
Design and Synthesis of New Boron-Based Benzo[c][1,2,5]oxadiazoles and Benzo[c][1,2,5]thiadiazoles as Potential Hypoxia Inhibitors



Abstract
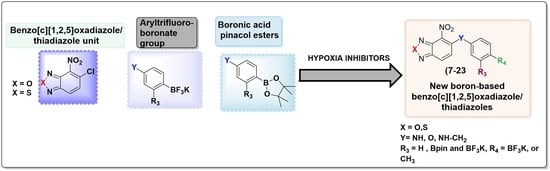
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
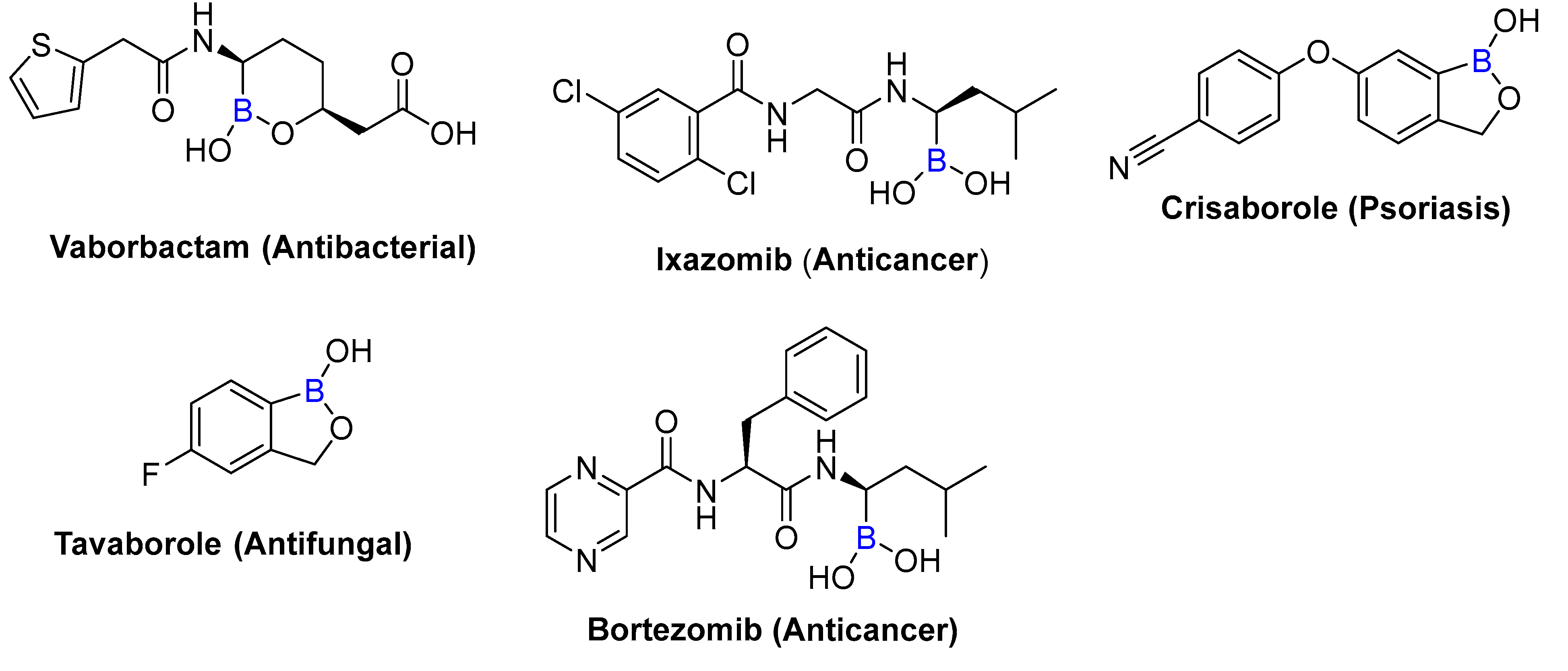
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Method for the Synthesis of (7), (8), (13) and (14)
3.1.1. 4-Nitro-N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)benzo[c][1,2,5] oxadiazol-5-amine (7)
3.1.2. 4-Nitro-N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)benzo[c][1,2,5] thiadiazol-5-amine (8)
3.2. General Method for the Synthesis of (9) and (10)
3.2.1. 4-Nitro-N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl)benzo[c][1,2,5] oxadiazol-5-amine (9)
3.2.2. 4-Nitro-N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl)benzo[c][1,2,5] thiadiazol-5-amine (10)
3.3. General Method for the Synthesis of (11) and (12)
3.3.1. 4-Nitro-5-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)benzo[c][1,2,5] oxadiazole (11)
3.3.2. 4-Nitro-5-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)benzo[c][1,2,5] thiadiazole (12)
3.3.3. N-(4-Methyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-4-nitrobenzo[c][1,2,5] oxadiazol-5-amine (13)
3.3.4. N-(4-Methyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-4-nitrobenzo[c][1,2,5] thiadiazol-5-amine (14)
3.4. General Method for Preparation of Trifluoroborate Salts (15–22)
3.4.1. Potassium Trifluoro (4-((4-Nitrobenzo[c][1,2,5] oxadiazol-5-yl)amino)phenyl) Borate (15)
3.4.2. Potassium Trifluoro (4-((4-Nitrobenzo[c][1,2,5] thiadiazol-5-yl)amino)phenyl) Borate (16)
3.4.3. Potassium Trifluoro (4-(((4-Nitrobenzo[c][1,2,5] oxadiazol-5-yl)amino)methyl)phenyl) Borate (17)
3.4.4. Potassium Trifluoro (4-(((4-Nitrobenzo[c][1,2,5] thiadiazol-5-yl)amino)methyl)phenyl) Borate (18)
3.4.5. Potassium Trifluoro (4-((4-Nitrobenzo[c][1,2,5] oxadiazol-5-yl)oxy)phenyl) Borate (19)
3.4.6. Potassium Trifluoro (4-((4-Nitrobenzo[c][1,2,5] thiadiazol-5-yl)oxy)phenyl) Borate (20)
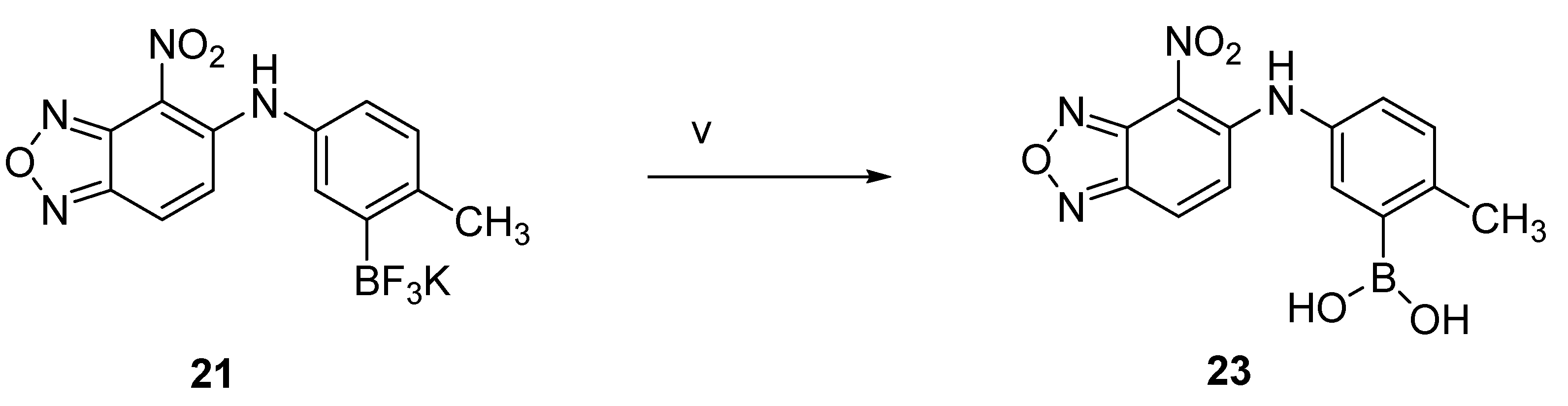
3.4.7. Potassium Trifluoro (2-Methyl-5-((4-nitrobenzo[c][1,2,5] oxadiazol-5-yl)amino)phenyl) Borate (21)
3.4.8. Potassium Trifluoro (2-Methyl-5-((4-nitrobenzo[c][1,2,5] thiadiazol-5-yl)amino)phenyl) Borate (22)
