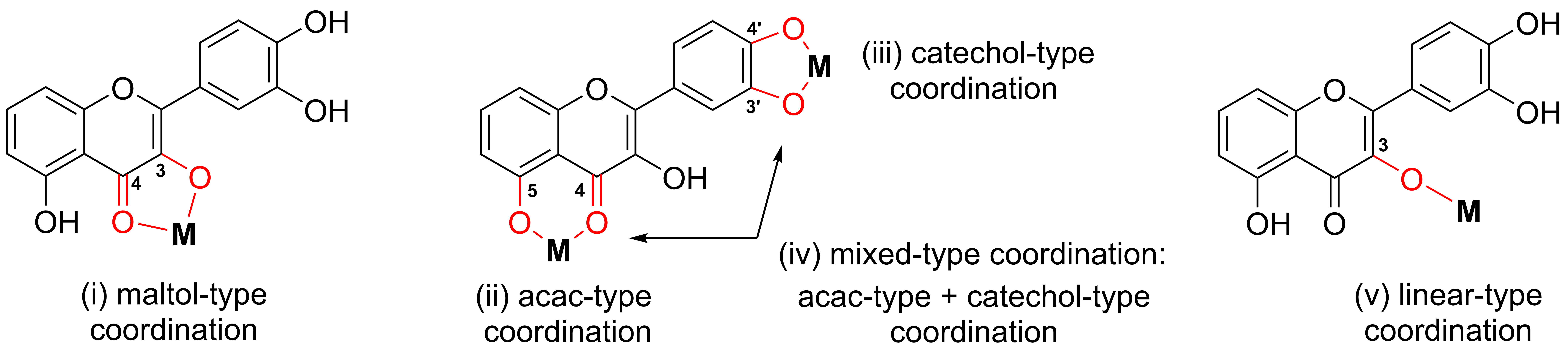
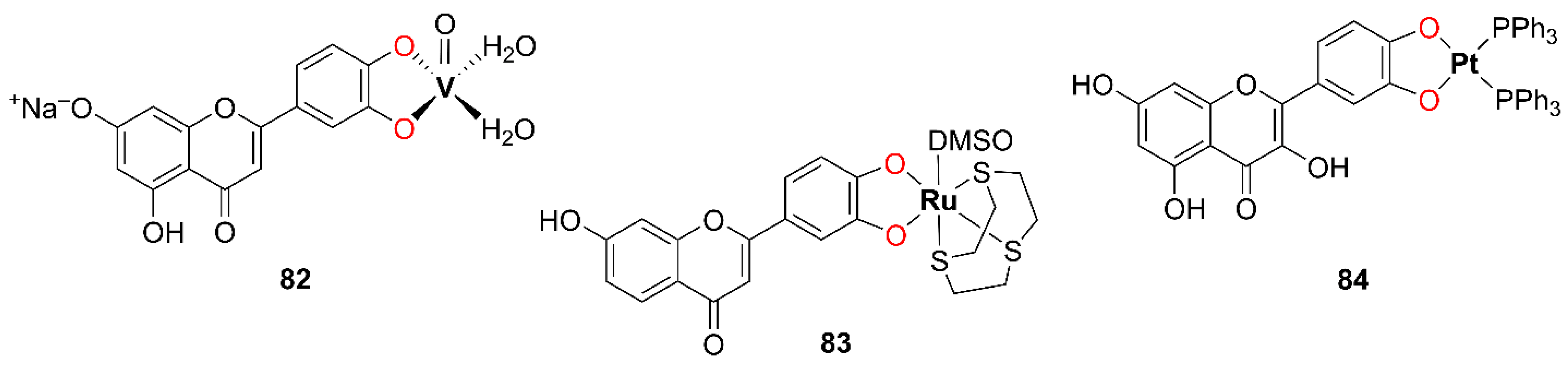
In metal complexes based on OH–F coordinated in a maltol-type manner, the ligand typically functions as a bidentate chelator, binding the metal center through two adjacent oxygen atoms located on the C-ring, namely the hydroxyl group at position C-3 and the carbonyl group at position C-4. This (O,O)-coordination mode results in the formation of a stable five-membered, envelope-like metallocycle, which critically influences the structural and electronic characteristics of the resulting complexes.
Coordination induces pronounced changes in the spectroscopic profiles of hydroxyflavones, which can be used to confirm a complex formation. In ultraviolet–visible (UV–Vis) spectra, band I, associated with a π → π* transition involving the B-ring, typically undergoes a bathochromic shift of 10–30 nm upon metal coordination. In infrared (IR) spectra, the C=O stretching vibration (originally observed at 1650–1665 cm−1 in the free ligand) shifts to lower wavenumbers (around 1610–1630 cm−1), while the broad O–H stretching band (3200–3500 cm−1) diminishes or disappears, indicating deprotonation and coordination via the C-3 hydroxyl group. Additionally, new bands appear in the 400–600 cm−1 region, corresponding to metal–ligand vibrations. In 1H NMR spectra, the disappearance of the characteristic signal for the hydroxyl proton at ~10–12 ppm further supports coordination, while the 13C nuclear magnetic resonance (NMR) signal of the carbonyl carbon shifts downfield by a few ppm, consistent with metal binding.
2.1.1. (O,O)-Coordinated Complexes: Simple Metal–Hydroxyflavone Chelates
In 2016, Dell’Anna et al. synthesized three square-planar Pt(II) complexes bearing triphenylphosphine ligands and (O,O) bidentate donor ligands: 3-hydroxyflavone, quercetin, and ethyl gallate [
37]. The complex containing ethyl gallate is not included in the present study. The complex with quercetin, coordinated in a catechol-type manner, is described in the following section, while complex
1 with 3-hydroxyflavone, exhibiting maltol-type coordination, is discussed here and shown in
Figure 5. A concentrated methanolic solution of KOH was added dropwise to a dichloromethane solution containing equimolar amounts of
cis-[PtCl
2(PPh
3)
2] and 3-hydroxyflavone under continuous stirring at room temperature. The reaction mixture was stirred overnight at an ambient temperature. The formed precipitate of KCl was removed by filtration, and the resulting filtrate was concentrated. Ethanol was added to the concentrated solution, followed by the addition of three equivalents of
n-pentane, which induced the precipitation of an orange solid. The solid was isolated by filtration, washed with cold
n-pentane, and dried under a vacuum.
The structure of complex 1 was proposed based on data obtained from NMR and IR spectroscopy, high-resolution mass spectrometry (HRMS), and elemental analysis (EA). The coordination mode in the solution was elucidated by NMR spectroscopy, while in the solid state it was determined using single-crystal X-Ray diffraction (XRD) analysis. The HRMS data supported the proposed molecular composition and structural formulation of the complex.
The single crystal obtained by slow diffusion of n-pentane into the tetrahydrofuran (THF) reaction solution was not of sufficient quality to allow complete structural refinement, likely due to the presence of photodegradation products. Upon standing in air, the complex gradually underwent photodegradation, yielding ortho-benzoyl-salicylic acid after five days, and subsequently, benzoic and salicylic acids upon prolonged exposure.
