
In Situ Synthesis of Hexadentate Cyclometalated Ir(III) Complexes as Photocatalysts for the Oxidation of Sulfides into Sulfoxides in Water

Abstract
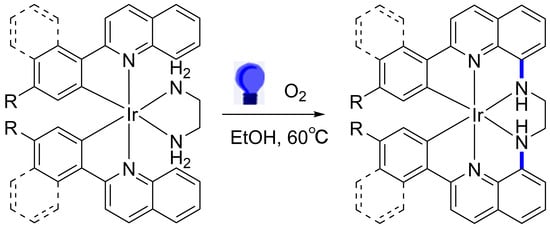
1. Introduction
2. Results and Discussion
2.1. Synthesis of Hexadentate Ir(III) Complexes
2.2. Photocatalysts for the Oxidation of Sulfides into Sulfoxides
3. Materials and Methods
3.1. Single-Crystal X-ray Crystallography
3.2. Synthesis and Characterization of Complexes
3.3. Procedure for Photooxidation of Sulfide into Sulfoxide
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- You, Y.; Nam, W. Photofunctional triplet excited states of cyclometalated Ir(iii) complexes: Beyond electroluminescence. Chem. Soc. Rev. 2012, 41, 7061–7084. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef]
- Lee, L.C.-C.; Lo, K.K.-W. Luminescent and Photofunctional Transition Metal Complexes: From Molecular Design to Diagnostic and Therapeutic Applications. J. Am. Chem. Soc. 2022, 144, 14420–14440. [Google Scholar] [CrossRef]
- Gorczyński, A.; Harrowfield, J.M.; Patroniak, V.; Stefankiewicz, A.R. Quaterpyridines as Scaffolds for Functional Metallosupramolecular Materials. Chem. Rev. 2016, 116, 14620–14674. [Google Scholar] [CrossRef]
- Chi, Y.; Chang, T.-K.; Ganesan, P.; Rajakannu, P. Emissive bis-tridentate Ir(III) metal complexes: Tactics, photophysics and applications. Coord. Chem. Rev. 2017, 346, 91–100. [Google Scholar] [CrossRef]
- Lee, Y.H.; Park, J.; Lee, J.; Lee, S.U.; Lee, M.H. Impact of Restricted Rotation of o-Carborane on Phosphorescence Efficiency. J. Am. Chem. Soc. 2015, 137, 8018–8021. [Google Scholar] [CrossRef]
- Esteruelas, M.A.; Lόpez, A.M.; Oñate, E.; San-Torcuato, A.; Tsai, J.-Y.; Xia, C. Preparation of Phosphorescent Iridium(III) Complexes with a Dianionic C,C,C,C-Tetradentate Ligand. Inorg. Chem. 2018, 57, 3720–3730. [Google Scholar] [CrossRef]
- Zhang, X.-M. Hydro(solvo)thermal in situ ligand syntheses. Coord. Chem. Rev. 2005, 249, 1201–1219. [Google Scholar] [CrossRef]
- Chen, X.-M.; Tong, M.-L. Solvothermal in Situ Metal/Ligand Reactions: A New Bridge between Coordination Chemistry and Organic Synthetic Chemistry. Acc. Chem. Res. 2007, 40, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Villafañe, F. ReI(CO)3 complexes with diimine ligands synthesized in situ. Coord. Chem. Rev. 2017, 339, 128–137. [Google Scholar] [CrossRef]
- Liu, C.-M.; Gao, S.; Kou, H.-Z. Dehydrogenative coupling of phenanthroline under hydrothermal conditions: Crystal structure of a novel layered vanadate complex constructed of 4,8,10-net sheets: [(2,2′-biphen)Co]V3O8. 5. Chem. Commun. 2001, 17, 1670–1671. [Google Scholar] [CrossRef]
- Viguri, M.E.; Huertos, M.A.; Pérez, J.; Riera, L.; Ara, I. Re-Mediated C-C Coupling of Pyridines and Imidazoles. J. Am. Chem. Soc. 2012, 134, 20326–20329. [Google Scholar] [CrossRef]
