

Phenyl–Pentafluorophenyl Interaction-Mediated Ir(C^N)2(N^N)-(Ni-Metallacycle) Dual Catalysis for Light-Driven C-S Cross-Coupling Synthesis

Abstract

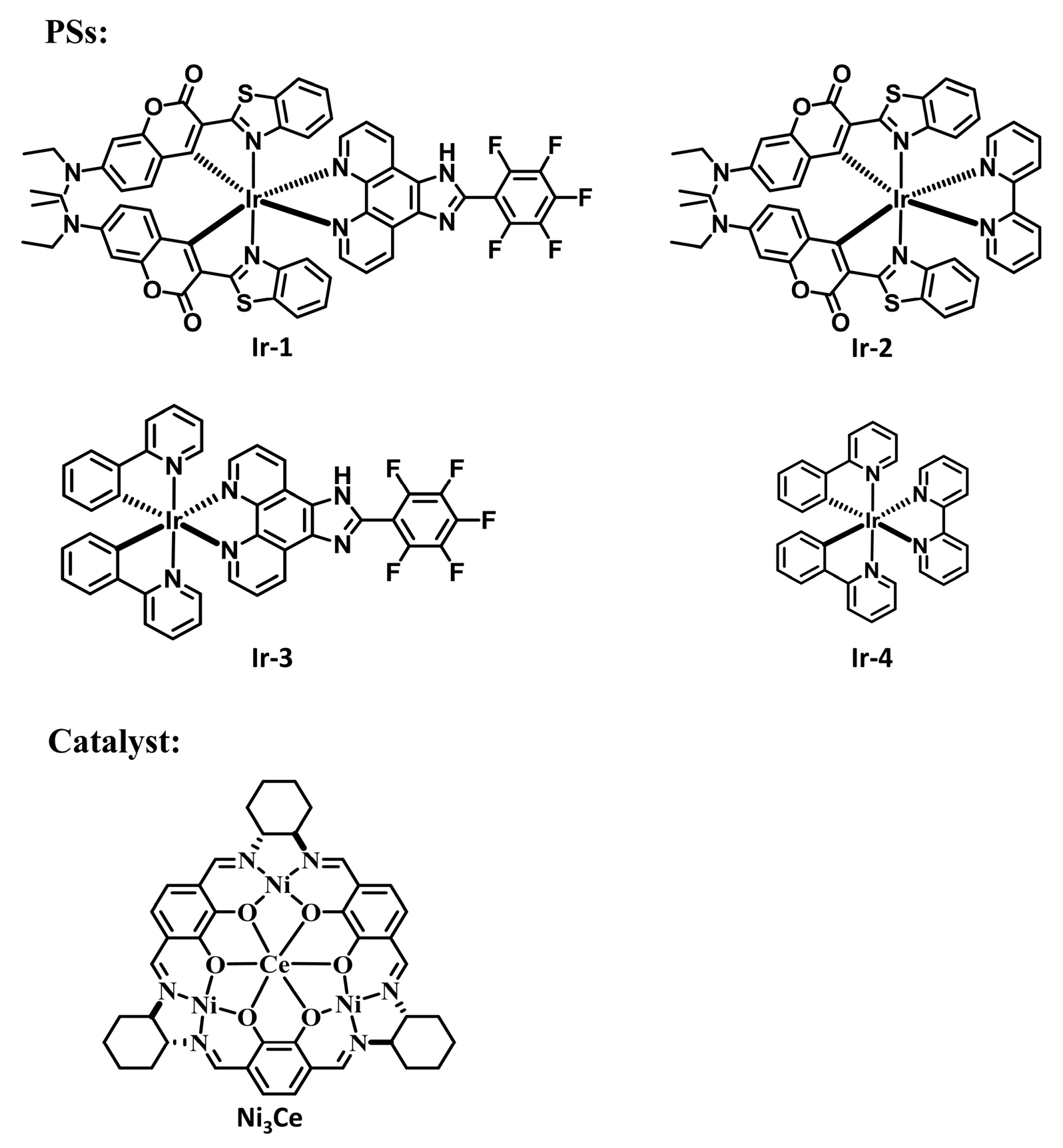
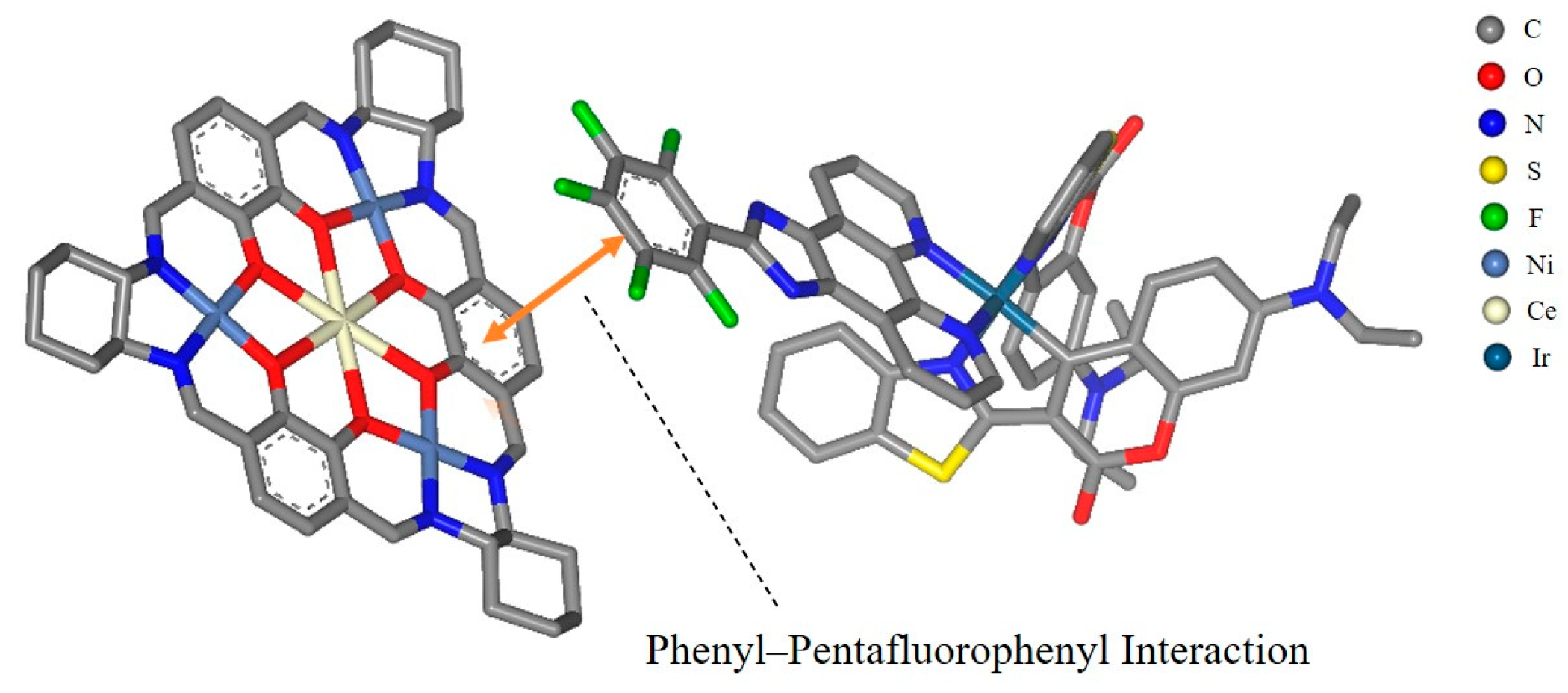
1. Introduction
2. Results
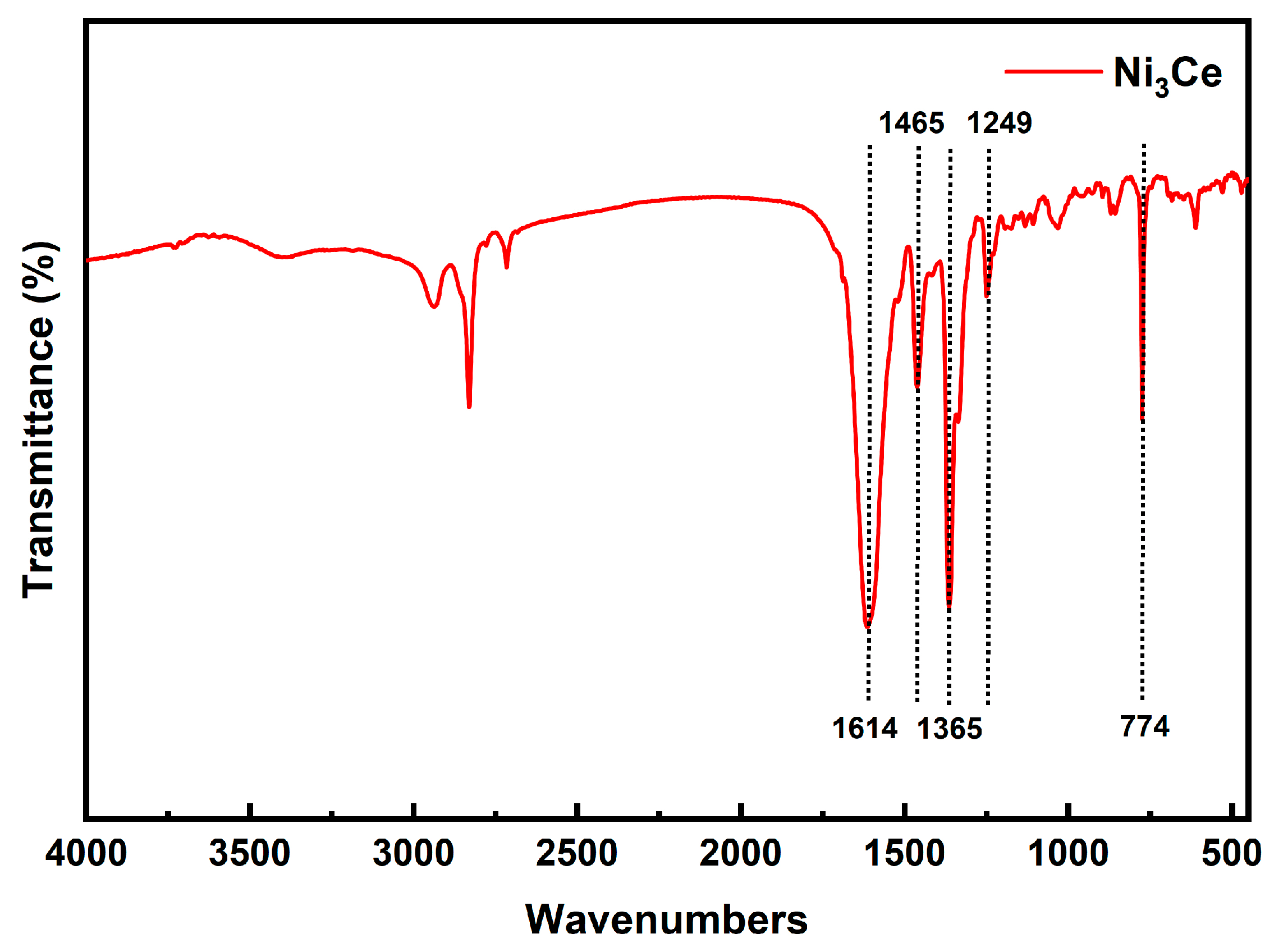
2.1. Fourier-Transform Infrared Spectroscopy of Ni3Ce
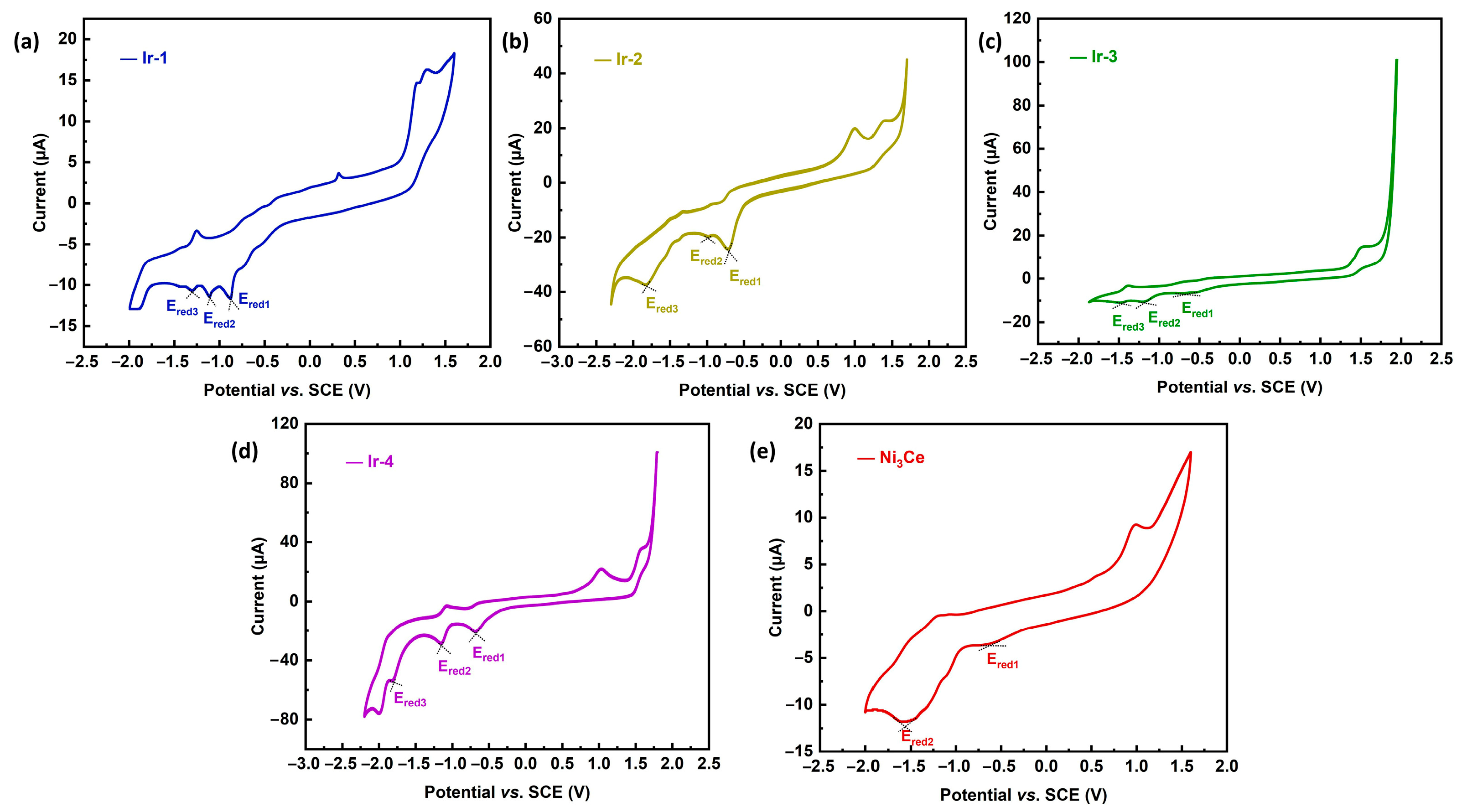
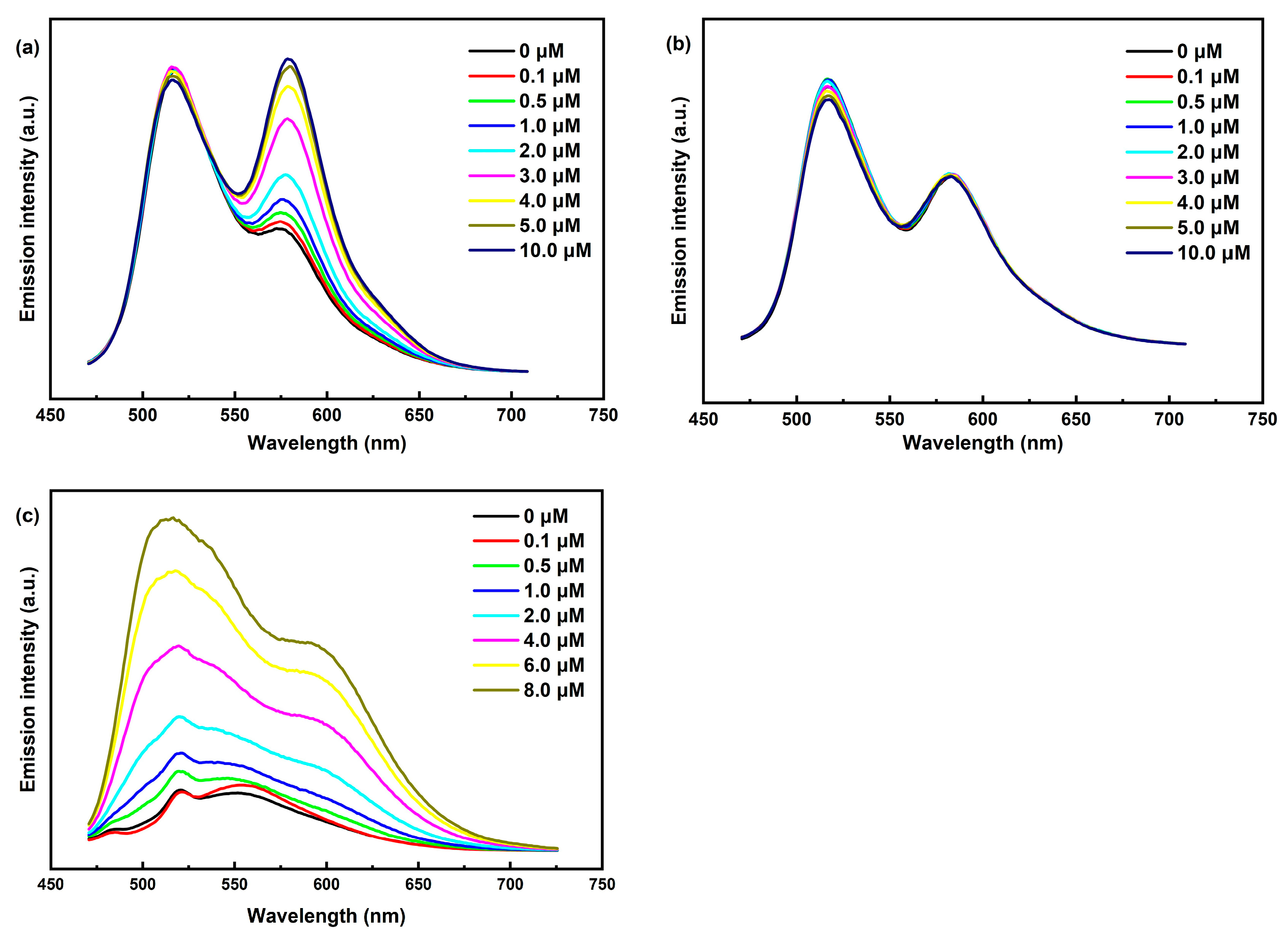
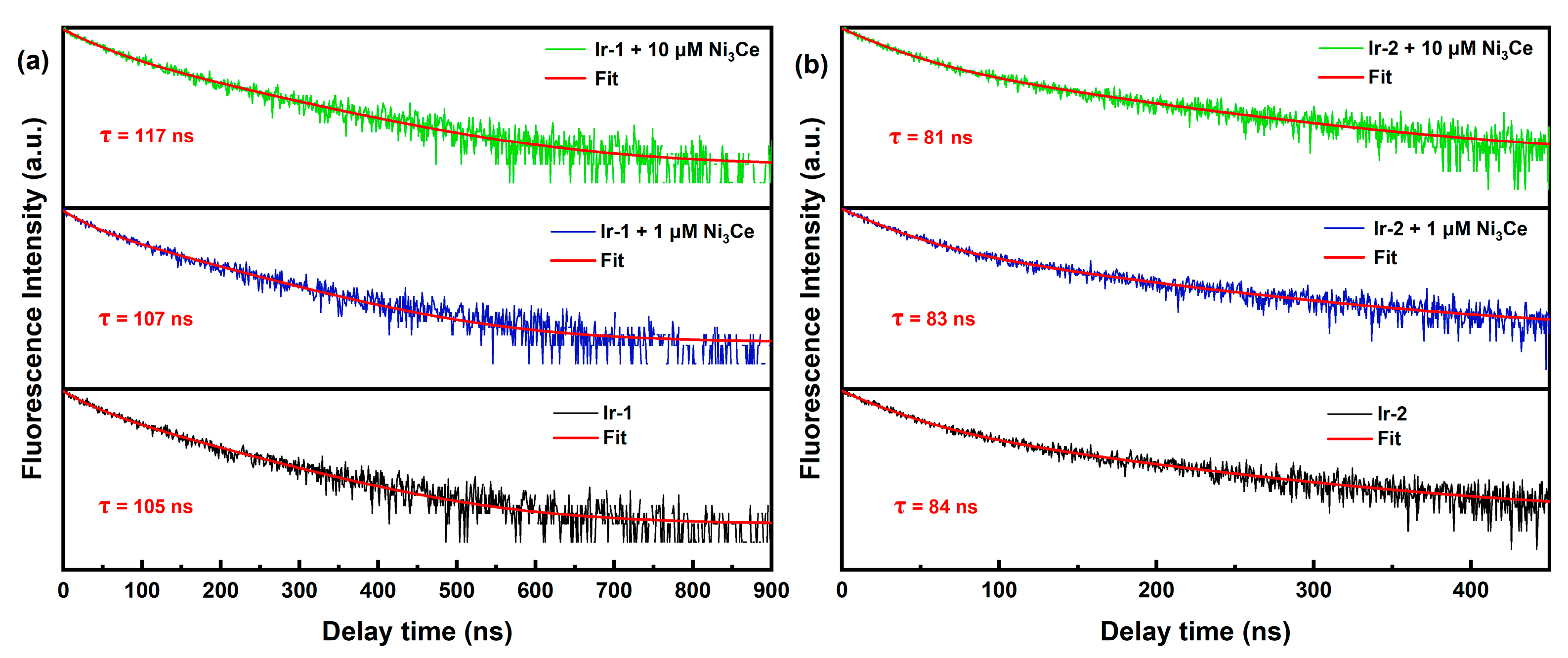
2.2. Electrochemical Characterization of Ni3Ce and Ir-Pss
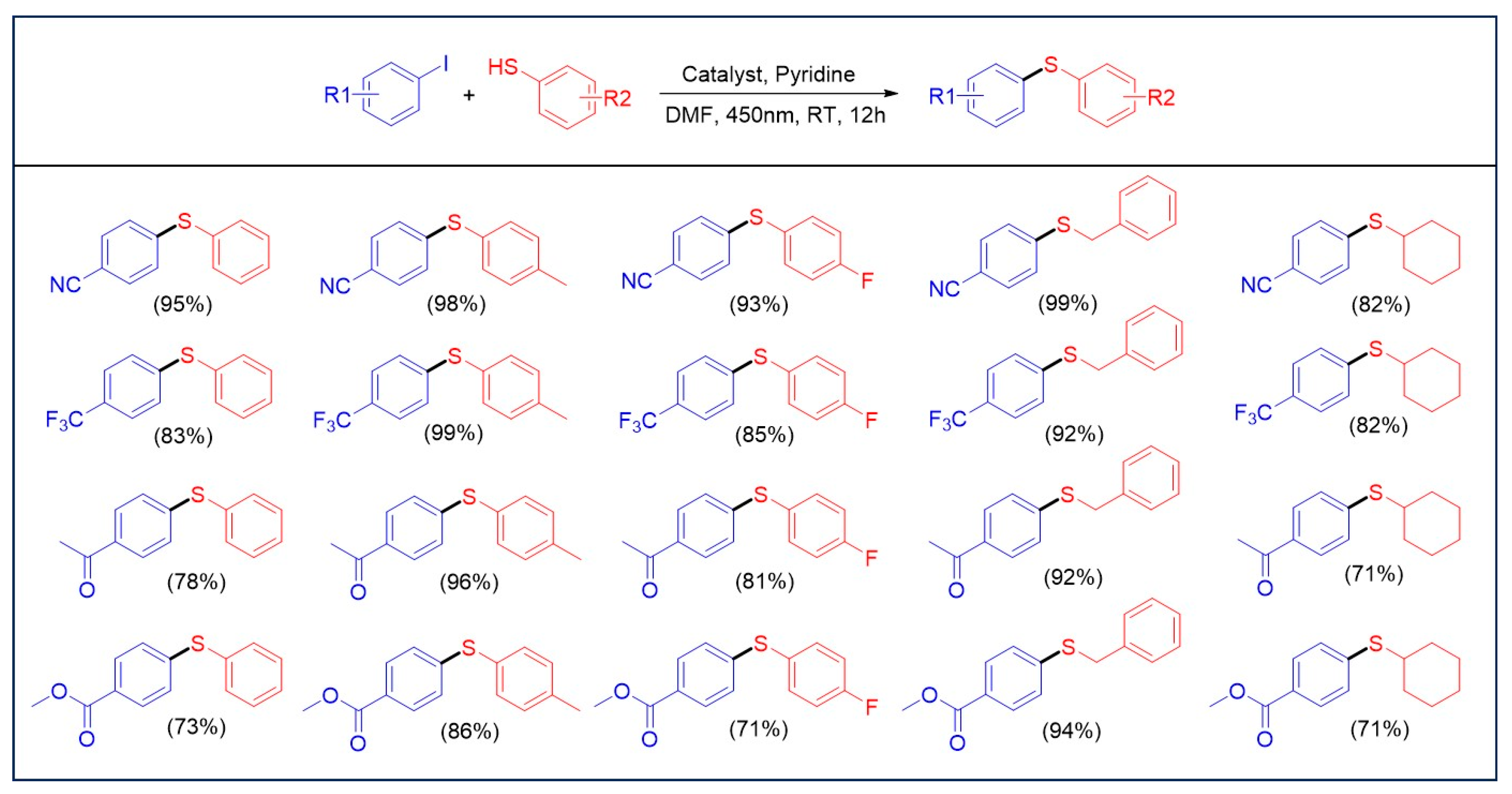
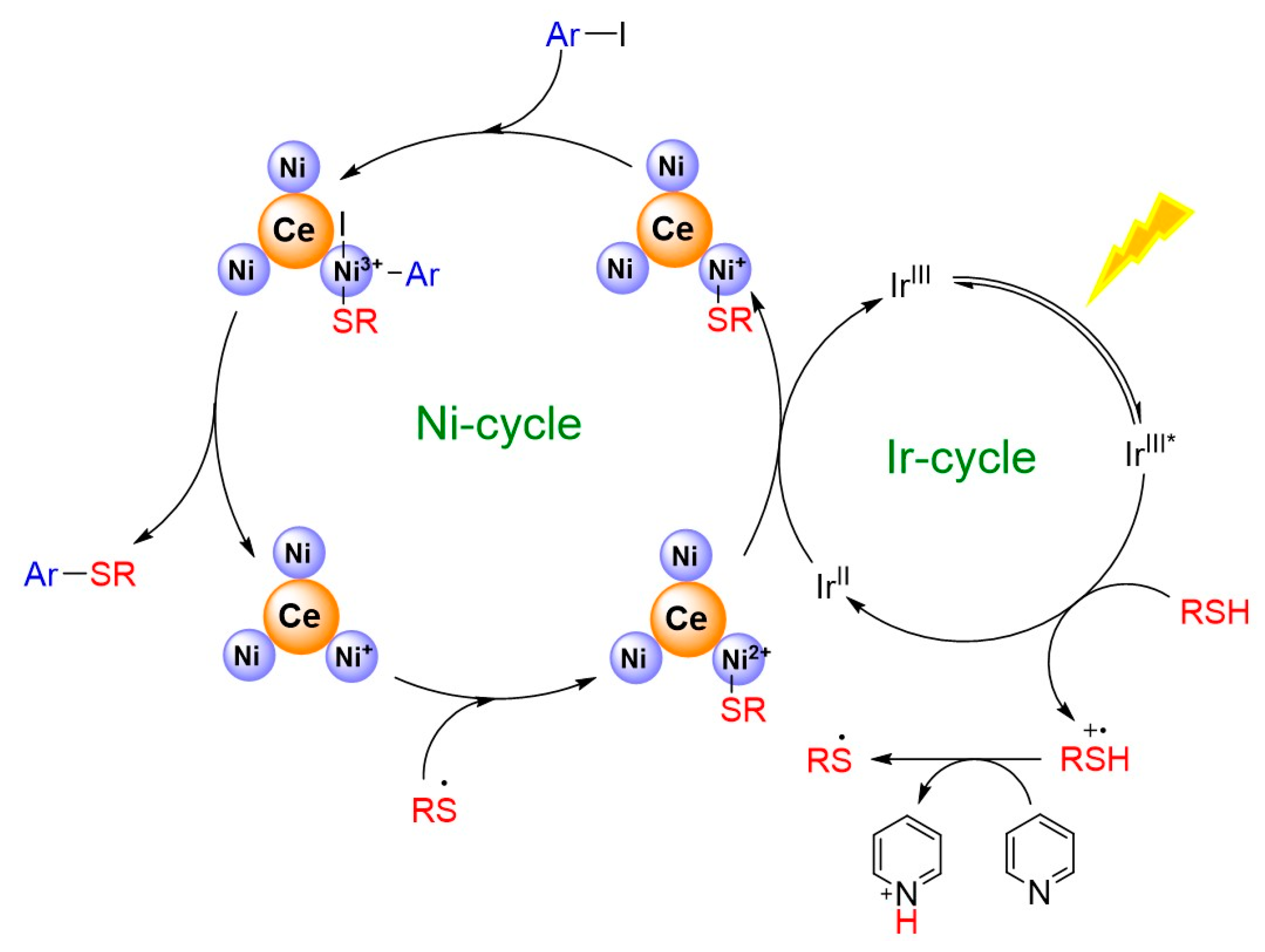
2.3. Photocatalytic C-S Cross-Coupling Reaction Studies
3. Materials and Methods
3.1. Reagents and Chemicals
3.2. Synthesis of Ni3Ce
3.3. Synthesis of 2-(2,3,4,5,6-Pentafluorophenyl)-1H-imidazo [4,5-F][1,10]phenanthroline (ppipH)
3.4. Synthesis of [Ir(coum)2(ppipH)](PF6) (Ir-1)
3.5. Synthesis of [Ir(coum)2(bpy)](PF6) (Ir-2)
3.6. Synthesis of [Ir(ppy)2(ppipH)](PF6) (Ir-3)
3.7. Material Characterization
3.8. General Procedure for the Photocatalytic Process
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gong, J.; Li, C.; Wasielewski, M.R. Advances in solar energy conversion. Chem. Soc. Rev. 2019, 48, 1862–1864. [Google Scholar] [CrossRef] [PubMed]
- Balzani, V.; Credi, A.; Venturi, M. Photochemical conversion of solar energy. Chemsuschem 2008, 1, 26–58. [Google Scholar] [CrossRef] [PubMed]