The cytotoxicity of complex
1 was evaluated using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay after 72 h of incubation against two tumor cell lines, U87 (human glioblastoma astrocytoma cell line), and MCF-7 (human breast adenocarcinoma cell line), without including any non-cancerous cell line to allow for the assessment of selectivity (
Table 1). Cisplatin was not tested under the same experimental conditions; instead, the literature values were used for comparison. The cytotoxicity of free 3-OH–F and its platinum (II) complex
1 was found to be similar on the U87 cell line, indicating only a minor synergistic effect upon complexation. In contrast, on the MCF-7 cell line, the platinum complex exhibited approximately twice the cytotoxic activity of the free ligand. Overall, the cytotoxicity of the complex on both cell lines was significantly lower than that of cisplatin; however, due to the reliance on data from the literature for cisplatin, the validity of this comparison remains uncertain. Theoretical calculations support the observation that the platinum complex readily releases its coordinated bioactive ligand in the solution, implying that the biological activity of the complex may largely depend on the cytotoxicity of its leaving groups. Antimicrobial activity of the synthesized complex was not evaluated.
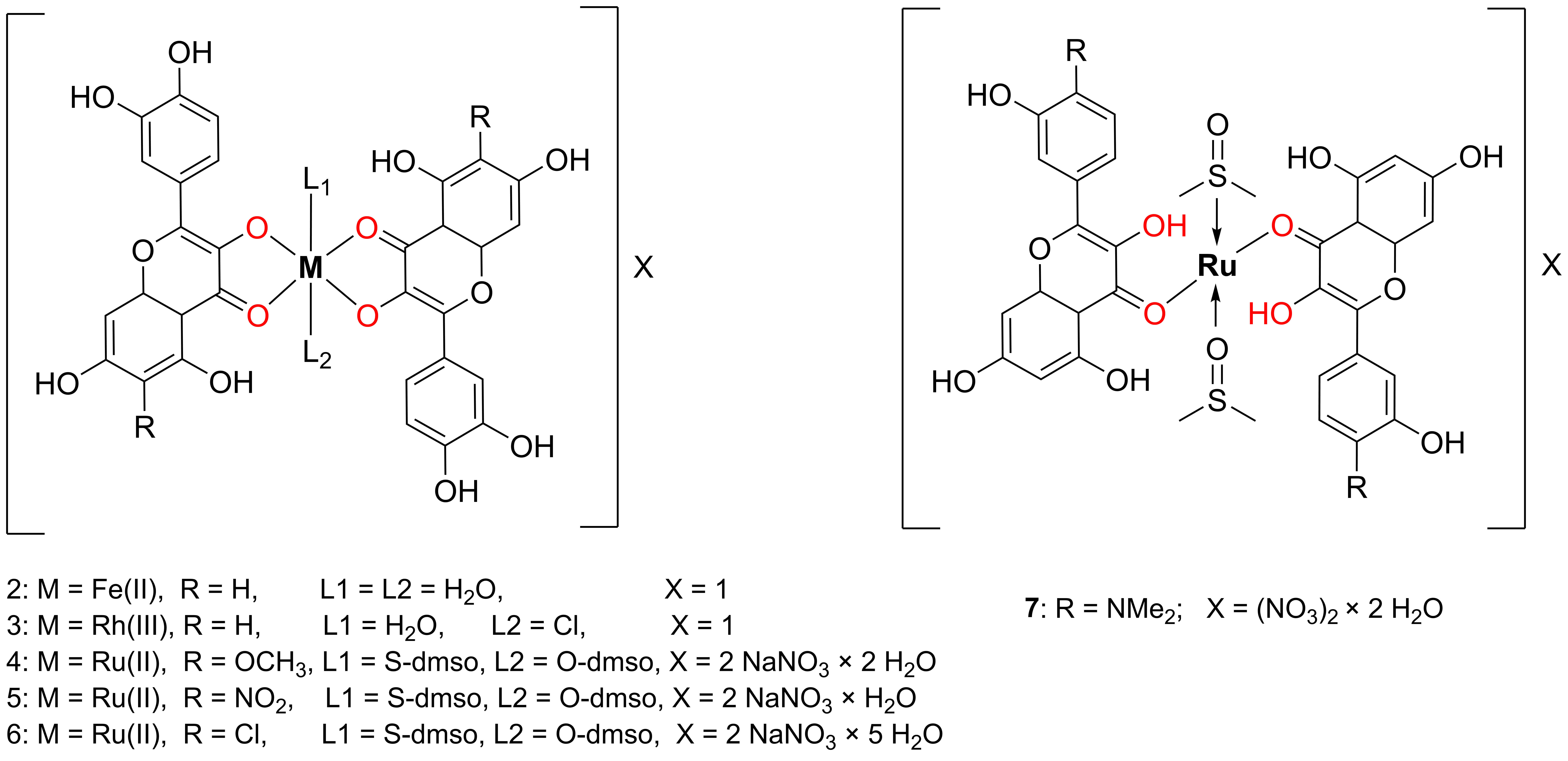
In 2016, Raza and coworkers synthesized an octahedral complex of Fe(II) with quercetin, compound
2, as presented in
Figure 6 [
38]. FeSO
4·7H
2O was refluxed and being stirred for 6 h with a double stoichiometric amount of quercetin in methanol. The reaction mixture was cooled, washed, and vacuum dried. The blackish-brown precipitate stable at room temperature was obtained.
The authors did not succeed in obtaining a crystal suitable for XRD analysis; therefore, the complex was characterized using other experimental techniques. Based on conductometric analysis (CA), complex 2 behaves as a nonelectrolyte, indicating its neutral nature. Two bidentately coordinated quercetin molecules and two water molecules are bound to a single Fe(II) ion. Upon addition of an Fe(II) salt to a methanolic solution of quercetin, a bathochromic shift in the UV–Vis absorption bands was observed, indicating a complex formation. The main changes in the IR spectrum upon complexation include a shift in the carbonyl stretching band from 1661 to 1646 cm−1, and the appearance of a new band at 630 cm−1, corresponding to Fe–O stretching. The presence of two water molecules in the complex was confirmed by both EA and thermogravimetric analyses (TGA). The absence of the NMR signal corresponding to the hydroxyl group at the C-3 position of quercetin indicates a maltol-type coordination mode of quercetin to the Fe(II) ion. Additionally, scanning electron microscopy (SEM) of complex 2 revealed a uniform matrix morphology with ice rock-like surface structures, and the average particle size was approximately 5 μm.
The interaction of the quercetin–iron (II) complex 2 with deoxyribonucleic acid (DNA) was investigated using UV–Vis spectroscopy, fluorescence spectroscopy (FS), and agarose gel electrophoresis. The results indicated that the complex moderately intercalates into DNA, effectively quenches the fluorescence of the classical intercalator ethidium bromide (EB), and competes for intercalative binding sites. In addition, the complex was shown to cleave DNA via an oxidative mechanism. These findings suggest that the complex is capable of directly interacting with genetic material, which may underlie its cytotoxic effects and support its potential as an anticancer agent.
The antibacterial activity of the Fe(II) complex
2 was evaluated using the well diffusion method after 24 h of incubation against two bacterial strains:
Staphylococcus aureus (Gram-positive, G+) and
Escherichia coli (Gram-negative, G−) (
Table 2). Penicillin sodium was used as the reference compound. The complex exhibited significantly stronger antibacterial activity compared to the free quercetin, as evidenced by larger inhibition zones, which may be explained by chelation theory [
39]. The antibacterial effect of the complex was shown to be concentration dependent. The inhibition zones produced by complex
2 were 4–8 times wider than those observed for free quercetin, although still 2–3 times narrower than those produced by the reference compound.
