
Theoretical Investigations of Para-Methoxystyrene/Styrene Polymerization Catalyzed by Cationic Methyl- and Dibenzobarrelene-Based α-Diimine Palladium Complexes



Abstract
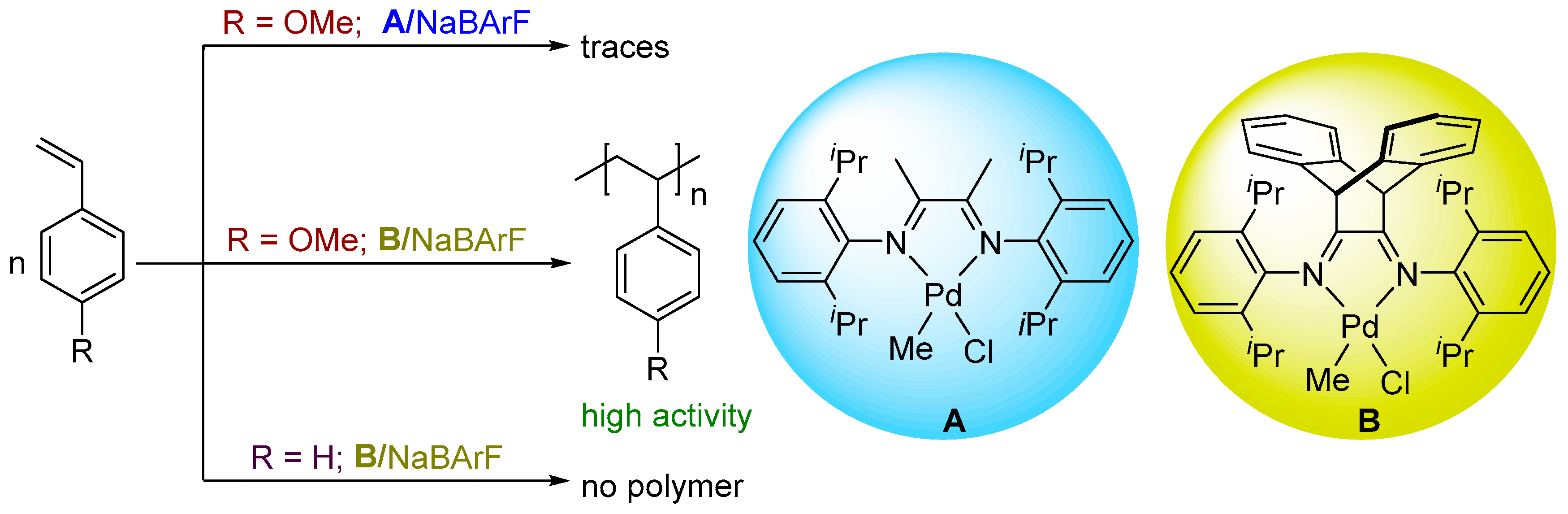
1. Introduction
2. Results and Discussion
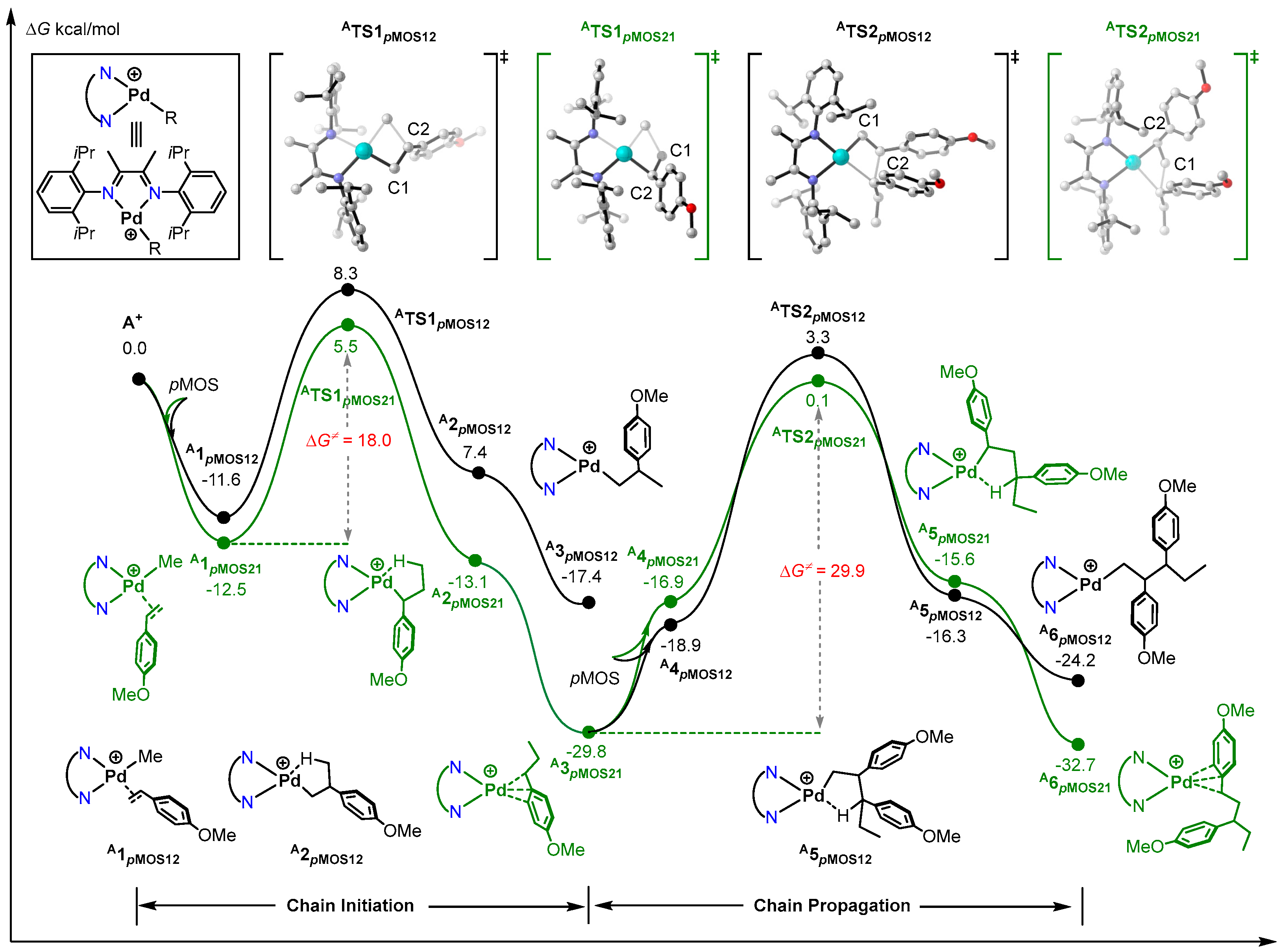
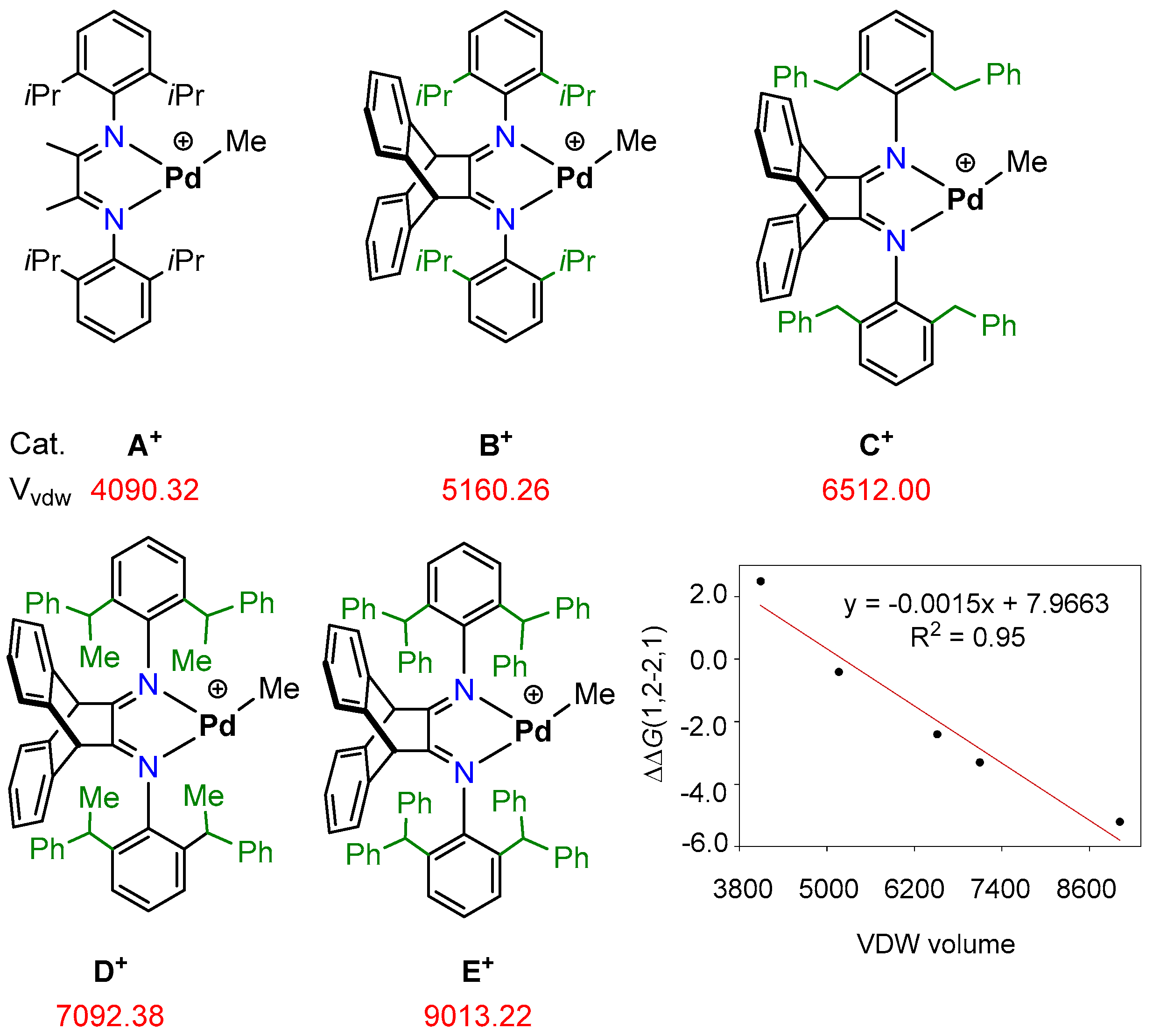
2.1. Mechanism of pMOS Polymerization by Cationic Species A+
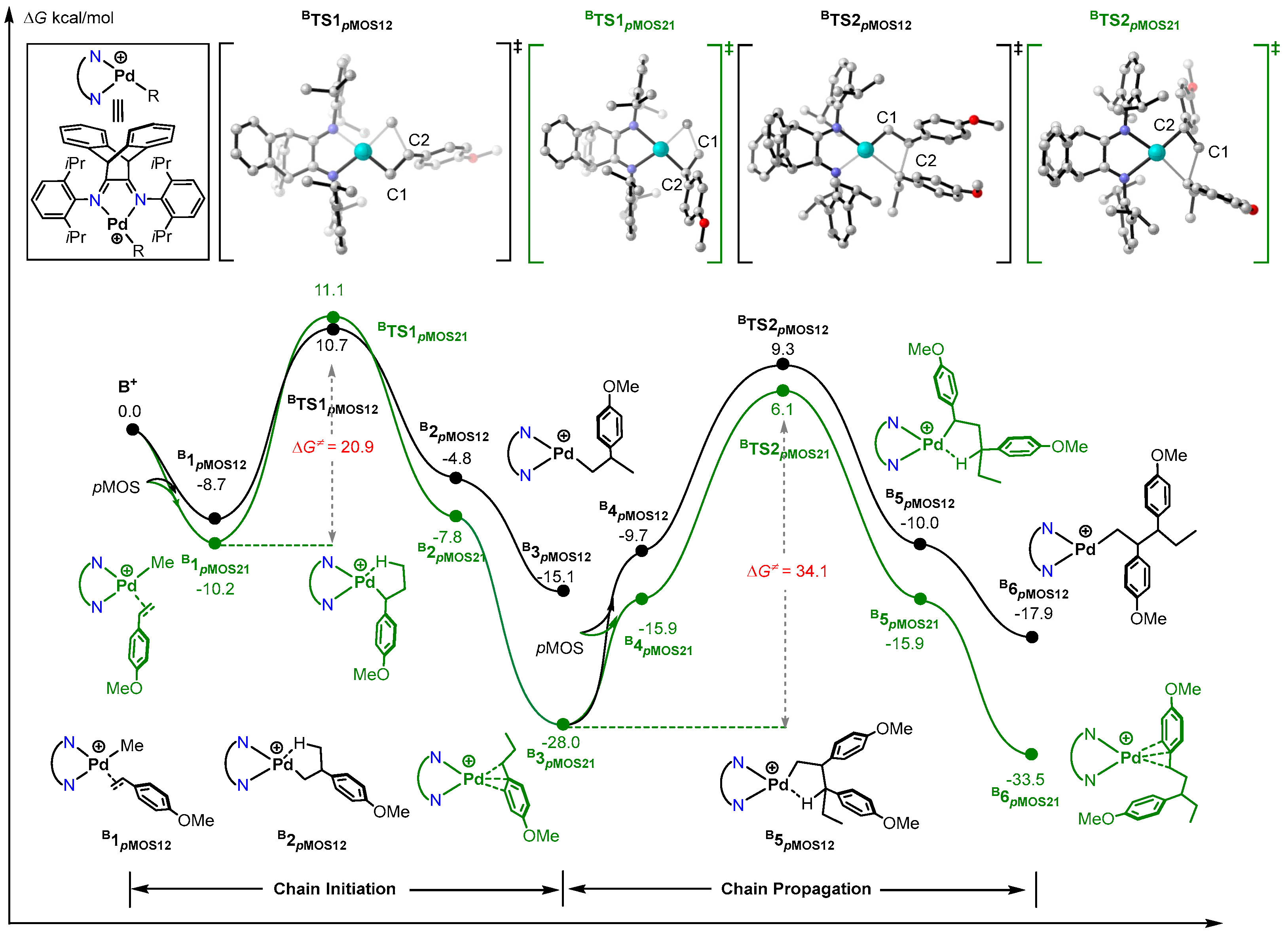
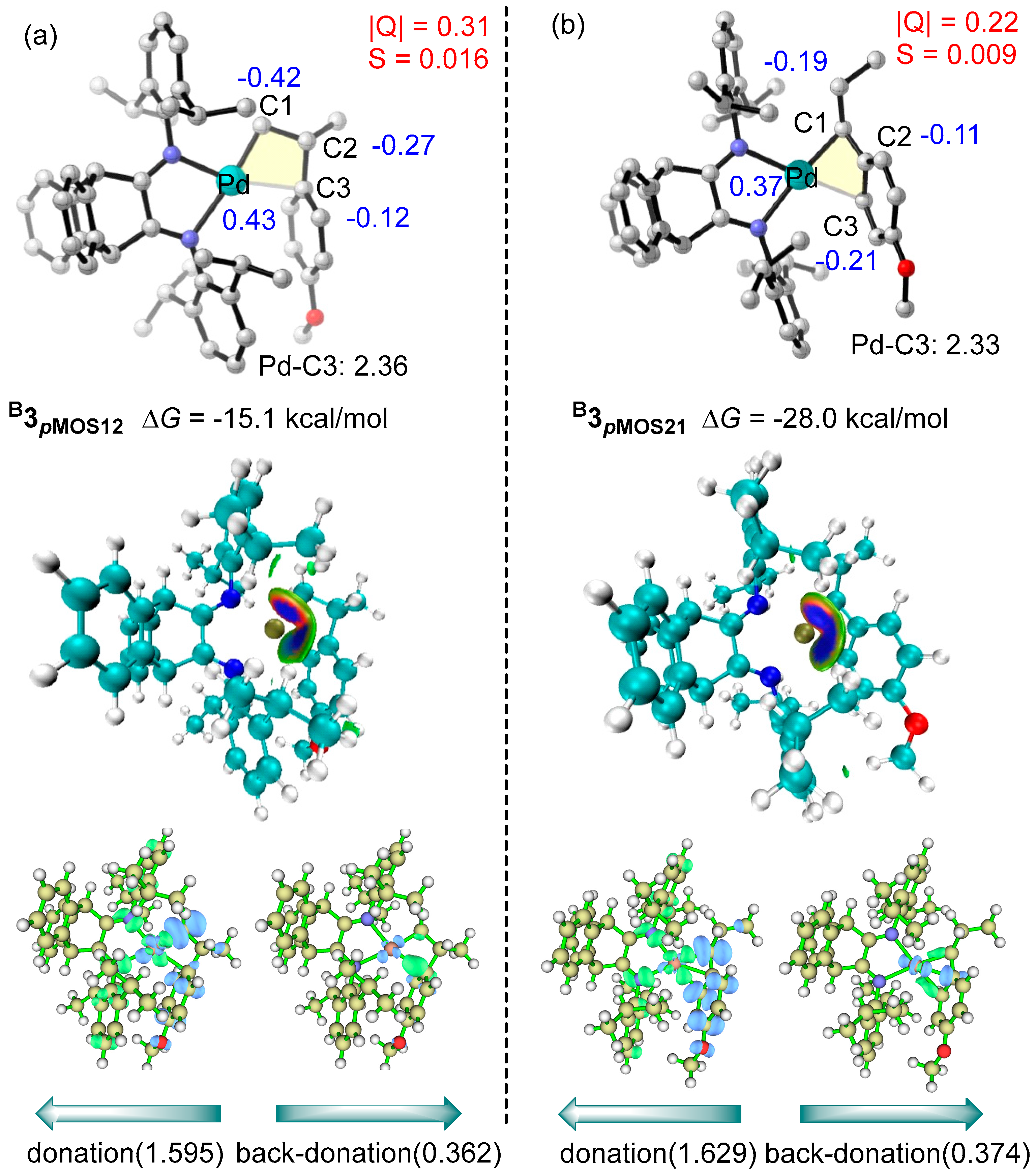
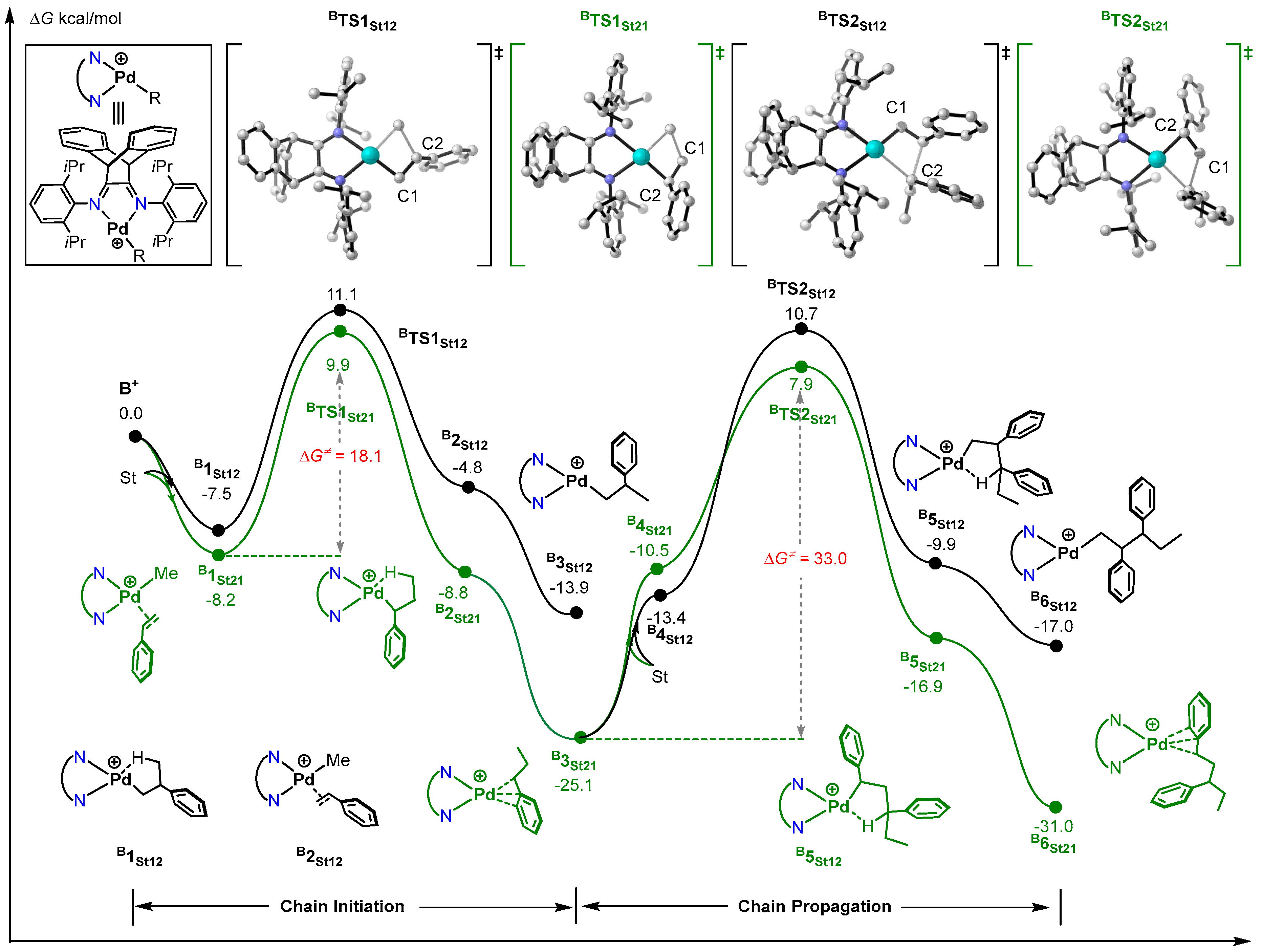
2.2. Mechanism of pMOS Polymerization by Cationic Species B+
2.3. Mechanism of Styrene Polymerization by Cationic Species B+
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zou, C.; Chen, C.L. Polar-functionalized, crosslinkable, self-healing and photoresponsive polyolefins. Angew. Chem. Int. Ed. 2020, 59, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Zou, C.; Chen, C. Material properties of functional polyethylenes from transition-metal-catalyzed ethylene–polar monomer copolymerization. Macromolecules 2022, 55, 1910–1922. [Google Scholar] [CrossRef]
- Mu, H.; Zhou, G.; Hu, X.; Jian, Z. Recent advances in nickel mediated copolymerization of olefin with polar monomers. Coord. Chem. Rev. 2021, 435, 213802–213818. [Google Scholar] [CrossRef]
- De, P.; Faust, R. Living carbocationic polymerization of p-methoxystyrene using p-methoxystyrene hydrochloride/SnBr4 initiating system: Determination of the absolute rate constant of propagation for ion pairs. Macromolecules 2004, 37, 7930–7937. [Google Scholar] [CrossRef]