- Skubi, K.L.; Blum, T.R.; Yoon, T.P. Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev. 2016, 116, 10035–10074. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.-J.; Si, D.-H.; Ye, S.; Dong, Y.-L.; Cao, R.; Huang, Y.-B. Photocoupled Electroreduction of CO2 over Photosensitizer-Decorated Covalent Organic Frameworks. J. Am. Chem. Soc. 2023, 145, 19856–19865. [Google Scholar] [CrossRef]
- Gangjee, A.; Zeng, Y.; Talreja, T.; McGuire, J.J.; Kisliuk, R.L.; Queener, S.F. Design and Synthesis of Classical and Nonclassical 6-Arylthio-2,4-diamino-5-ethylpyrrolo[2,3-d]pyrimidines as Antifolates. J. Med. Chem. 2007, 50, 3046–3053. [Google Scholar] [CrossRef]
- Le Grand, B.; Pignier, C.; Létienne, R.; Cuisiat, F.; Rolland, F.; Mas, A.; Vacher, B. Sodium Late Current Blockers in Ischemia Reperfusion: Is the Bullet Magic? J. Med. Chem. 2008, 51, 3856–3866. [Google Scholar] [CrossRef]
- Sun, Z.-Y.; Botros, E.; Su, A.-D.; Kim, Y.; Wang, E.; Baturay, N.Z.; Kwon, C.-H. Sulfoxide-Containing Aromatic Nitrogen Mustards as Hypoxia-Directed Bioreductive Cytotoxins. J. Med. Chem. 2000, 43, 4160–4168. [Google Scholar] [CrossRef]
- Eichman, C.C.; Stambuli, J.P. Transition Metal Catalyzed Synthesis of Aryl Sulfides. Molecules 2011, 16, 590–608. [Google Scholar] [CrossRef]
- Sayah, M.; Organ, M.G. Potassium Isopropoxide: For Sulfination It is the Only Base You Need! Chem. Eur. J. 2013, 19, 16196–16199. [Google Scholar] [CrossRef]
