
Nano-ZrO2@C, Nano-(ZrC, ZrO2)@C and Nano-ZrC@C Composites Prepared by Plasma-Assisted Carbonization of Zr-Loaded Iminodiacetate-Functionalized Styrene-Divinylbenzene Copolymers

 , , , , , ,
, , , , , ,  ,
, 
Abstract
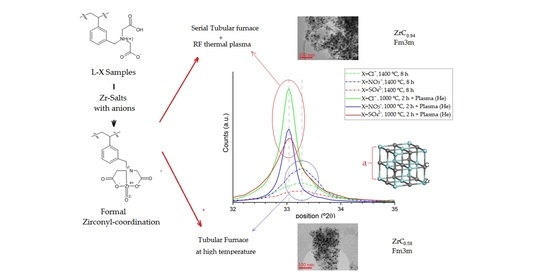
:
1. Introduction
2. Results and Discussion
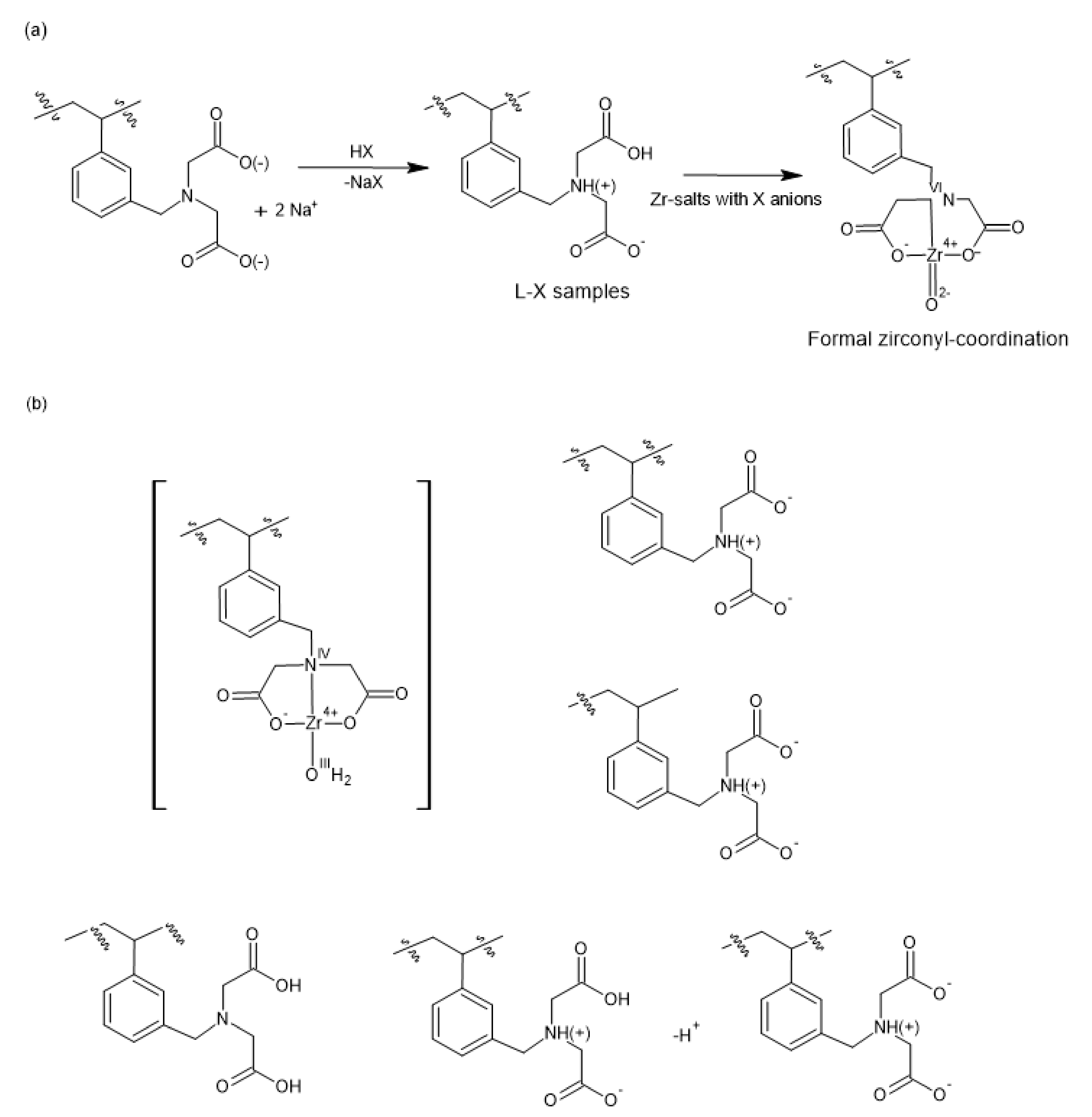
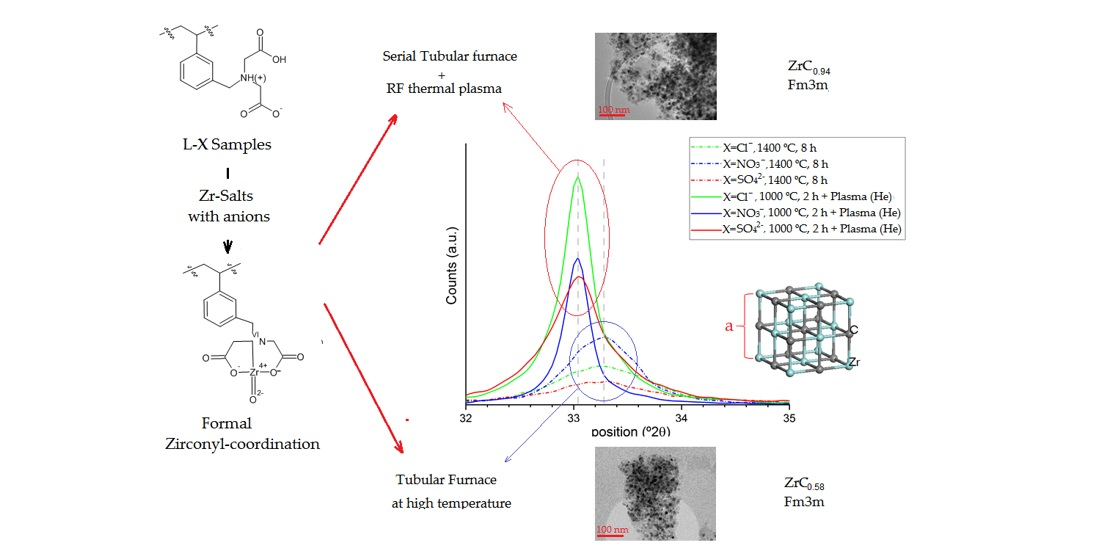
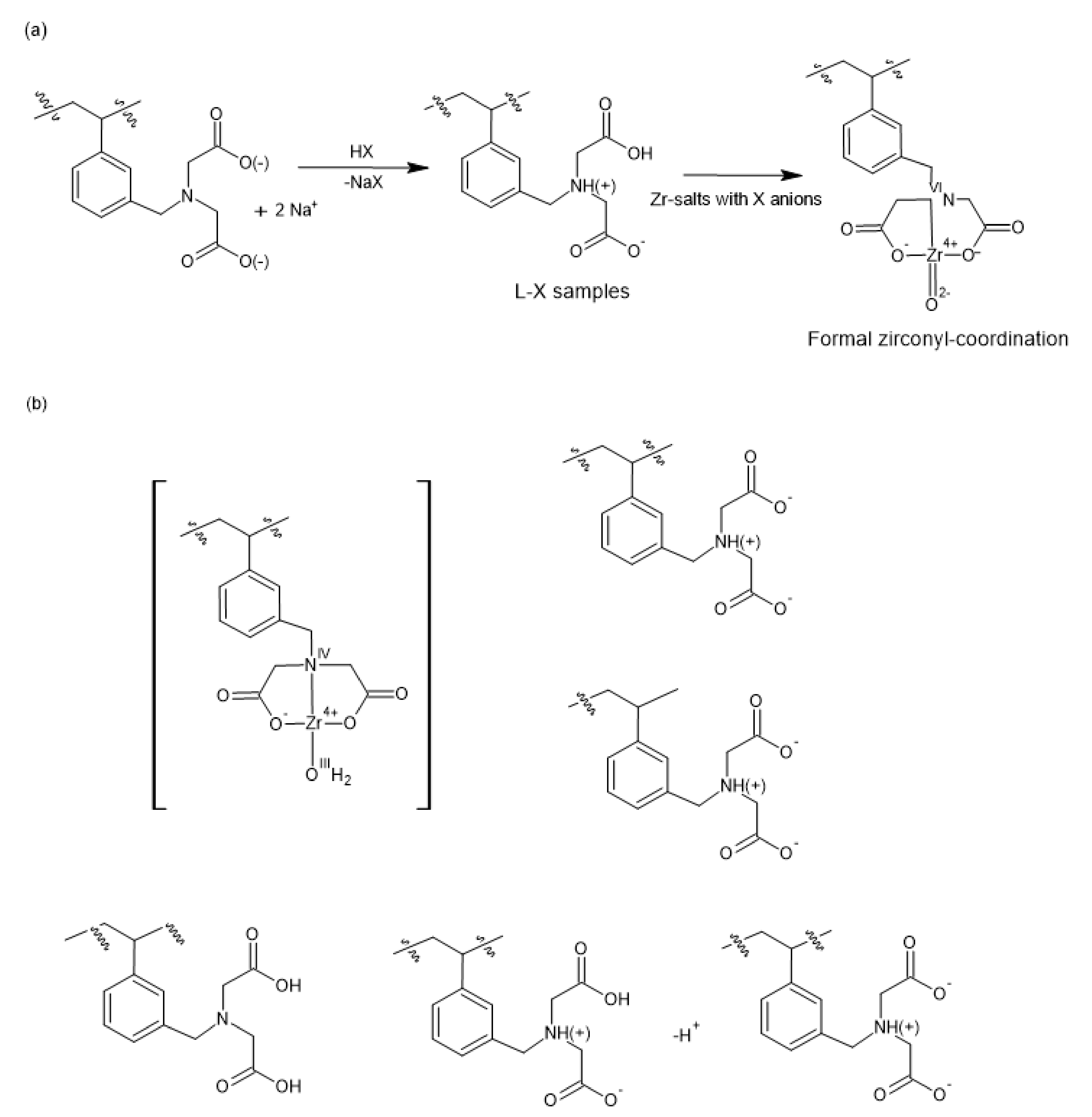