3.4.9. (2-Methyl-5-((4-nitrobenzo[c][1,2,5]oxadiazol-5-yl)amino)phenyl)boronic acid (23)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rankin, E.B.; Nam, J.-M.; Giaccia, A.J. Hypoxia: Signaling the metastatic cascade. Trends Cancer 2016, 2, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Samanta, P.; Sarkar, R.; Biswas, S.; Saha, P.; Hajra, S.; Bhowmik, A. Targeting HIF-1α by Natural and Synthetic Compounds: A Promising Approach for Anti-Cancer Therapeutics Development. Molecules 2022, 27, 5192. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Zhao, S.; Han, J.; Zheng, L.; Yang, Z.; Zhao, L. Hypoxia-inducible factor-1α induces multidrug resistance protein in colon cancer. OncoTargets Ther. 2015, 8, 1941. [Google Scholar] [CrossRef] [PubMed]
- Das, B.C.; Madhukumar, A.V.; Anguiano, J.; Mani, S. Design, synthesis and biological evaluation of 2H-benzo [b][1, 4] oxazine derivatives as hypoxia targeted compounds for cancer therapeutics. Bioorg. Med. Chem. Lett. 2009, 19, 4204–4206. [Google Scholar] [CrossRef]
- Reddy, S.B.; Williamson, S.K. Tirapazamine: A novel agent targeting hypoxic tumor cells. Expert Opin. Investig. Drugs 2009, 18, 77–87. [Google Scholar] [CrossRef]
- Albadari, N.; Deng, S.; Li, W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin. Drug Discov. 2019, 14, 667–682. [Google Scholar] [CrossRef]
- Das, B.C.; Nandwana, N.K.; Das, S.; Nandwana, V.; Shareef, M.A.; Das, Y.; Saito, M.; Weiss, L.M.; Almaguel, F.; Hosmane, N.S. Boron Chemicals in Drug Discovery and Development: Synthesis and Medicinal Perspective. Molecules 2022, 27, 2615. [Google Scholar] [CrossRef]
- Song, S.; Gao, P.; Sun, L.; Kang, D.; Kongsted, J.; Poongavanam, V.; Zhan, P.; Liu, X. Recent developments in the medicinal chemistry of single boron atom-containing compounds. Acta Pharm. Sin. B 2021, 11, 3035–3059. [Google Scholar] [CrossRef]
- Das, B.C.; Shareef, M.A.; Das, S.; Nandwana, N.K.; Das, Y.; Saito, M.; Weiss, L.M. Boron-Containing Heterocycles as Promising Pharmacological Agents. Bioorg. Med. Chem. 2022, 63, 116748. [Google Scholar] [CrossRef]
- Das, B.C.; Thapa, P.; Karki, R.; Schinke, C.; Das, S.; Kambhampati, S.; Banerjee, S.K.; Van Veldhuizen, P.; Verma, A.; Weiss, L.M. Boron chemicals in diagnosis and therapeutics. Future Med. Chem. 2013, 5, 653–676. [Google Scholar] [CrossRef]
- Almi, I.; Belaidi, S.; Melkemi, N.; Bouzidi, D. Chemical reactivity, drug-likeness and structure activity/property relationship studies of 2, 1, 3-benzoxadiazole derivatives as anti-cancer activity. J. Bionanosci. 2018, 12, 49–57. [Google Scholar] [CrossRef]