- Kojima, K.; Sawamoto, M.; Higashimura, T. Living cationic polymerization of p-methoxystyrene by the hydrogen iodide/zinc iodide and hydrogen iodide/iodine initiating systems: Effects of tetrabutylammonium halides in a polar solvent. Macromolecules 1990, 23, 948–953. [Google Scholar] [CrossRef]
- Kawamura, T.; Uryu, T.; Matsuzaki, K. Stereoregularity of polystyrene derivatives, 1. poly(methylstyrene)s and poly-(methoxystyrene)s obtained with ziegler catalyst, cationic catalyst, or radical initiators. Makromol. Chem. 1982, 183, 125–141. [Google Scholar] [CrossRef]
- Iio, K.; Kobayashi, K.; Matsunaga, M. Radical polymerization of allyl alcohol and allyl acetate. Polym. Adv. Technol. 2007, 18, 953–958. [Google Scholar] [CrossRef]
- Chen, M.; Chen, C.L. Direct and tandem routes for the copolymerization of ethylene with polar functionalized internal olefins. Angew. Chem., Int. Ed. 2020, 59, 1206–1210. [Google Scholar] [CrossRef]
- Mitsushige, Y.; Yasuda, H.; Carrow, B.P.; Ito, S.; Kobayashi, M.; Tayano, T.; Watanabe, Y.; Okuno, Y.; Hayashi, S.; Kuroda, J.; et al. Methylene-bridged bisphosphine monoxide ligands for palladium-catalyzed copolymerization of ethylene and polar monomers. ACS Macro Lett. 2018, 7, 305–311. [Google Scholar] [CrossRef]