2.1. Preparation and Properties of Zr-Loaded Ion-Exchangers (L-Zr-X, X=Chloride (Cl), Nitrate (N) and Sulfate (S))
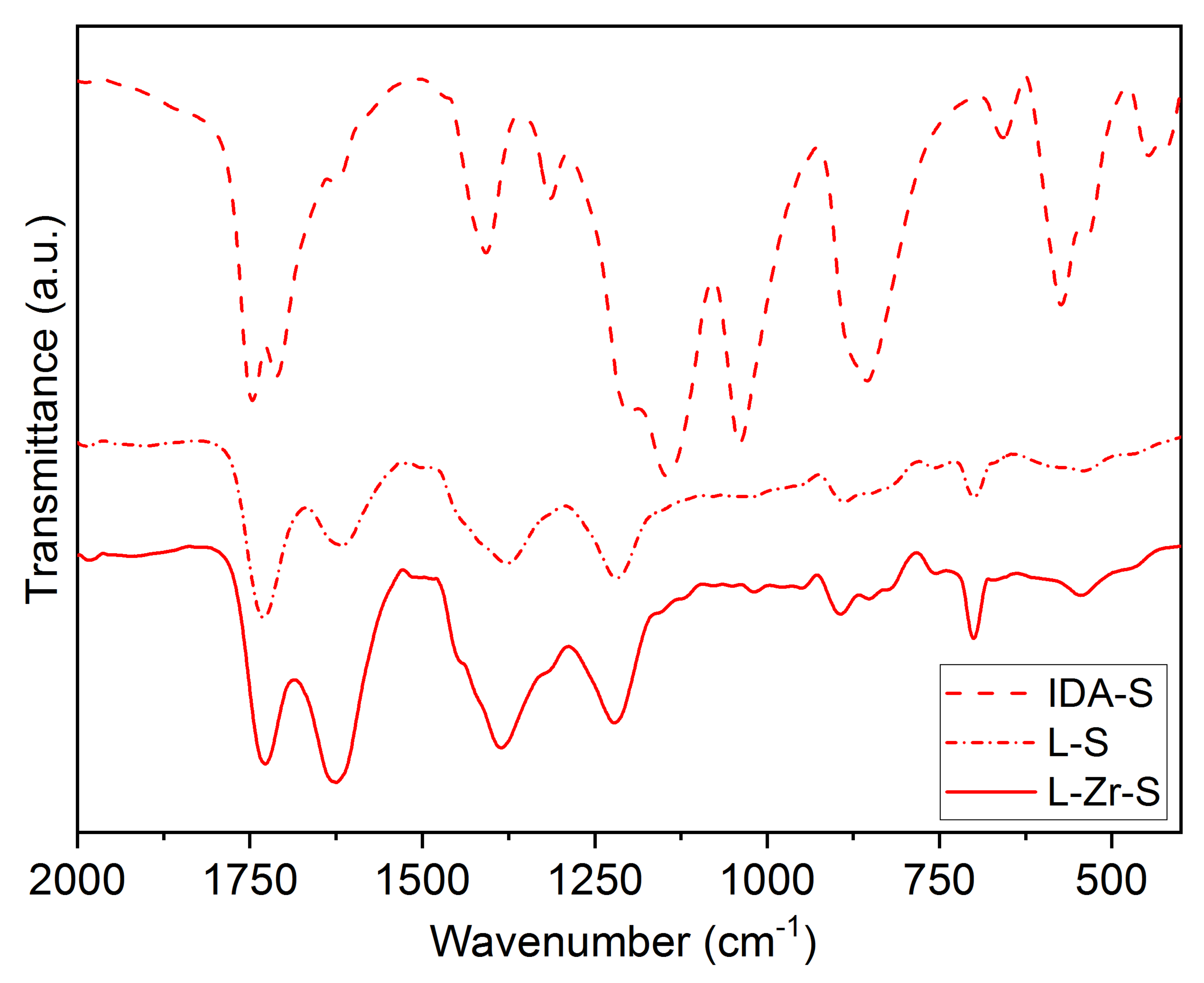
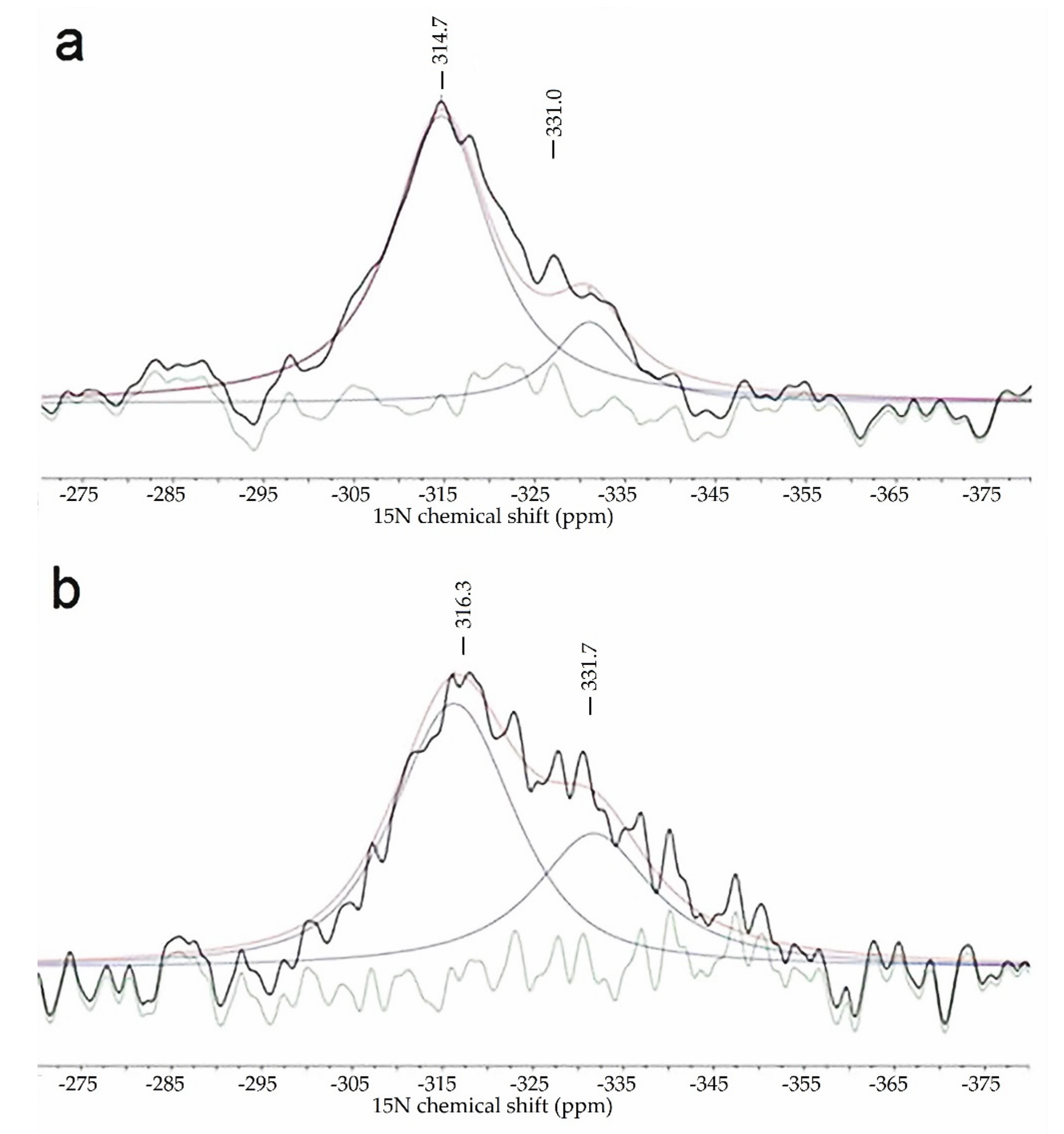