- Dutta, T.; Pal, K.; Koner, A.L. Cellular metabolic activity marker via selective turn-ON detection of transporter protein using nitrobenzoxadiazole-based fluorescent reporter. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Schininà, B.; Martorana, A.; Colabufo, N.A.; Contino, M.; Niso, M.; Perrone, M.G.; De Guidi, G.; Catalfo, A.; Rappazzo, G.; Zuccarello, E. 4-Nitro-2, 1, 3-benzoxadiazole derivatives as potential fluorescent sigma receptor probes. RSC Adv. 2015, 5, 47108–47116. [Google Scholar] [CrossRef]
- Fiorentino, F.; De Angelis, M.; Menna, M.; Rovere, A.; Caccuri, A.M.; D’Acunzo, F.; Palamara, A.T.; Nencioni, L.; Rotili, D.; Mai, A. Anti-influenza A virus activity and structure–activity relationship of a series of nitrobenzoxadiazole derivatives. J. Enzym. Inhib. Med. Chem. 2021, 36, 2128–2138. [Google Scholar] [CrossRef] [PubMed]
- Siciliano, G.; Di Paolo, V.; Rotili, D.; Migale, R.; Pedini, F.; Casella, M.; Camerini, S.; Dalzoppo, D.; Henderson, R.; Huijs, T. The Nitrobenzoxadiazole Derivative NBDHEX Behaves as Plasmodium falciparum Gametocyte Selective Inhibitor with Malaria Parasite Transmission Blocking Activity. Pharmaceuticals 2022, 15, 168. [Google Scholar] [CrossRef]
- Silva, V.A.; Lafont, F.; Benhelli-Mokrani, H.; Breton, M.L.; Hulin, P.; Chabot, T.; Paris, F.; Sakanyan, V.; Fleury, F. Rapid diminution in the level and activity of DNA-dependent protein kinase in cancer cells by a reactive nitro-benzoxadiazole compound. Int. J. Mol. Sci. 2016, 17, 703. [Google Scholar] [CrossRef]
- Sakanyan, V.; Angelini, M.; Le Béchec, M.; Lecocq, M.F.; Benaiteau, F.; Rousseau, B.; Gyulkhandanyan, A.; Gyulkhandanyan, L.; Logé, C.; Reiter, E. Screening and discovery of nitro-benzoxadiazole compounds activating epidermal growth factor receptor (EGFR) in cancer cells. Sci. Rep. 2014, 4, 1–11. [Google Scholar] [CrossRef]
- De Luca, A.; Carpanese, D.; Rapanotti, M.C.; Viguria, T.M.S.; Forgione, M.A.; Rotili, D.; Fulci, C.; Iorio, E.; Quintieri, L.; Chimenti, S. The nitrobenzoxadiazole derivative MC3181 blocks melanoma invasion and metastasis. Oncotarget 2017, 8, 15520. [Google Scholar] [CrossRef]
- Fulci, C.; Rotili, D.; De Luca, A.; Stella, L.; Morozzo della Rocca, B.; Forgione, M.; Di Paolo, V.; Mai, A.; Falconi, M.; Quintieri, L. A new nitrobenzoxadiazole-based GSTP1-1 inhibitor with a previously unheard of mechanism of action and high stability. J. Enzym. Inhib. Med. Chem. 2017, 32, 240–247. [Google Scholar] [CrossRef]
- Rotili, D.; De Luca, A.; Tarantino, D.; Pezzola, S.; Forgione, M.; Della Rocca, B.M.; Falconi, M.; Mai, A.; Caccuri, A.M. Synthesis and structure–activity relationship of new cytotoxic agents targeting human glutathione-S-transferases. Eur. J. Med. Chem. 2015, 89, 156–171. [Google Scholar] [CrossRef]