- Fernández-Rodríguez, M.A.; Shen, Q.; Hartwig, J.F. A General and Long-Lived Catalyst for the Palladium-Catalyzed Coupling of Aryl Halides with Thiols. J. Am. Chem. Soc. 2006, 128, 2180–2181. [Google Scholar] [CrossRef]
- Hartwig, J.F. Evolution of a Fourth Generation Catalyst for the Amination and Thioetherification of Aryl Halides. Acc. Chem. Res. 2008, 41, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Tyson, E.L.; Ament, M.S.; Yoon, T.P. Transition Metal Photoredox Catalysis of Radical Thiol-Ene Reactions. J. Org. Chem. 2013, 78, 2046–2050. [Google Scholar] [CrossRef]
- Oderinde, M.S.; Frenette, M.; Robbins, D.W.; Aquila, B.; Johannes, J.W. Photoredox Mediated Nickel Catalyzed Cross-Coupling of Thiols With Aryl and Heteroaryl Iodides via Thiyl Radicals. J. Am. Chem. Soc. 2016, 138, 1760–1763. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, E.; Weike, C.; Mayerhofer, V.J.; Funes-Ardoiz, I.; Teskey, C.J. Dual Photoredox/Cobaloxime Catalysis for Cross-Dehydrogenative α-Heteroarylation of Amines. Org. Lett. 2021, 23, 5378–5382. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, C.; Yi, H.; Meng, Q.; Bian, C.; Chen, H.; Jian, J.-X.; Wu, L.-Z.; Lei, A. External Oxidant-Free Oxidative Cross-Coupling: A Photoredox Cobalt-Catalyzed Aromatic C–H Thiolation for Constructing C–S Bonds. J. Am. Chem. Soc. 2015, 137, 9273–9280. [Google Scholar] [CrossRef]