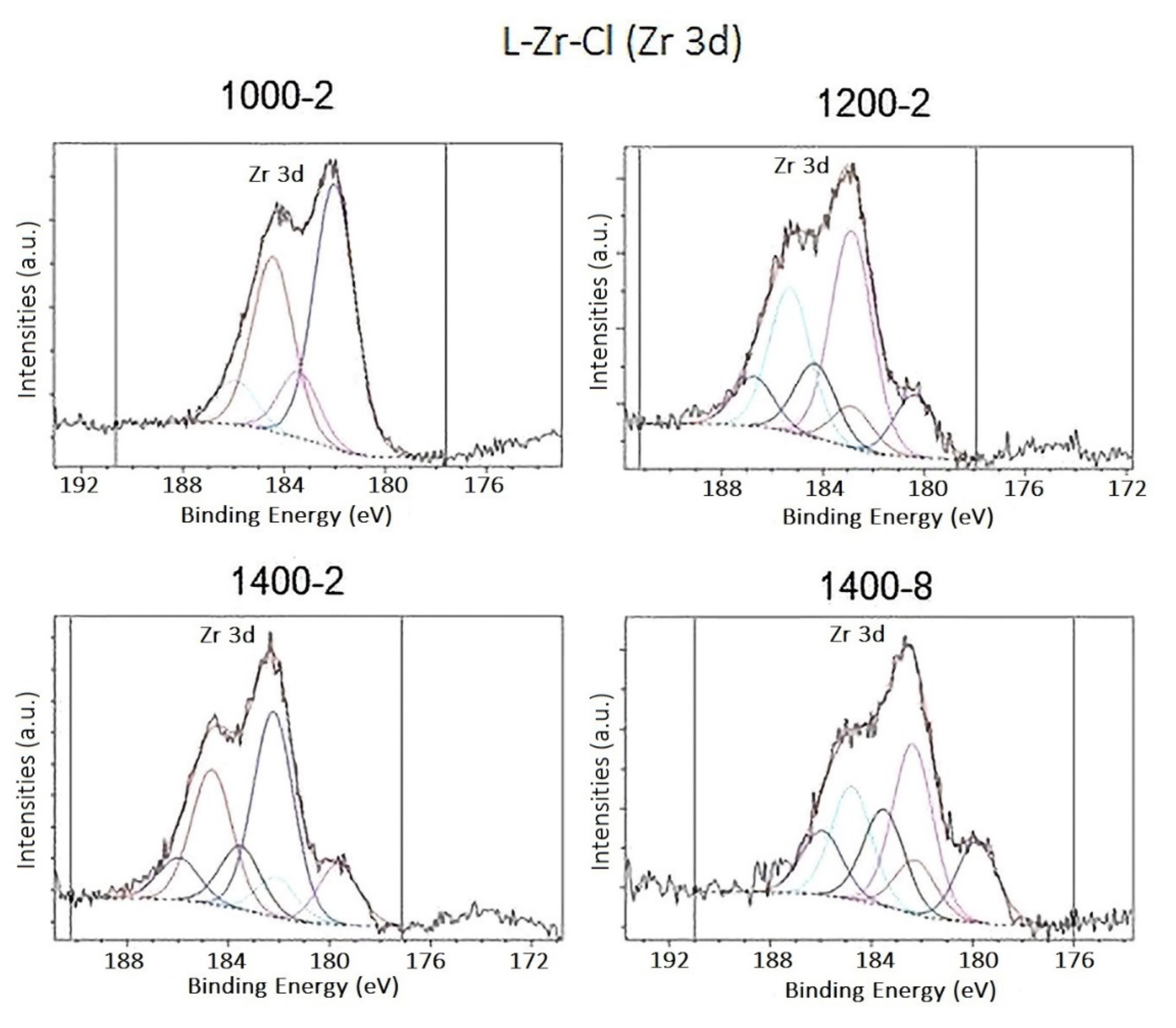
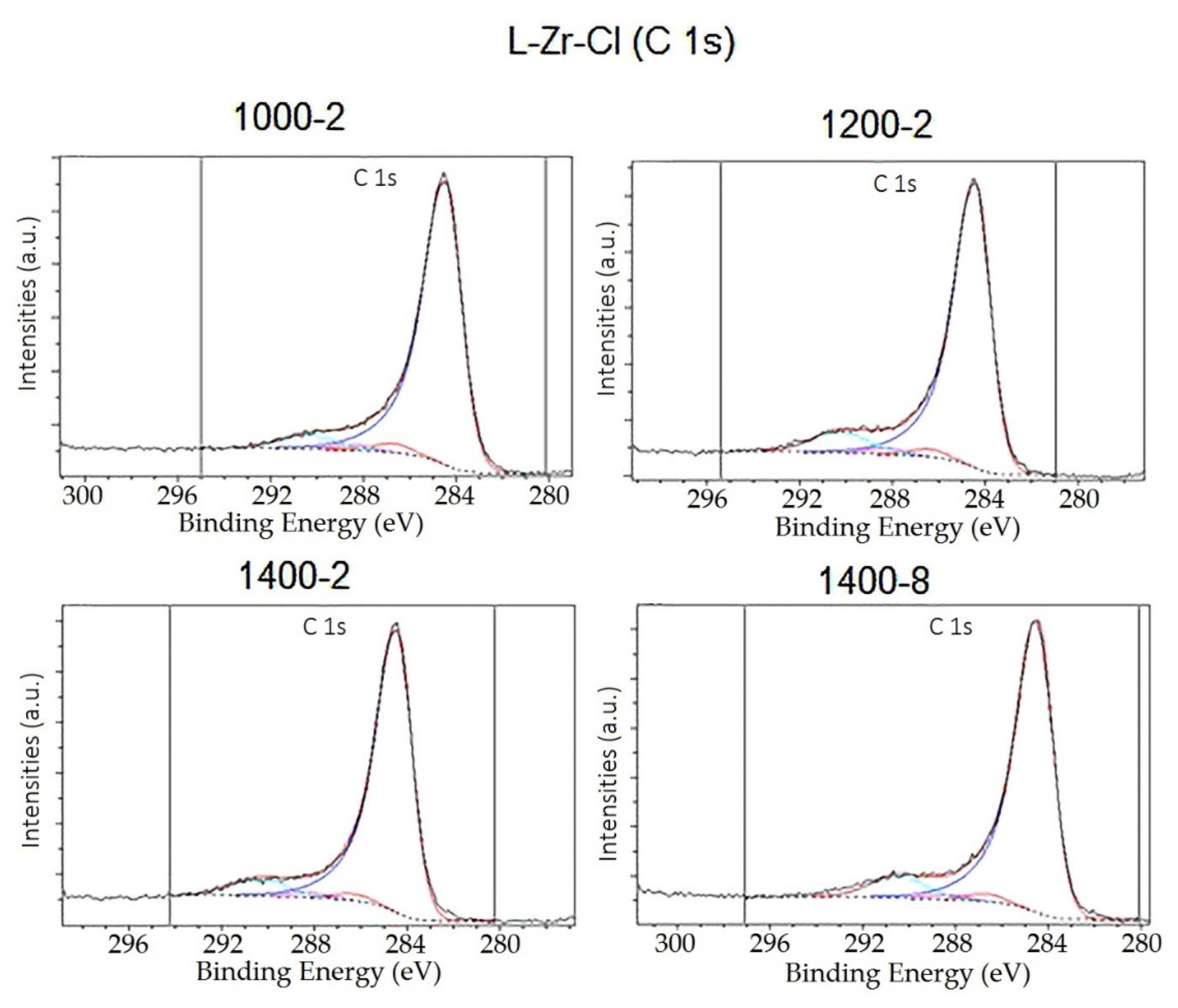