Figure 6.
Proposed structures of Fe(II), Rh(III), and Ru(II) complexes with quercetin and its derivatives, as reported by Raza et al., Sahyon et al., and Singh et al., respectively [
38,
40,
41].
Figure 6.
Proposed structures of Fe(II), Rh(III), and Ru(II) complexes with quercetin and its derivatives, as reported by Raza et al., Sahyon et al., and Singh et al., respectively [
38,
40,
41].
In 2022, Sahyon et al. synthesized the Rh(III) complex
3 presented in
Figure 3 by reacting quercetin with RhCl
3·3H
2O in an ethanolic NaOH solution under stirring at room temperature [
40]. After the solvent was evaporated, a dark red microcrystalline complex precipitated. The product was then filtered, washed with an ethanol/diethyl ether (1:1) mixture, and dried under anhydrous conditions.
CA revealed that the quercetin-based Rh(III) complex 3 behaves as a non-electrolyte in the solution. TGA confirmed the presence of one water molecule in the coordination sphere of the complex, in agreement with the results of EA. The coordination mode was elucidated by comparing the IR spectra of free quercetin and the synthesized Rh(III) complex 3. A shift in the carbonyl stretching frequency from 1664 cm−1 in free quercetin to 1620 cm−1 in complex 3 indicates the involvement of the carbonyl group in metal coordination. Among the hydroxyl groups, the C-3 hydroxyl was identified as the primary donor site due to its increased acidity attributed to its proximity to the carbonyl group and position within an oxygen-containing heterocyclic ring.
Rhodium(III) is a d6 transition metal with an 1A1g ground state. In the synthesized complex 3, the Rh(III) center adopts a hexa-coordinated, low-spin octahedral geometry, as evidenced by UV–Vis spectroscopy and magnetic susceptibility measurements. The diamagnetic character of the complex confirmed a t2g6 electron configuration, consistent with d2sp3 hybridization. Powder XRD (PXRD) analysis further supported this structure, showing similar bond lengths and bond angles close to 90º, indicating a quasi-ideal octahedral geometry. Good agreement was observed between the experimental PXRD data and theoretical calculations. The complex binds strongly to calf thymus DNA via a surface interaction with the phosphate backbone, inducing secondary structure changes (Kb = 2.76 × 106 M−1), which may underlie its antiproliferative effect.
The cytotoxic potential of complex
3 was assessed using the MTT assay against five human cancer cell lines: cervical adenocarcinoma cell line (HeLa), hepatocellular carcinoma cell line (HepG2), colorectal adenocarcinoma cell line (Caco-2), prostate adenocarcinoma cell line (PC-3), and MCF-7 (breast), as well as one normal human lung fibroblast cell line (WI-38). Cisplatin was used as the reference drug (
Table 3). In all tumor cell lines, complex
3 exhibited significantly higher cytotoxicity than free quercetin (4- to 15-fold); however, quercetin itself demonstrated markedly greater selectivity toward cancer cells. Compared to cisplatin, the complex was 2–4 times less cytotoxic but demonstrated an approximately 6-fold greater selectivity for cancer cells over normal cells. The highest selectivity indices (SI), calculated as the ratio of IC
50 values for normal to tumor cells, were observed for HepG-2 (liver) and HeLa (cervix) cell lines, with values of 4.8 and 4.3, respectively.
Mechanistic studies suggest that complex 3 inhibits HeLa cell proliferation by inducing apoptosis, evidenced by cell cycle arrest in the pre-G1 phase, most likely due to inhibited DNA replication. This arrest may occur through the activation of the p53 tumor suppressor gene, as indicated by decreased levels of the anti-apoptotic Bcl-2 protein. Additionally, a reduction in MMP-9 levels was observed, supporting the complex’s antiproliferative and anti-metastatic effects. Based on these findings, a mechanistic hypothesis can be proposed: the complex 3 binds to the DNA surface, preventing replication in cancer cells and promoting p53 expression. Increased p53 levels can lead to the inhibition of both Bcl-2 and MMP-9, followed by the activation of caspase-9. Caspase-9 then activates caspase-3, initiating apoptosis. This apoptotic process results in the pre-G1 cell cycle arrest and decreased proliferation of HeLa cells, as reflected in the reduced proportion of cells in the G2/M phase.
To the best of our knowledge the antimicrobial activity of complex 3 was not investigated.
In 2017, Singh et al. synthesized a series of quercetin derivatives bearing para-substituted B-rings and subsequently prepared their [Ru
II(DMSO)
2(querc)
2] complexes (
4–
7,
Figure 6) [
41]. The distorted octahedral geometry structure of the complexes was proposed based on spectroscopic data (IR, NMR, and HRMS) and supported by density functional theory (DFT) calculations. In the resulting complexes
4–
7, two dimethyl sulfoxide (DMSO) ligands were coordinated monodentately, one via the sulfur atom and the other via the oxygen atom, reflecting the ambidentate nature of DMSO.
The cytotoxicity of the synthesized complexes
4–
7 was evaluated using the MTT assay after 24 h of exposure against MCF-7 (breast) cell line (
Table 4). Among the tested compounds, complex
4, featuring a methoxy substituent on the B-ring, exhibited the highest cytotoxic activity against the MCF-7 cancer cell line (IC
50 = 16 μM). In ligand–complex pairs, the IC
50 values were consistently lower for the metal complexes
4–
7 compared to their corresponding quercetin-based ligands, suggesting a potential synergistic effect between the metal center and the coordinated ligand.