- Perepichka, I.; Kundu, S.; Hearne, Z.; Li, C.-J. Efficient merging of copper and photoredox catalysis for the asymmetric cross-dehydrogenative-coupling of alkynes and tetrahydroisoquinolines. Org. Biomol. Chem. 2015, 13, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Ling, B.; Yao, S.; Ouyang, S.; Bai, H.; Zhai, X.; Zhu, C.; Li, W.; Xie, J. Nickel-Catalyzed Highly Selective Radical C−C Coupling from Carboxylic Acids with Photoredox Catalysis. Angew. Chem. Int. Ed. 2024, 63, e202405866. [Google Scholar] [CrossRef] [PubMed]
- Xuan, J.; Zeng, T.-T.; Chen, J.-R.; Lu, L.-Q.; Xiao, W.-J. Room Temperature C—P Bond Formation Enabled by Merging Nickel Catalysis and Visible-Light-Induced Photoredox Catalysis. Chem. Eur. J. 2015, 21, 4962–4965. [Google Scholar] [CrossRef]
- Terrett, J.A.; Cuthbertson, J.D.; Shurtleff, V.W.; MacMillan, D.W.C. Switching on elusive organometallic mechanisms with photoredox catalysis. Nature 2015, 524, 330–334. [Google Scholar] [CrossRef]
- Oderinde, M.S.; Jones, N.H.; Juneau, A.; Frenette, M.; Aquila, B.; Tentarelli, S.; Robbins, D.W.; Johannes, J.W. Highly Chemoselective Iridium Photoredox and Nickel Catalysis for the Cross-Coupling of Primary Aryl Amines with Aryl Halides. Angew. Chem. Int. Ed. 2016, 55, 13219–13223. [Google Scholar] [CrossRef]
- Li, Y.-L.; Li, A.-J.; Huang, S.-L.; Vittal, J.J.; Yang, G.-Y. Polypyridyl Ru(ii) or cyclometalated Ir(iii) functionalized architectures for photocatalysis. Chem. Soc. Rev. 2023, 52, 4725–4754. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.L.; Kapper, S.C.; Zeng, T.; Thompson, M.E.; Kubiak, C.P. Improving Photocatalysis for the Reduction of CO2 through Non-covalent Supramolecular Assembly. J. Am. Chem. Soc. 2019, 141, 14961–14965. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, F.; Zhang, B.; Zhou, X.; Yu, F.; Sun, L. Visible Light-Driven Water Oxidation Promoted by Host–Guest Interaction between Photosensitizer and Catalyst with A High Quantum Efficiency. J. Am. Chem. Soc. 2015, 137, 4332–4335. [Google Scholar] [CrossRef]
- Wu, Q.-J.; Si, D.-H.; Wu, Q.; Dong, Y.-L.; Cao, R.; Huang, Y.-B. Boosting Electroreduction of CO2 over Cationic Covalent Organic Frameworks: Hydrogen Bonding Effects of Halogen Ions. Angew. Chem. Int. Ed. 2023, 62, e202215687. [Google Scholar] [CrossRef]
- Guo, H.; Si, D.-H.; Zhu, H.-J.; Chen, Z.-A.; Cao, R.; Huang, Y.-B. Boosting CO2 Electroreduction over a Covalent Organic Framework in the Presence of Oxygen. Angew. Chem. Int. Ed. 2024, 63, e202319472. [Google Scholar] [CrossRef] [PubMed]
- Reichenbächer, K.; Süss, H.I.; Hulliger, J. Fluorine in crystal engineering—“the little atom that could”. Chem. Soc. Rev. 2005, 34, 22–30. [Google Scholar] [CrossRef]
- Sun, Y.; Lei, Y.; Liao, L.; Hu, W. Competition between Arene–Perfluoroarene and Charge-Transfer Interactions in Organic Light-Harvesting Systems. Angew. Chem. Int. Ed. 2017, 56, 10352–10356. [Google Scholar] [CrossRef]