- Omori, H.; Hiroto, S.; Takeda, Y.; Fliegl, H.; Minakata, S.; Shinokubo, H. Ni(II) 10-Phosphacorrole: A Porphyrin Analogue Containing Phosphorus at the Meso Position. J. Am. Chem. Soc. 2019, 141, 4800–4805. [Google Scholar] [CrossRef]
- Vallavoju, N.; Sivaguru, J. Supramolecular photocatalysis: Combining confinement and non-covalent interactions to control light initiated reactions. Chem. Soc. Rev. 2014, 43, 4084–4101. [Google Scholar] [CrossRef]
- Espinal-Viguri, M.; Fombona, S.; Álvarez, D.; Díaz, J.; Menéndez, M.I.; Lόpez, R.; Pérez, J.; Riera, L. Regiochemistry Control by Bipyridine Substituents in the Deprotonation of ReI and MoII N-Alkylimidazole Complexes. Chem. Eur. J. 2019, 25, 9253–9265. [Google Scholar] [CrossRef]
- Sipos, G.; Drinkel, E.E.; Dorta, R. The Emergence of Sulfoxides as Efficient Ligands in Transition Metal Catalysis. Chem. Soc. Rev. 2015, 44, 3834–3860. [Google Scholar] [CrossRef] [PubMed]
- Nishiguchi, S.; Izumi, T.; Kouno, T.; Sukegawa, J.; Ilies, L.; Nakamura, E. Synthesis of Esomeprazole and Related Proton Pump Inhibitors through Iron-Catalyzed Enantioselective Sulfoxidation. ACS Catal. 2018, 8, 9738–9743. [Google Scholar] [CrossRef]
- Kinen, C.O.; Rossi, L.I.; de Rossi, R.H. The Development of an Environmentally Benign Sulfide Oxidation Procedure and Its Assessment by Green Chemistry Metrics. Green Chem. 2009, 11, 223–228. [Google Scholar] [CrossRef]
- Zhang, P.F.; Wang, Y.; Li, H.; Antonietti, M. Metal-Free Oxidation of Sulfides by Carbon Nitride with Visible Light Illumination at Room Temperature. Green Chem. 2012, 14, 1904–1908. [Google Scholar] [CrossRef]
- Miller, B.L.; Williams, T.D.; Schöneich, C. Mechanism of Sulfoxide Formation through Reaction of Sulfur Radical Cation Complexes with Superoxide or Hydroxide Ion in Oxygenated Aqueous Solution. J. Am. Chem. Soc. 1996, 118, 11014–11025. [Google Scholar] [CrossRef]
- Xing, C.; Deng, J.; Tan, R.; Gao, M.; Hao, P.; Yin, D.; Yin, D. Cooperative Chiral Salen TiIV Catalyst Supported on Ionic Liquid-Functionalized Graphene Oxide Accelerates Asymmetric Sulfoxidation in Water. Catal. Sci. Technol. 2017, 7, 5944–5952. [Google Scholar] [CrossRef]
- Heydari-turkmani, A.; Zakavi, S. The First Solid State Porphyrin-Weak Acid Molecular Complex: A Novel Metal Free, Nanosized and Porous Photocatalyst for Large Scale Aerobic Oxidations in Water. J. Catal. 2018, 364, 394–405. [Google Scholar] [CrossRef]
- Wei, L.-Q.; Ye, B.-H. Cyclometalated Ir–Zr Metal–Organic Frameworks as Recyclable Visible-Light Photocatalysts for Sulfide Oxidation into Sulfoxide in Water. ACS Appl. Mater. Interfaces 2019, 11, 41448–41457. [Google Scholar] [CrossRef]