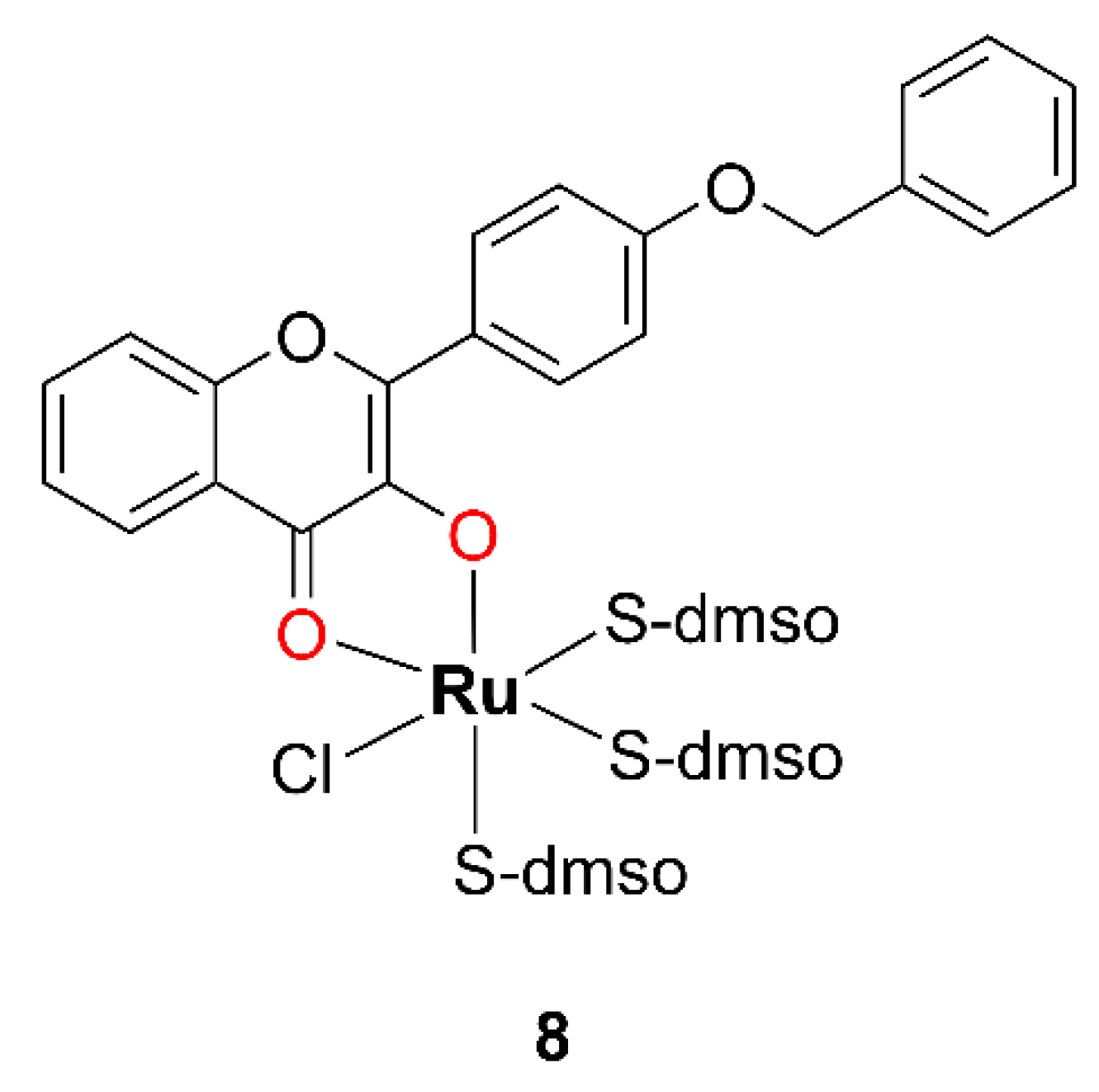
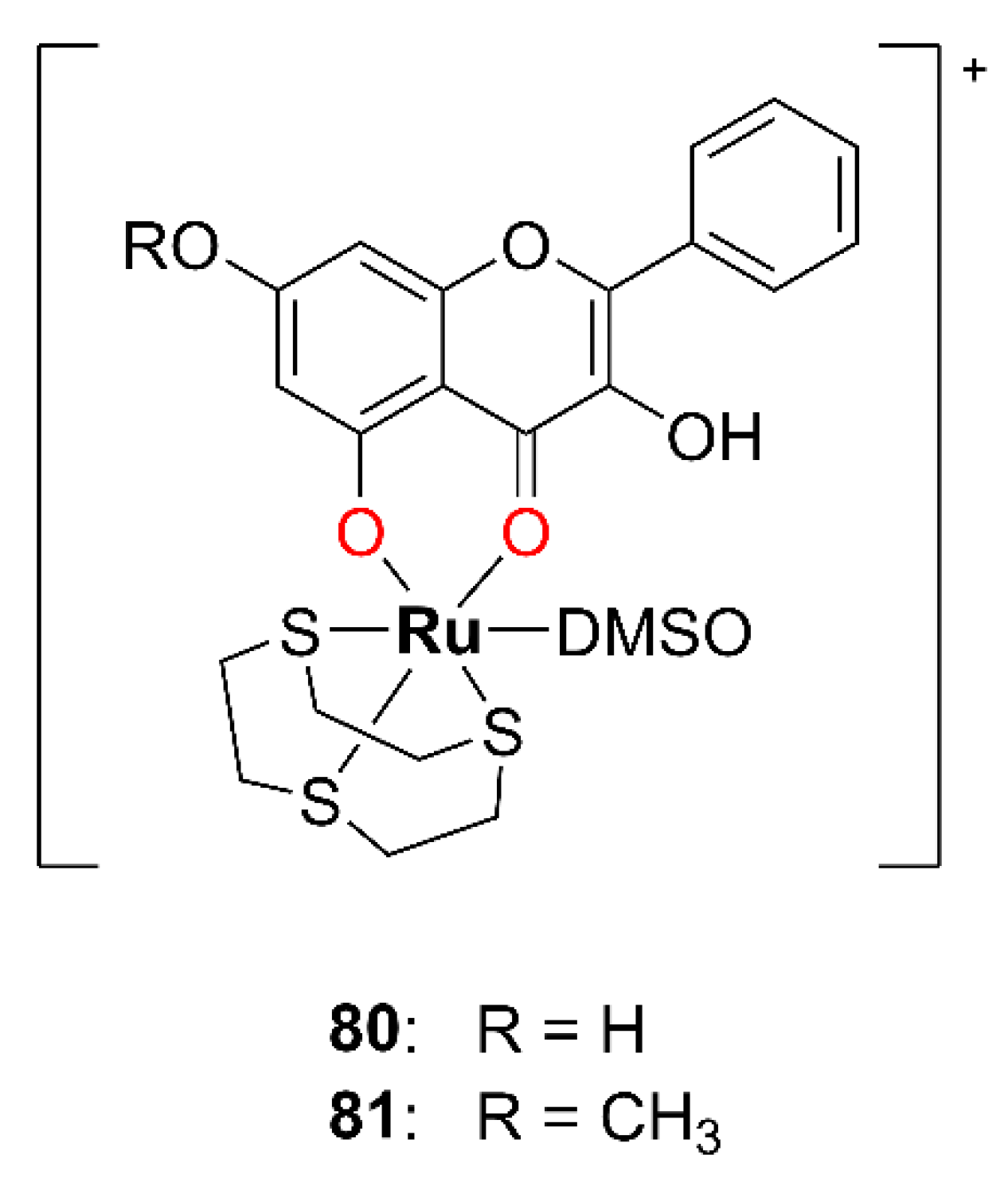
In 2010, Prajapati and coworkers synthesized two Ru(II) complexes with DMSO [
42]. One of these complexes featured a chalcone ligand coordinated in a bidentate (O,O) fashion and is therefore not considered in this study. The other complex, compound
8, which incorporates a 3-OH–F derivative, is relevant to the present discussion and is shown in
Figure 7.
The complex 8 was synthesized by the dropwise addition of a methanolic solution of cis-[RuIICl2(dmso)4] to a methanolic solution of the 3-OH–F derivative containing an equimolar amount of Et3N, adjusted to pH around 9.0. The resulting reaction mixture was stirred on a steam bath for 30 min. Subsequently, the solution was concentrated to one fourth of its original volume and left to stand at room temperature for 24 h. The resulting dark reddish-brown crystalline solid was collected by filtration, washed sequentially with methanol and diethyl ether, and finally dried under a vacuum. According to the authors, complex 8 is reported to be air-stable. CA indicates that it is a neutral species. The evidence for the complex 8 formation includes the appearance of a characteristic S=O stretching band around 1050 cm−1 and a Ru–S vibration band at approximately 430 cm−1. The proposed structure of complex 8 was further confirmed by XRD analysis, which revealed a monoclinic crystal system containing four discrete complex molecules arranged in a chain, with no solvent-accessible voids in the crystal packing.