- Ota, Y.; Ito, S.; Kobayashi, M.; Kitade, S.; Sakata, K.; Tayano, T.; Nozaki, K. Crystalline isotactic polar polypropylene from the palladium-catalyzed copolymerization of propylene and polar monomers. Angew. Chem. Int. Ed. 2016, 55, 7505–7509. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, C.; Mecking, S.; Jian, Z. Ultrahigh branching of main-chain-functionalized polyethylenes by inverted insertion selectivity. Angew. Chem. Int. Ed. 2020, 59, 14296–14302. [Google Scholar] [CrossRef]
- Chen, J.; Gao, Y.; Marks, T.J. Early transition metal catalysis for olefin-polar monomer copolymerization. Angew. Chem. Int. Ed. 2020, 59, 14726–14735. [Google Scholar] [CrossRef] [PubMed]
- Keyes, A.; Basbug Alhan, H.E.; Ordonez, E.; Ha, U.; Beezer, D.B.; Dau, H.; Liu, Y.S.; Tsogtgerel, E.; Jones, G.R.; Harth, E. Olefins and vinyl polar monomers: Bridging the gap for next generation materials. Angew. Chem. Int. Ed. 2019, 58, 12370–12391. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.F.; Yan, Y.; Solan, G.A.; Ma, Y.; Sun, W.H. Recent advancements in N-ligated group 4 molecular catalysts for the (co)polymerization of ethylene. Coord. Chem. Rev. 2020, 411, 213254. [Google Scholar] [CrossRef]
- Wang, Y.; Lai, J.; Gao, R.; Gou, Q.; Li, B.; Zheng, G.; Guo, Z. Recent advances in nickel catalysts with industrial exploitability for copolymerization of ethylene with polar monomers. Polymers 2024, 16, 1676. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Zheng, H.; Gao, H.; Cheng, Z.; Feng, C.; Yang, J.; Gao, H. Recent advances in synthesis of non-alternating polyketone generated by copolymerization of carbon monoxide and ethylene. Int. J. Mol. Sci. 2024, 25, 1348. [Google Scholar] [CrossRef]