- Botta, C.; Cariati, E.; Cavallo, G.; Dichiarante, V.; Forni, A.; Metrangolo, P.; Pilati, T.; Resnati, G.; Righetto, S.; Terraneo, G.; et al. Fluorine-induced J-aggregation enhances emissive properties of a new NLO push-pull chromophore. J. Mater. Chem. C 2014, 2, 5275–5279. [Google Scholar] [CrossRef]
- Lin, S.-M.; Velayudham, M.; Tsai, C.-H.; Chang, C.-H.; Lee, C.-C.; Luo, T.-T.; Thanasekaran, P.; Lu, K.-L. A Molecular Triangle as a Precursor Toward the Assembly of a Jar-Shaped Metallasupramolecule. Organometallics 2014, 33, 40–44. [Google Scholar] [CrossRef]
- Han, Y.-F.; Li, H.; Jin, G.-X. Host-guest chemistry with bi- and tetra-nuclear macrocyclic metallasupramolecules. Chem. Commun. 2010, 46, 6879–6890. [Google Scholar] [CrossRef]
- Liu, J.-X.; Zhang, X.-B.; Li, Y.-L.; Huang, S.-L.; Yang, G.-Y. Polyoxometalate functionalized architectures. Coord. Chem. Rev. 2020, 414, 213260. [Google Scholar] [CrossRef]
- Tritton, D.N.; Bodedla, G.B.; Tang, G.; Zhao, J.; Kwan, C.-S.; Leung, K.C.-F.; Wong, W.-Y.; Zhu, X. Iridium motif linked porphyrins for efficient light-driven hydrogen evolution via triplet state stabilization of porphyrin. J. Mater. Chem. A 2020, 8, 3005–3010. [Google Scholar] [CrossRef]
- Yoshizawa, M.; Klosterman, J.K.; Fujita, M. Functional Molecular Flasks: New Properties and Reactions within Discrete, Self-Assembled Hosts. Angew. Chem. Int. Ed. 2009, 48, 3418–3438. [Google Scholar] [CrossRef]