Preliminary cytotoxicity screening of complex
8 was performed using the MTT assay after 48 and 72 h of incubation against Dalton’s lymphoma cell line, a murine transplantable T-cell lymphoma (DL) (
Table 5). The IC
50 value of complex
8 was found to be in the nanomolar range, indicating significantly higher cytotoxicity compared to the corresponding free OH–F ligand
L8. This enhanced activity of complex
8 may suggest a synergistic effect between the coordinated ligand
L8 and the Ru(II) metal center in mediating the cytotoxic response.
Ruthenium–DMSO complexes have attracted considerable interest due to their promising selectivity toward solid tumor metastases and relatively low toxicity to healthy tissues [
43], which has led some of these compounds to enter clinical trials [
44].
In 2020, Gantsho and coworkers synthesized 18 Re(I) carbonyl complexes incorporating tropolone, 3-OH–F, and three differently substituted quinolones as bidentate ligands [
45]. Since tropolone and quinolones are not members of the OH–F family, only five Re(I) tricarbonyl complexes
9–
13 containing 3-OH–F will be discussed here (
Figure 8). Starting from [NEt
4]
2[Re(CO)
3Br
3], the addition of AgNO
3 at pH 2.2 yielded the triaqua complex
fac-[Re(CO)
3(H
2O)
3]
+. Subsequent substitution of two labile aqua ligands with the bidentate 3-OH–F resulted in complex
9. In methanolic solution, one of the coordinated water molecules is replaced by methanol, resulting in the complex
10. Complexes
11–
13 were synthesized by introducing one equivalent of a monodentate phosphine ligand. All complexes
9–
13 were isolated as yellow precipitates or thin, needle-like crystals, which were unsuitable for XRD analysis. Structural characterization was performed using EA along with IR, UV–Vis, and NMR spectroscopy.
Complexes 9 and 10 exhibit a high tendency toward the substitution of the coordinated water or methanol ligand. In complexes 11–13, elevated temperatures and an excess of the phosphine ligand promote the efficient substitution of the axially positioned carbonyl ligand trans to the phosphine. The kinetics of the substitution of complex 10 with triphenylphosphine (PPh3), 1,3,5-triaza-7-phosphaadamantane (PTA), and tricyclohexylphosphine (PCy3) were studied, and all reactions proceeded via a single-step, first-order process, with rates directly dependent on the concentration of the entering phosphine ligand.
A cytotoxicity assessment was performed exclusively for complex
11 due to the limited solubility of the other complexes in this series. The evaluation employed a fluorometric cell viability assay based on resazurin after 48 h of incubation with HeLa (cervix) as the tumor cell line, and human retinal pigment epithelial cells immortalized with hTERT (RPE-1) as the normal cell line. Cisplatin served as a positive control (
Table 6). The cytotoxicity of complex
11 against HeLa cells was comparable to that of cisplatin. The SI of the complex, calculated as the ratio of IC
50 values for RPE-1 and HeLa cells, was 1.5, indicating a limited selectivity toward tumor cells under the tested conditions. In contrast, cisplatin exhibited an SI of approximately five. The authors suggest that the moderate cytotoxicity observed for complex
11 may be attributed to its high kinetic stability.
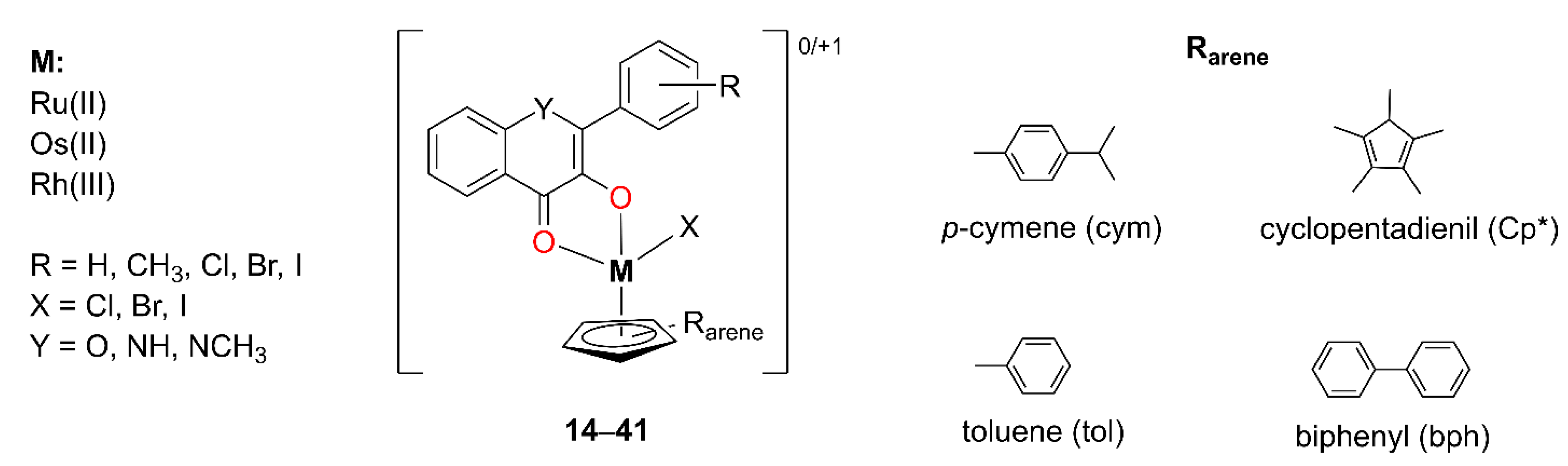
Kurzwernhart et al. synthesized 28 distinct metal complexes
14–
41 derived from 3-OH–F derivatives with Ru(II), Os(II), and Rh(III) as metals [
46,
47,
48,
49]. The complexes adopt a pseudo-octahedral “piano-stool” geometry, characterized by slight variations in M–O bond lengths and a moderately twisted phenyl substituent on the ligand. The general structure of the complexes is presented in
Figure 9, while the individual ligand structures and corresponding cytotoxicity data are summarized in
Table 7. The cytotoxicity of complexes
14–
41 was evaluated by an MTT assay after 96 h of exposure using three human cancer cell lines: the ovarian carcinoma cell line (CH1), the colorectal adenocarcinoma cell line (SW480), and the lung carcinoma epithelial cell line (A549). The positive control value for cisplatin was obtained from the literature.
Complexes 14–23, with the general formula [RuII(Cl)(cym)(3-OH–F)], differ in both the nature and position of substituents on the B-ring of the 3-OH–F scaffold. For several of these complexes (15, 17, 18, and 21), single-crystal XRD data were obtained. Complexes bearing para-substituted B-rings exhibited the highest cytotoxic activity. In contrast, the reduced activity observed for ortho-substituted derivatives may be attributed to steric effects, as the phenyl ring in these compounds adopts a significantly more twisted conformation. This structural distortion is likely to impair molecular recognition and diminish the binding affinity to biological targets. Notably, all complexes showed a stronger inhibition of topoisomerase IIα compared to the corresponding free OH–F derivatives, which was attributed to their multitargeted mechanism of action. Furthermore, the extent of topoisomerase IIα inhibition correlated well with the observed cytotoxicity.