- Xu, M.; Yu, F.; Li, P.; Xu, G.; Zhang, S.; Wang, F. Enhancing chain initiation efficiency in the cationic allyl-nickel catalyzed (co)polymerization of ethylene and methyl acrylate. Inorg. Chem. 2020, 59, 4475–4482. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, W.; Xu, G.; Pang, W.; Li, Y.; Tan, C.; Wang, F. Bulky iminophosphine-based nickel and palladium catalysts bearing 2,6-dibenzhydryl groups for ethylene oligo-/polymerization. Appl. Organomet. Chem. 2020, 34, 5428. [Google Scholar] [CrossRef]
- Guironnet, D.; Roesle, P.; Runzi, T.; Gottker Schnetmann, I.; Mecking, S. Insertion polymerization of acrylate. J. Am. Chem. Soc. 2009, 131, 422–423. [Google Scholar] [CrossRef]
- Jian, Z.; Mecking, S. Insertion homo- and copolymerization of diallyl ether. Angew. Chem. Int. Ed. 2015, 54, 15845–15849. [Google Scholar] [CrossRef]
- Saki, Z.; D’Auria, I.; Dall’anese, A.; Milani, B.; Pellecchia, C. Copolymerization of ethylene and methyl acrylate by pyridylimino Ni(II) catalysts affording hyperbranched poly(ethylene-co-methyl acrylate)s with tunable structures of the ester groups. Macromolecules 2020, 53, 9294–9305. [Google Scholar] [CrossRef]