- Peng, H.-L.; Li, Y.; Chen, X.-Y.; Li, L.-P.; Ke, Z.; Ye, B.-H. Visible-Light-Induced Amination of Quinoline at the C8 Position via a Postcoordinated Interligand-Coupling Strategy under Mild Conditions. Inorg. Chem. 2021, 60, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.-F.; Peng, H.-L.; Zhang, X.-P.; Ye, B.-H. Discrepancy between Proline and Homoproline in Chiral Recognition and Diastereomeric Photoreactivity with Iridium(III) Complexes. Inorg. Chem. 2021, 60, 5423–5431. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-K.; Li, L.-P.; Zhou, H.-Y.; Xiong, M.-F.; Fan, J.-Y.; Ye, B.-H. Switching the Photoreactions of Ir(III) Diamine Complexes between C-N Coupling and Dehydrogenation under Visible Light Irradiation. Inorg. Chem. 2022, 61, 20834–20847. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.-F.; Liu, G.-F.; Ye, B.-H. Solvent-Induced Umpolung Reaction from Dioxygenation to C-S Coupling in Bis(2-phenylquinoline) Iridium(III) Thiolate Complexes. Inorg. Chem. 2023, 62, 11654–11664. [Google Scholar] [CrossRef]
- Matsuo, N. Benzo[h]quinolin-10-yl-N Iridium(III) Complexes. Bull. Chem. Soc. Jpn. 1974, 47, 767–768. [Google Scholar]
- Yao, S.-Y.; Ou, Y.L.; Ye, B.-H. Asymmetric Synthesis of Enantiomerically Pure Mono- and Binuclear Bis(cyclometalated) Iridium(III) Complexes. Inorg. Chem. 2016, 55, 6018–6026. [Google Scholar] [CrossRef]
- Zhang, K.Y.; Li, S.P.-Y.; Zhu, N.; Or, I.W.-S.; Cheung, M.S.-H.; Lam, Y.-W.; Lo, K.K.-W. Structure, Photophysical and Electrochemical Properties, Biomolecular Interactions, and Intracellular Uptake of Luminescent Cyclometalated Iridium(III) Dipyridoquinoxaline Complexes. Inorg. Chem. 2010, 49, 2530–2540. [Google Scholar] [CrossRef] [PubMed]
- Lamansky, S.; Djurovich, P.; Murphy, D.; Abdel-Razzaq, F.; Lee, H.-E.; Adachi, C.; Burrows, P.E.; Forrest, S.R.; Thompson, M.E. Highly Phosphorescent Bis-Cyclometalated Iridium Complexes: Synthesis, Photophysical Characterization, and Use in Organic Light Emitting Diodes. J. Am. Chem. Soc. 2001, 123, 4304–4312. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Liu, R.; Qiu, Y.; Duan, L. High-performance yellow and orange-emitting diodes based on novel sublimable cationic iridium(III) complexes by ligand control. J. Mater. Chem. C 2018, 6, 5630–5638. [Google Scholar] [CrossRef]
- Bonesi, S.M.; Manet, I.; Freccero, M.; Fagnoni, M.; Albini, A. Photosensitized Oxidation of Sulfides: Discriminating between the Singlet-Oxygen Mechanism and Electron Transfer Involving Superoxide Anion or Molecular Oxygen. Chem. Eur. J. 2006, 12, 4844–4857. [Google Scholar] [CrossRef] [PubMed]
- Baptista, M.S.; Cadet, J.; Di Mascio, P.; Ghogare, A.A.; Greer, A.; Hamblin, M.R.; Lorente, C.; Nunez, S.C.; Ribeiro, M.S.; Thomas, A.H.; et al. Type I and Type II Photosensitized Oxidation Reactions: Guidelines and Mechanistic Pathways. Photochem. Photobiol. 2017, 93, 912–919. [Google Scholar] [CrossRef]
- Baciocchi, E.; Giacco, T.D.; Elisei, F.; Gerini, M.F.; Guerra, M.; Lapi, A.; Liberali, P. Electron Transfer and Singlet Oxygen Mechanisms in the Photooxygenation of Dibutyl Sulfide and Thioanisole in MeCN Sensitized by N-Methylquinolinium Tetrafluoborate and 9,10-Dicyanoanthracene. The Probable Involvement of a Thiadioxirane Intermediate in Electron Transfer Photooxygenations. J. Am. Chem. Soc. 2003, 125, 16444–16454. [Google Scholar] [PubMed]