In a subsequent set of compounds (
24–
33, [Ru
II(X)(R
arene)(3-OH–F)]), the aromatic ligand coordinated to the metal center (R
arene) was varied. The results showed that changes in lipophilicity were more strongly influenced by the 3-OH–F ligand than by the R
arene moiety [
40]. Finally, in complexes
34–
41([Os
II/Rh
III(Cl)(R
arene)(3-OH–F)]
0/+1), ruthenium (II) was replaced by osmium (II) or rhodium (III) as the central metal ion, but this did not significantly alter the observed cytotoxicity [
41].
The complexes 14–41 synthesized by Kurzwernhart et al. exhibited the highest cytotoxic activity against the CH1 (ovarian) cell line, while lower activity was observed against the SW480 (colon) and the lowest toward A549 (lung) cells, which are generally more resistant. Most IC50 values for the CH1 cell line were in the low micromolar range (≤7.9 μM), with compounds 17, 25, 29, 35, and 37 showing nanomolar potency. These five compounds share structural features including a chloride leaving group and a para-substituted B-ring of the OH–F moiety bearing either a chlorine or methyl substituent. These results suggest that both the nature and position of substituents on the B-ring, as well as the identity of the leaving group, play a critical role in enhancing cytotoxicity.
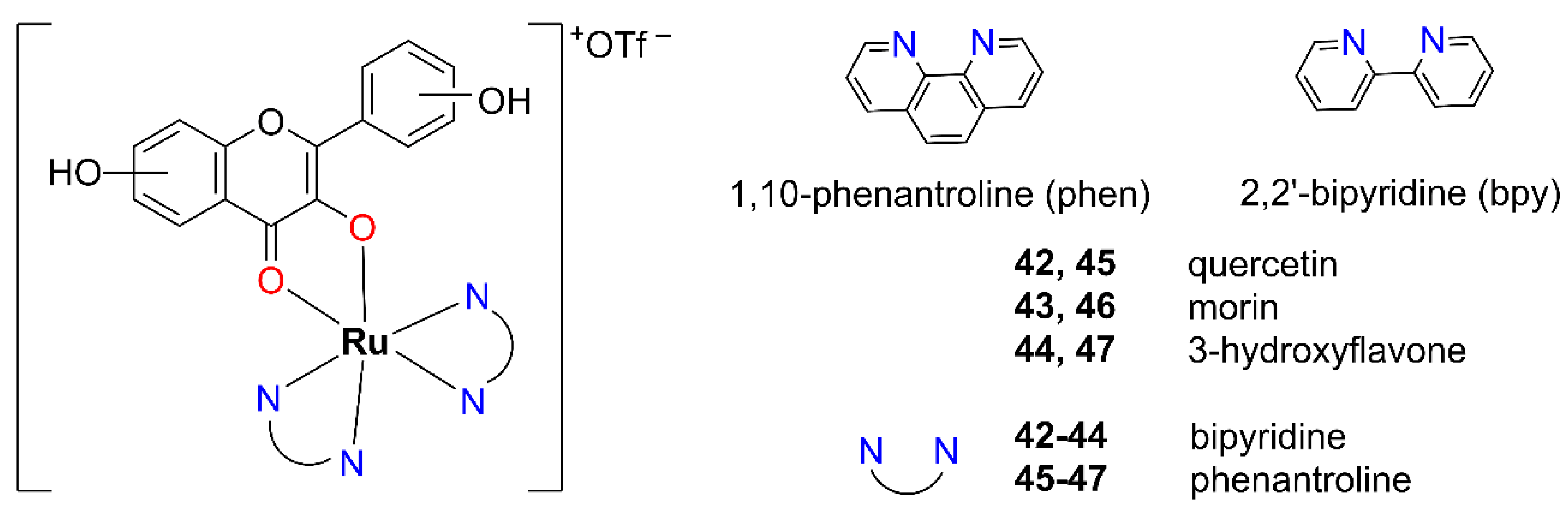
2.1.2. Mixed-Ligand Systems: Incorporating Both (O,O) and (N,N) Donor Atoms
Starting from
cis-[Ru
II(bpy)
2Cl
2] or [Ru
II(phen)
2(CO
3)], Zahirović et al. synthesized a series of octahedral cationic ruthenium (II) complexes
42–
47 with OH–F ligands [
50]. In these complexes, the diimine ligands, 2,2′-bipyridine (bpy) or 1,10-phenanthroline (phen), are coordinated in a bidentate manner through nitrogen atoms, while the OH–F ligand is coordinated via two oxygen atoms in a maltol-type binding mode (
Figure 10). The resulting complexes are low-spin, diamagnetic species consistent with a t
2g6 electronic configuration of the Ru(II) center.
Given that the synthesized complexes 42–47 lack good leaving groups, the most probable mode of DNA interaction is intercalation. This is supported by their cationic nature and the presence of planar π-extended aromatic systems. On the other hand, the efficiency of π–π stacking interactions with nucleobases tends to decrease with the increasing number of hydroxyl groups on the OH–F ligand.
The cytotoxicity of these types of Ru(II) complexes
42–
47 was evaluated using the MTT assay after 72 h of incubation on four human cancer cell lines: colorectal adenocarcinoma cell line (SW620), HepG2 (liver), MCF-7 (breast), and HeLa (cervix) (
Table 8). Unfortunately, the measurement errors were sufficiently high enough to raise concerns regarding the reliability and significance of the obtained results. However, two results stood out as significant: the bipyridine-based complex
44 exhibited nanomolar cytotoxicity against SW620 cells (IC
50 = 0.75 μM), while the phenanthroline-based complex
47 showed micromolar activity against the MCF-7 cell line (IC
50 = 8.32 μM). Both of these complexes are derivatives of 3-OH–F.
Antimicrobial activity was assessed by the disk diffusion method against four Gram-positive bacterial strains (
Staphylococcus aureus ATCC 25923,
Enterococcus faecalis ATCC 19433,
Streptococcus β-hemolytic group A, and methicillin-resistant
Staphylococcus aureus (MRSA)). Vancomycin was used as the reference drug for all Gram-positive strains, except for MRSA, where gentamicin served as the control. Four Gram-negative bacterial strains were also tested (
Klebsiella pneumoniae ATCC 1705,
Acinetobacter baumannii ATCC BAA-747,
Pseudomonas aeruginosa, and
Escherichia coli), with gentamicin as the reference drug. Additionally, the fungal strain
Candida albicans was included, with nystatin used as the control. The results are presented in
Table 9. Interestingly, complex
44 exhibited zones of inhibition against Gram-positive bacteria comparable in width to that of vancomycin. In the case of MRSA, the zone of inhibition was significantly wider than that of gentamicin. Against the tested fungal strain, the inhibition zone observed for
44 was identical to that measured for nystatin. Comparison of the inhibition zones of the synthesized metal complexes with those of the free hydroxyflavone ligands indicates a synergistic effect of coordination, enhancing antimicrobial activity against both Gram-positive bacteria and the fungal strain
Candida albicans. Based on the obtained results, a significant improvement in the antimicrobial efficacy of complex
44 was observed against the tested Gram-positive bacteria and
Candida albicans strains, relative to the uncoordinated 3-hydroxyflavone ligand.