- Chistyakov, V.; Semenyuk, Y.P.; Morozov, P.; Prazdnova, E.; Chmykhalo, V.; Kharchenko, E.Y.; Kletskii, M.; Borodkin, G.; Lisovin, A.; Burov, O. Synthesis and biological properties of nitrobenzoxadiazole derivatives as potential nitrogen (ii) oxide donors: SOX induction, toxicity, genotoxicity, and DNA protective activity in experiments using Escherichia coli-based lux biosensors. Russ. Chem. Bull. 2015, 64, 1369–1377. [Google Scholar] [CrossRef]
- Neto, B.A.; Lapis, A.A.; da Silva Júnior, E.N.; Dupont, J. 2, 1, 3-Benzothiadiazole and Derivatives: Synthesis, Properties, Reactions, and Applications in Light Technology of Small Molecules. Eur. J. Org. Chem. 2013, 2013, 228–255. [Google Scholar] [CrossRef]
- Mataka, S.; Takahashi, K.; Imura, T.; Tashiro, M. Reduction of 4, 7-diphenyl-1, 2, 5-thia (oxa) diazolo [3, 4-c] pyridines affording 2, 5-diphenyl-3, 4-diaminopyridines and ring closure of the diamines to fluorescent azaheterocycles. J. Heterocycl. Chem. 1982, 19, 1481–1488. [Google Scholar] [CrossRef]
- Balasankar, T.; Gopalakrishnan, M.; Nagarajan, S. Synthesis and antibacterial activity of some 5-(4-biphenylyl)-7-aryl [3, 4-d][1, 2, 3]-benzothiadiazoles. Eur. J. Med. Chem. 2005, 40, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Malanga, G.; Reiter, R.D.; Garay, E. Update on tizanidine for muscle spasticity and emerging indications. Expert Opin. Pharmacother. 2008, 9, 2209–2215. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-L.; Chen, X.-Y.; Gao, Y.; Gao, L.-X.; Sheng, L.; Zhu, J.; Xu, L.; Ding, Z.-Z.; Zhang, C.; Li, J.-Y. Benzo [c][1, 2, 5] thiadiazole derivatives: A new class of potent Src homology-2 domain containing protein tyrosine phosphatase-2 (SHP2) inhibitors. Bioorganic Med. Chem. Lett. 2017, 27, 5154–5157. [Google Scholar] [CrossRef]
- dos Santos, B.F.; Pereira, C.F.; Pinz, M.P.; de Oliveira, A.R.; Brand, G.; Katla, R.; Wilhelm, E.A.; Luchese, C.; Domingues, N.L. Efficient palladium-catalyzed C-S cross-coupling reaction of benzo-2, 1, 3-thiadiazole at C-5-position: A potential class of AChE inhibitors. Appl. Organomet. Chem. 2020, 34, e5650. [Google Scholar] [CrossRef]
- da Costa Rodrigues, K.; de Oliveira, R.L.; da Silva Chaves, J.; da Rocha, V.M.E.; Dos Santos, B.F.; Fronza, M.G.; de Campos Domingues, N.L.; Savegnago, L.; Wilhelm, E.A.; Luchese, C. A new arylsulfanyl-benzo-2, 1, 3-thiadiazoles derivative produces an anti-amnesic effect in mice by modulating acetylcholinesterase activity. Chem.-Biol. Interact. 2022, 351, 109736. [Google Scholar] [CrossRef]