- Chen, Y.; Zhu, C.; Guo, Z.; Liu, W.; Yang, X. Asymmetric Synthesis of Hydroquinolines with α,α-Disubstitution through Organocatalyzed Kinetic Resolution. Angew. Chem. Int. Ed. 2021, 60, 5268–5272. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Liu, W.; Zhao, F.; Chen, Y.; Tao, H.; He, Y.-P.; Yang, X. Kinetic Resolution of 2,2-Disubstituted Dihydroquinolines through Chiral Phosphoric Acid-Catalyzed C6-Selective Asymmetric Halogenations. Organ. Lett. 2021, 23, 4104–4108. [Google Scholar] [CrossRef] [PubMed]
- Mitra, R.; Zhu, H.; Grimme, S.; Niemeyer, J. Functional Mechanically Interlocked Molecules: Asymmetric Organocatalysis with a Catenated Bifunctional Brønsted Acid. Angew. Chem. Int. Ed. 2017, 56, 11456–11459. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Entry | Catalyst | DFSC b | Yield (%) c |
| 1 | 1 | - | 97 |
| 2 | 3 | - | 92 |
| 3 | 4 | - | 100 |
| 4 | 4a | - | 79 |
| 5 | 4 | N2 | 0 |
| 6 | 4 | dark | 0 |
| 7 | - | - | 0 |
| 8 | 4 | 2 eq. DABCO | 24 |
| 9 | 4 | 2 eq. BQ | 81 |
| 10 | 4 | 2 eq. DMB | 90 |
| 11 | 4 | 2 eq. iPrOH | 98 |
| Complex | 1a | 3a | 3 |
|---|---|---|---|
| Molecular formula | C32H26F8IrN4P | C34H32F6IrN4O2P | C34H28F6IrN4O2P |
| Temp/K | 150 | 150 | 150 |
| Mr | 841.74 | 865.80 | 861.77 |
| Crystal system | triclinic | triclinic | monoclinic |
| Space group | P-1 | P-1 | P21/n |
| a/Å | 8.3615(3) | 10.6791(3) | 10.58160(10) |
| b/Å | 10.6980(5) | 12.5175(4) | 18.5399(2) |
| c/Å | 19.2560(7) | 13.8170(4) | 17.3118(2) |
| α/° | 76.136(4) | 81.202(2) | 90 |
| β/° | 84.487(3) | 84.247(2) | 90.3740(10) |
| γ/° | 77.048(3) | 82.229(2) | 90 |
| V/Å3 | 1628.11(12) | 1802.52(9) | 3396.19(6) |
| Z | 2 | 2 | 4 |
| Dc (g cm−3) | 1.717 | 1.595 | 1.685 |
| Μ (mm−1) | 9.072 | 8.171 | 8.674 |
| aR1[I > 2σ(I)] | 0.0365 | 0.0329 | 0.0392 |
| bωR2 (all data) | 0.0930 | 0.0807 | 0.1009 |
| GOF | 1.055 | 1.039 | 1.062 |
| CCDC No. | 2291984 | 2291986 | 2291985 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, J.-Y.; Yao, S.-Y.; Ye, B.-H. In Situ Synthesis of Hexadentate Cyclometalated Ir(III) Complexes as Photocatalysts for the Oxidation of Sulfides into Sulfoxides in Water. Inorganics 2024, 12, 73. https://doi.org/10.3390/inorganics12030073
Fan J-Y, Yao S-Y, Ye B-H. In Situ Synthesis of Hexadentate Cyclometalated Ir(III) Complexes as Photocatalysts for the Oxidation of Sulfides into Sulfoxides in Water. Inorganics. 2024; 12(3):73. https://doi.org/10.3390/inorganics12030073
Chicago/Turabian StyleFan, Jing-Yan, Su-Yang Yao, and Bao-Hui Ye. 2024. "In Situ Synthesis of Hexadentate Cyclometalated Ir(III) Complexes as Photocatalysts for the Oxidation of Sulfides into Sulfoxides in Water" Inorganics 12, no. 3: 73. https://doi.org/10.3390/inorganics12030073
APA StyleFan, J.-Y., Yao, S.-Y., & Ye, B.-H. (2024). In Situ Synthesis of Hexadentate Cyclometalated Ir(III) Complexes as Photocatalysts for the Oxidation of Sulfides into Sulfoxides in Water. Inorganics, 12(3), 73. https://doi.org/10.3390/inorganics12030073