Gençkal et al. synthesized heteroleptic complexes of Co(II), Ni(II), and Cu(II) with quercetin and diimine (phen or bpy) ligands (
48–
51,
Figure 11) [
51]. A methanolic solution of metal (II) chloride x-hydrate was added dropwise to a methanolic solution of quercetin and sodium hydroxide under constant stirring at room temperature. The reaction mixture was stirred for an additional hour, after which a methanolic solution of phen or bpy was added. The mixture was then refluxed for approximately 4 h. Upon completion, it was allowed to cool to room temperature, and the resulting solid was filtered, washed with methanol, and dried under ambient conditions.
According to experimental measurements, two distinct types of complexes were obtained: an octahedral [Co
II/Ni
II(queH-1)Cl(phen)(H
2O)]∙2H
2O (
48–
49), and a square-pyramidal geometry for [Cu
II(queH-1)Cl(phen/bpy)]∙xH
2O (
50–
51), as illustrated in
Figure 11.
EA confirms the proposed molecular formulas of the complexes 48–51. CA indicates that the complexes are neutral in nature and thus behave as non-electrolytes. Their stability was verified by repeating the CA after 24 and 48 h, with consistent results. The magnetic susceptibility (χ) values for the Ni(II) and Co(II) complexes are consistent with mononuclear octahedral coordination, while those for the Cu(II) complexes support the assignment of a mononuclear square-pyramidal geometry for complexes 50 and 51. In the UV–Vis spectrum, the characteristic absorption band of free quercetin at 371 nm, assigned to the B-ring, undergoes a bathochromic shift of approximately 50 nm upon complexation, supporting a maltol-type coordination mode of the quercetin ligand. The band maximum corresponding to the carbonyl stretching vibration shifted to lower wavenumbers in all metal complexes, indicating coordination through the adjacent hydroxyl group. Since the hydroxyl group at the C3 position is significantly more acidic than the one at C5, it is expected to preferentially participate in metal coordination. Furthermore, the IR spectra reveal new bands in the 433–417 cm−1 and 652–643 cm−1 regions, corresponding to M–N and M–O bond vibrations, respectively, confirming the formation of metal–ligand bonds through nitrogen and oxygen donor atoms.
The cytotoxic activity of the compounds 48–51 was initially evaluated using Sulforhodamine B (SRB) and ATP-based cell viability assays after 48 h of incubation against four different human cancer cell lines: A549 (lung), PC-3 (prostate), HeLa (cervix), and MCF-7 (breast). Improved results were obtained using the ATP assay, which is generally more sensitive than the SRB assay. Among them, the Cu(II) complexes 50 and 51 exhibited the most pronounced cytotoxicity toward the MCF-7 breast cancer cell line. Consequently, an additional breast cancer cell line, namely the human triple-negative breast cancer cell line (MDA-MB-231), was selected for further evaluation. At higher concentrations, some cytotoxic activity was also observed for the free ligands, quercetin, phen, and bpy, in breast cancer cell lines, although this activity was significantly lower compared to that of the Cu(II) complexes. Complex 50, containing phen, showed high cytotoxicity after 48 h of exposure in both MCF-7 and MDA-MB-231 cell lines, with IC50 values in the low micromolar range. One of the mechanisms underlying the cytotoxic effect of complex 50 was identified as apoptosis induction, as evidenced by an approximately 40% increase in apoptotic cells, mediated through the Caspase-3/7-dependent pathway. These findings identify complex 50 as a promising candidate for further development as an antitumor agent targeting breast cancer.
In 2014, Wang et al. synthesized four M(II) complexes
52–
55 (M = Cu(II) or Zn(II)) incorporating the OH–F kaempferol as an (O,O) donor ligand, along with bpy or phen as (N,N) donor ligands, as depicted in
Figure 11 [
52]. A methanolic solution of MCl
2 was added to a methanolic solution containing equimolar amounts of kaempferol and NaOH at an ambient temperature. Subsequently, bpy or phen, dissolved in methanol, was added dropwise. The reaction mixture was filtered, and the resulting yellow precipitate was washed with ethanol and dried under a vacuum. Since single crystals suitable for XRD analysis could not be obtained, the structures of the complexes were proposed based on EA, HRMS, UV–Vis, IR, and NMR spectroscopy. A fluorometric competitive experiment using EB as a probe revealed moderate intercalation by the complexes. The increased viscosity of CT-DNA in the presence of the metal complexes
52–
55 further supported these findings. Notably, the complexes
52–
55, especially complex
52, were able to induce the cleavage of plasmid DNA (pUC19) in a dose-dependent manner.
Kaempferol and its complexes (
52–
55) were tested for cytotoxic activity against the MDA-MB-231 (breast) cancer cell line using the MTT assay after 24 h of incubation (
Table 10). All complexes showed significantly improved cytotoxicity compared to the free ligand, indicating a potential synergistic interaction between kaempferol and the coordinated metal ions.