- Liao, G.; Xiao, Z.; Chen, X.; Du, C.; Zhong, L.; Cheung, C.S.; Gao, H. Fast and regioselective polymerization of para-alkoxystyrene by palladium catalysts for precision production of high-molecular weight polystyrene derivatives. Macromolecules 2020, 53, 256–266. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Independent gradient model based on hirshfeld partition: A new method for visual study of interactions in chemical systems. J. Comput. Chem. 2022, 43, 539–555. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Mitoraj, M.P.; Michalak, A.; Ziegler, T. A combined charge and energy decomposition scheme for bond analysis. J. Chem. Theory Comput. 2009, 5, 962–975. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A. Van der waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and accurate Ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Huang, W.; Xi, Y.; Pan, D.; Fan, L.; Fang, K.; Huang, G.; Chen, Y. Palladium-catalyzed enantioselective multicomponent cross-coupling of trisubstituted olefins. J. Am. Chem. Soc. 2024, 146, 16892–16901. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Uchikura, T.; Kato, S.; Makino, Y.; Fujikawa, M.J.; Yamanaka, M.; Akiyama, T. Chiral phosphoric acid-palladium(II) complex catalyzed asymmetric desymmetrization of biaryl compounds by C(sp3)-H activation. J. Am. Chem. Soc. 2023, 145, 15906–15911. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, A.W.; Böhme, M.; Dapprich, S.; Gobbi, A.; Höllwarth, A.; Jonas, V.; Köhler, K.F.; Stegmann, R.; Veldkamp, A.; Frenking, G. A set of f-polarization functions for pseudo-potential basis sets of the transition metals Sc-Cu, Y-Ag and La-Au. Chem. Phys. Lett. 1993, 208, 111–114. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions-the IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B. 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.Q.; Karunananda, M.K.; Zeng, T.; Yang, S.; Liu, Z.; Houk, K.N.; Liu, P.; Engle, K.M. Redox-paired alkene difunctionalization enables skeletally divergent synthesis. J. Am.Chem. Soc. 2023, 145, 12351–12359. [Google Scholar] [CrossRef]
- Ni, H.Q.; Alturaifi, T.M.; Rodphon, W.; Scherschel, N.F.; Yang, S.; Wang, F.; Engle, K.M. Anti-selective cyclopropanation of nonconjugated alkenes with diverse pronucleophiles via directed nucleopalladation. J. Am. Chem. Soc. 2024, 146, 24503–24514. [Google Scholar] [CrossRef]
- Andrae, D.; Haüβermann, U.; Dolg, M.; Stoll, H.; Preuβ, H. Energy-adjusted ab initio pseudopotentials for the 2nd and 3rd row transition-elements. Theor. Chim. Acta. 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Zhang, P.; Guo, C.Q.; Yao, W.; Lu, C.J.; Li, Y.Z.; Paton, R.S.; Liu, R.R. Pd-catalyzed asymmetric amination of enamines: Expedient synthesis of structurally diverse N-C atropisomers. ACS Catal. 2023, 13, 7680–7690. [Google Scholar] [CrossRef]
- Biosca, M.; Saltó, J.; Magre, M.; Norrby, P.-O.; Pàmies, O.; Diéguez, M. An Improved Class of Phosphite-Oxazoline Ligands forPd-Catalyzed Allylic Substitution Reactions. ACS Catal. 2019, 9, 6033–6048. [Google Scholar] [CrossRef]