- Syntrivanis, L.-D.; Tiefenbacher, K. Reactivity Inside Molecular Flasks: Acceleration Modes and Types of Selectivity Obtainable. Angew. Chem. Int. Ed. 2024, 63, e202412622. [Google Scholar] [CrossRef]
- Xue, R.; Liu, Y.-S.; Huang, S.-L.; Yang, G.-Y. Recent Progress of Covalent Organic Frameworks Applied in Electrochemical Sensors. ACS Sens. 2023, 8, 2124–2148. [Google Scholar] [CrossRef] [PubMed]
- Nagae, H.; Sakamoto, K.; Fujiwara, S.; Schindler, T.; Kon, Y.; Sato, K.; Okuda, J.; Mashima, K. Aerobic oxygenation of α-methylene ketones under visible-light catalysed by a CeNi3 complex with a macrocyclic tris(salen)-ligand. Chem. Commun. 2021, 57, 11169–11172. [Google Scholar] [CrossRef]
- Chen, W.; Chen, Y.; Li, J.; Zhang, S.; Zhang, D.; Li, D.; Wang, S.; Yu, F.; Chen, Y.; Zhang, J. Layer stacked polyimide with great built-in electronic field for fast lithium-ion storage based on strong p-p stacking effect. Energy Storage Mater. 2024, 68, 103349. [Google Scholar] [CrossRef]
- Takizawa, S.-y.; Pérez-Bolívar, C.; Anzenbacher, P., Jr.; Murata, S. Cationic Iridium Complexes Coordinated with Coumarin Dyes—Sensitizers for Visible-Light-Driven Hydrogen Generation. Eur. J. Inorg. Chem. 2012, 2012, 3975–3979. [Google Scholar] [CrossRef]
- Sebata, S.; Takizawa, S.-y.; Ikuta, N.; Murata, S. Photofunctions of iridium(iii) complexes in vesicles: Long-lived excited states and visible-light sensitization for hydrogen evolution in aqueous solution. Dalton Trans. 2019, 48, 14914–14925. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