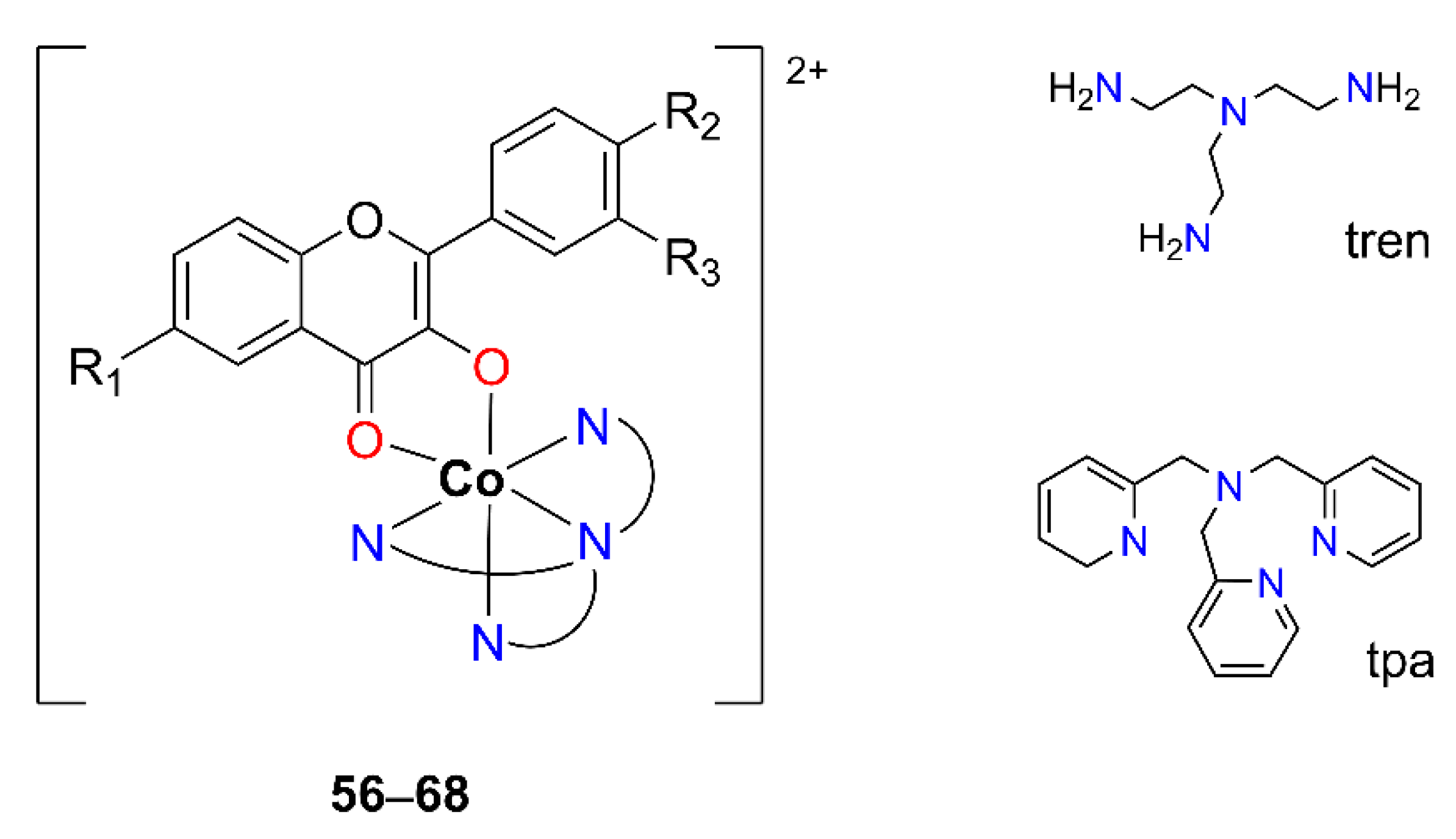
In 2021, Kozsup et al. reported the synthesis and characterization of a series of sixteen octahedral Co(III) complexes containing derivatized OH–F and tetradentate nitrogen ligands [
53], namely tris(2-aminoethyl)amine (tren) or tris(2-pyridylmethyl)amine (tpa). Thirteen compounds of these (
56–
68, [Co
III(4N)(OH–F)]
2+) were obtained by maltol-type coordination (
Figure 12,
Table 11), using variously substituted OH–F-ligands (3-hydroxyflavone (flavH), 6-F-3′-NO
2-3-hydroxyflavone (NO
2FflavH), 3′-NO
2-3-hydroxyflavone (NO
2flavH), 4′-Me-3-hydroxyflavone (MeflavH), 6-Cl-4′-OMe-3-hydroxyflavone (ClO-MeflavH), 6-Br-3-hydroxyflavone (BrflavH), and 4′-iPr-3-hydroxyflavone (iPrflavH). Two additional complexes (
69 and
70,
Figure 13) were obtained by the acac method and will be discussed in the following
Section 2.2. One complex, ([Co(tren)(nar)](Cl)(ClO
4)), which contains naringenin (nar) from the flavanone family, will not be a subject of this review.
The [Co(4N)(flav)]2+ type of compounds (56–70) was obtained in an overnight reaction by mixing the stoichiometric amount of the metal precursor ([Co(4N)Cl2]Cl (4N = tren or tpa)) and the ligand in MeOH at 60 °C with an equivalent of NaOH as a base. Most of the complexes were crystallized from water solutions after the addition of a counterion, ClO4−.
The full characterization of a series of compounds
56–
70 included NMR, IR, and HRMS measurements, as well as crystal structure and a cyclic voltammetric (CV) analysis. Characterization data confirmed acac coordination mode (O,O) of the OH–F ligand to the metal ion and octahedral geometry in all cases. Prior to biological study, the Co(III) species were tested for their stability in a D
2O:DMSO = 5:1 (
v/
v) mixture revealing no spectral changes in ambient conditions. Additionally, their electrochemical profile was determined using CV. The redox study indicated the influence of the substituents on the values of reduction potential as the presence of electron-donating substituents on the OH–F further shifted the cobalt complex’s reduction potential toward more negative values. The nature of the substituents demonstrated the greatest impact on the cytotoxic activity of the compounds (
Table 11).
The cytotoxicity of most complexes (
56–
65), as well as the free flavone ligands, was evaluated after 72 h of incubation against two human cancer cell lines, epithelial carcinoma cell line (A431) and A549 (lung), under both normoxic (21% O
2) and hypoxic (1% O
2) conditions and end-point SRB assay (
Table 12). The A549 cell line was generally more sensitive to the tested compounds than A431, with the exception of compound
60, for which the EC
50 values were lower in the A431 cells. Among the free ligands, MeflavH exhibited no cytotoxicity against either cell line, while flavH showed slight activity only against A549 under normoxic conditions. The ligands NO
2flavH, NO
2FflavH, and BrflavH were also exclusively active against A549, but their cytotoxic effects in this cell line were significant. Overall, the results indicate that the Co(III) species derived from the [Co(tpa)Cl
2]Cl precursor exhibit significantly stronger cytotoxic effects compared to those obtained from [Co(tren)Cl
2]Cl, highlighting the influence of substituent variation in OH–F-based ligands.







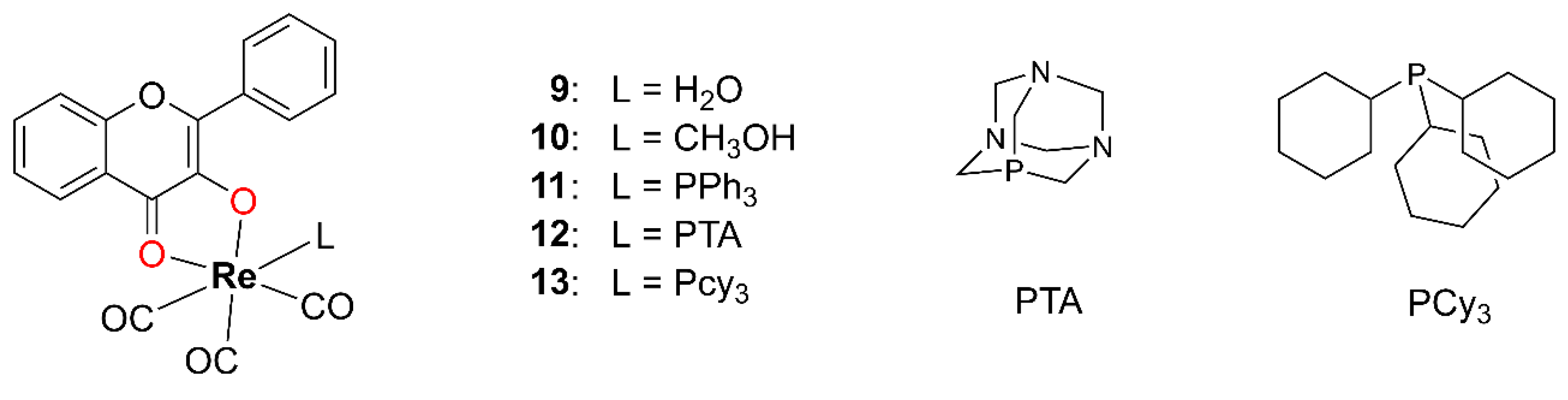



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}