- Yan, M.; Kang, X.; Li, S.; Xu, X.; Luo, Y.; He, S.; Chen, C. Mechanistic Studies on Nickel-Catalyzed Ethylene Polymerization: Ligand Effects and Quantitative Structure–Activity Relationship Model. Organometallics 2022, 41, 3212–3218. [Google Scholar] [CrossRef]
- Seo, S.; Shin, T.; Choi, I.; Kim, H. Origin of Stereoselectivity in Pd-Catalyzed Asymmetric Allylic Substitutions with Trost-Type Mixed Bidentate Phosphorus Ligands. ACS Catal. 2024, 14, 11113–11120. [Google Scholar] [CrossRef]
- Legault, C.Y. CYLView, Version 1.0b; Université de Sherbrooke: Sherbrooke, QC, Canada, 2020. Available online: http://www.cylview.org (accessed on 11 April 2023).
- Liu, C.; Qin, Z.X.; Ji, C.L.; Hong, X.; Szostak, M. Highly-chemoselective step-down reduction of carboxylic acids to aromatic hydrocarbons via palladium catalysis. Chem. Sci. 2019, 10, 5736–5742. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Kang, X.; Dai, S. Stable ultrahighly branched polyethylene synthesis via externally robust chain-walking polymerization. ACS Catal. 2024, 14, 13531–13541. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | 1pMOS21 | TS1pMOS12 (TS1pMOS21) | P12 (P21) | ∆∆P | ∆G (1,2-) | ∆G (2,1-) | ∆∆G (1,2–2,1) |
|---|---|---|---|---|---|---|---|
| A+ | −12.5 | 8.3 (5.5) | −17.4 (−29.8) | 12.4 | 20.5 | 18.0 | 2.5 |
| B+ | −10.2 | 10.7 (11.1) | −15.1 (−28.0) | 12.9 | 20.9 | 21.3 | −0.4 |