|---|---|---|
| Entry | Variation from Standard Conditions a | Yield b (%) |
| 1 | Ir-1 | 99 |
| 2 | No Ir-1 | N.D. |
| 3 | No Ni3Ce | N.D. |
| 4 | No light | N.D. |
| 5 | No base | 80 |
| 6 | Air | 33 |
| 7 | Ir-2 | 75 |
| 8 | Ir-3 | 82 |
| 9 | Ir-4 | 52 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.-F.; Zhang, Z.-X.; Li, J.; Huang, S.-L. Phenyl–Pentafluorophenyl Interaction-Mediated Ir(C^N)2(N^N)-(Ni-Metallacycle) Dual Catalysis for Light-Driven C-S Cross-Coupling Synthesis. Inorganics 2025, 13, 229. https://doi.org/10.3390/inorganics13070229
Chen J-F, Zhang Z-X, Li J, Huang S-L. Phenyl–Pentafluorophenyl Interaction-Mediated Ir(C^N)2(N^N)-(Ni-Metallacycle) Dual Catalysis for Light-Driven C-S Cross-Coupling Synthesis. Inorganics. 2025; 13(7):229. https://doi.org/10.3390/inorganics13070229
Chicago/Turabian StyleChen, Jun-Feng, Zhan-Xin Zhang, Jing Li, and Sheng-Li Huang. 2025. "Phenyl–Pentafluorophenyl Interaction-Mediated Ir(C^N)2(N^N)-(Ni-Metallacycle) Dual Catalysis for Light-Driven C-S Cross-Coupling Synthesis" Inorganics 13, no. 7: 229. https://doi.org/10.3390/inorganics13070229
APA StyleChen, J.-F., Zhang, Z.-X., Li, J., & Huang, S.-L. (2025). Phenyl–Pentafluorophenyl Interaction-Mediated Ir(C^N)2(N^N)-(Ni-Metallacycle) Dual Catalysis for Light-Driven C-S Cross-Coupling Synthesis. Inorganics, 13(7), 229. https://doi.org/10.3390/inorganics13070229