- Nandwana, V.; Nandwana, N.K.; Das, Y.; Saito, M.; Panda, T.; Das, S.; Almaguel, F.; Hosmane, N.S.; Das, B.C. The role of microbiome in brain development and neurodegenerative diseases. Molecules 2022, 27, 3402. [Google Scholar] [CrossRef]
- Yang, J.; Das, B.C.; Aljitawi, O.; Kumar, A.; Das, S.; Van Veldhuizen, P. Magmas Inhibition in Prostate Cancer: A Novel Target for Treatment-Resistant Disease. Cancers 2022, 14, 2732. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, M.; García-Mendoza, E.; Farfán-García, E.D.; Das, B.C.; Ciprés-Flores, F.J.; Trujillo-Ferrara, J.G.; Tamay-Cach, F.; Soriano-Ursúa, M.A. Not all boronic acids with a five-membered cycle induce tremor, neuronal damage and decreased dopamine. Neurotoxicology 2017, 62, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Das, B.C.; Nandwana, N.K.; Ojha, D.P.; Das, S.; Evans, T. Synthesis of a boron-containing amidoxime reagent and its application to synthesize functionalized oxadiazole and quinazolinone derivatives. Tetrahedron Lett. 2022, 92, 153657. [Google Scholar] [CrossRef] [PubMed]
- CijiangáHe, J. A novel procedure for the synthesis of borylated quinolines and its application in the development of potential boron-based homeodomain interacting protein kinase 2 (HIPK2) inhibitors. RSC Adv. 2022, 12, 24187–24191. [Google Scholar]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, S.; Shareef, M.A.; Das, B.C. Design and Synthesis of New Boron-Based Benzo[c][1,2,5]oxadiazoles and Benzo[c][1,2,5]thiadiazoles as Potential Hypoxia Inhibitors. Inorganics 2023, 11, 34. https://doi.org/10.3390/inorganics11010034
Das S, Shareef MA, Das BC. Design and Synthesis of New Boron-Based Benzo[c][1,2,5]oxadiazoles and Benzo[c][1,2,5]thiadiazoles as Potential Hypoxia Inhibitors. Inorganics. 2023; 11(1):34. https://doi.org/10.3390/inorganics11010034
Chicago/Turabian StyleDas, Sasmita, Mohammed Adil Shareef, and Bhaskar C. Das. 2023. "Design and Synthesis of New Boron-Based Benzo[c][1,2,5]oxadiazoles and Benzo[c][1,2,5]thiadiazoles as Potential Hypoxia Inhibitors" Inorganics 11, no. 1: 34. https://doi.org/10.3390/inorganics11010034
APA StyleDas, S., Shareef, M. A., & Das, B. C. (2023). Design and Synthesis of New Boron-Based Benzo[c][1,2,5]oxadiazoles and Benzo[c][1,2,5]thiadiazoles as Potential Hypoxia Inhibitors. Inorganics, 11(1), 34. https://doi.org/10.3390/inorganics11010034