| C+ | −15.4 | 4.5 (6.9) | −20.6 (−30.5) | 9.9 | 19.9 | 22.3 | −2.4 |
| D+ | −6.2 | 13.3 (16.7) | −10.3 (−17.1) | 6.8 | 19.5 | 22.8 | −3.3 |
| E+ | −8.5 | 11.3 (16.5) | −14.4 (−22.8) | 8.3 | 19.8 | 25.0 | −5.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, L.; Luo, Y.; Wen, X.; Zhang, W.; Zhou, G. Theoretical Investigations of Para-Methoxystyrene/Styrene Polymerization Catalyzed by Cationic Methyl- and Dibenzobarrelene-Based α-Diimine Palladium Complexes. Inorganics 2024, 12, 315. https://doi.org/10.3390/inorganics12120315
Zhu L, Luo Y, Wen X, Zhang W, Zhou G. Theoretical Investigations of Para-Methoxystyrene/Styrene Polymerization Catalyzed by Cationic Methyl- and Dibenzobarrelene-Based α-Diimine Palladium Complexes. Inorganics. 2024; 12(12):315. https://doi.org/10.3390/inorganics12120315
Chicago/Turabian StyleZhu, Ling, Yi Luo, Xin Wen, Wenzhen Zhang, and Guangli Zhou. 2024. "Theoretical Investigations of Para-Methoxystyrene/Styrene Polymerization Catalyzed by Cationic Methyl- and Dibenzobarrelene-Based α-Diimine Palladium Complexes" Inorganics 12, no. 12: 315. https://doi.org/10.3390/inorganics12120315
APA StyleZhu, L., Luo, Y., Wen, X., Zhang, W., & Zhou, G. (2024). Theoretical Investigations of Para-Methoxystyrene/Styrene Polymerization Catalyzed by Cationic Methyl- and Dibenzobarrelene-Based α-Diimine Palladium Complexes. Inorganics, 12(12), 315. https://doi.org/10.3390/inorganics12120315