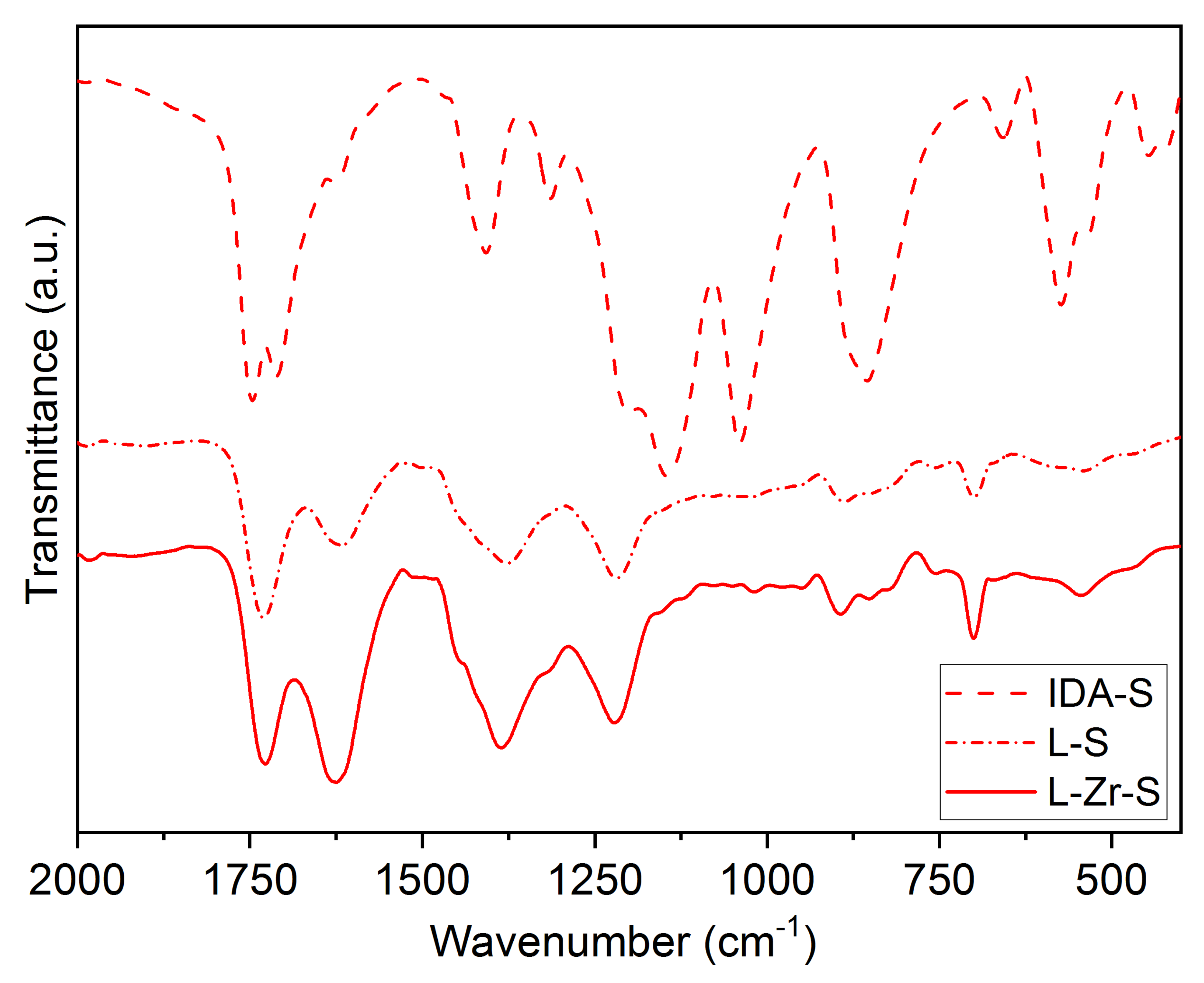
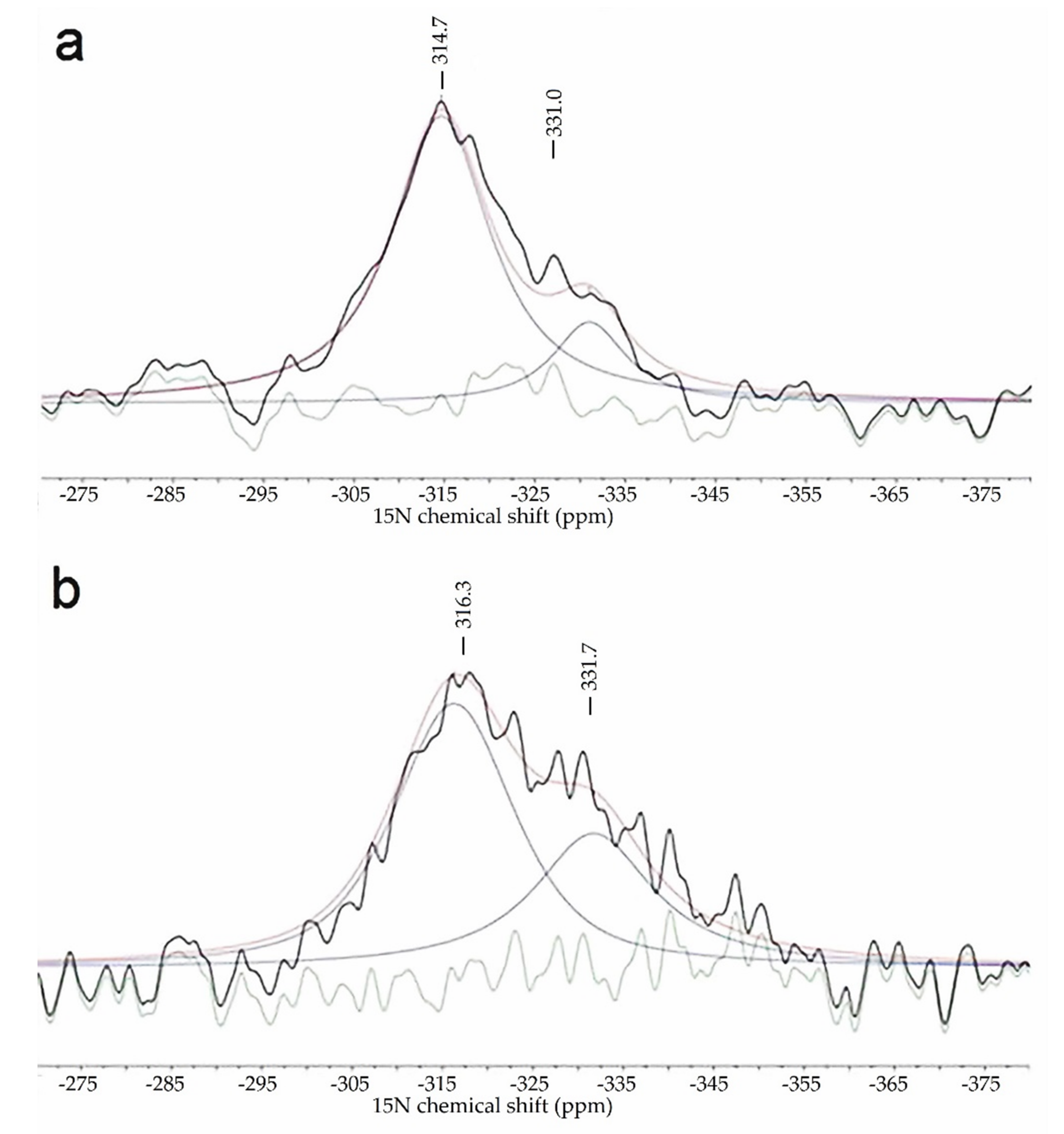
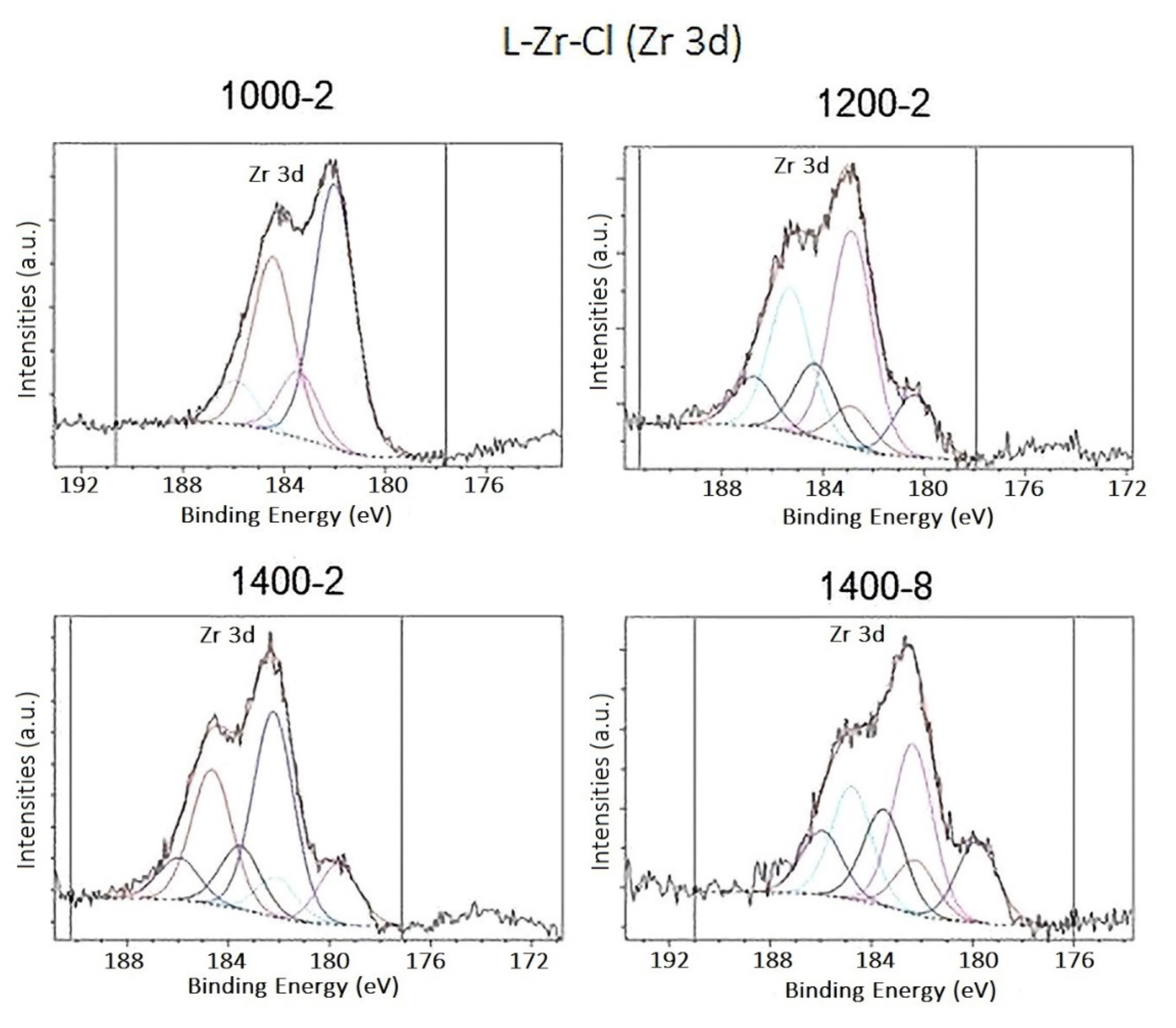
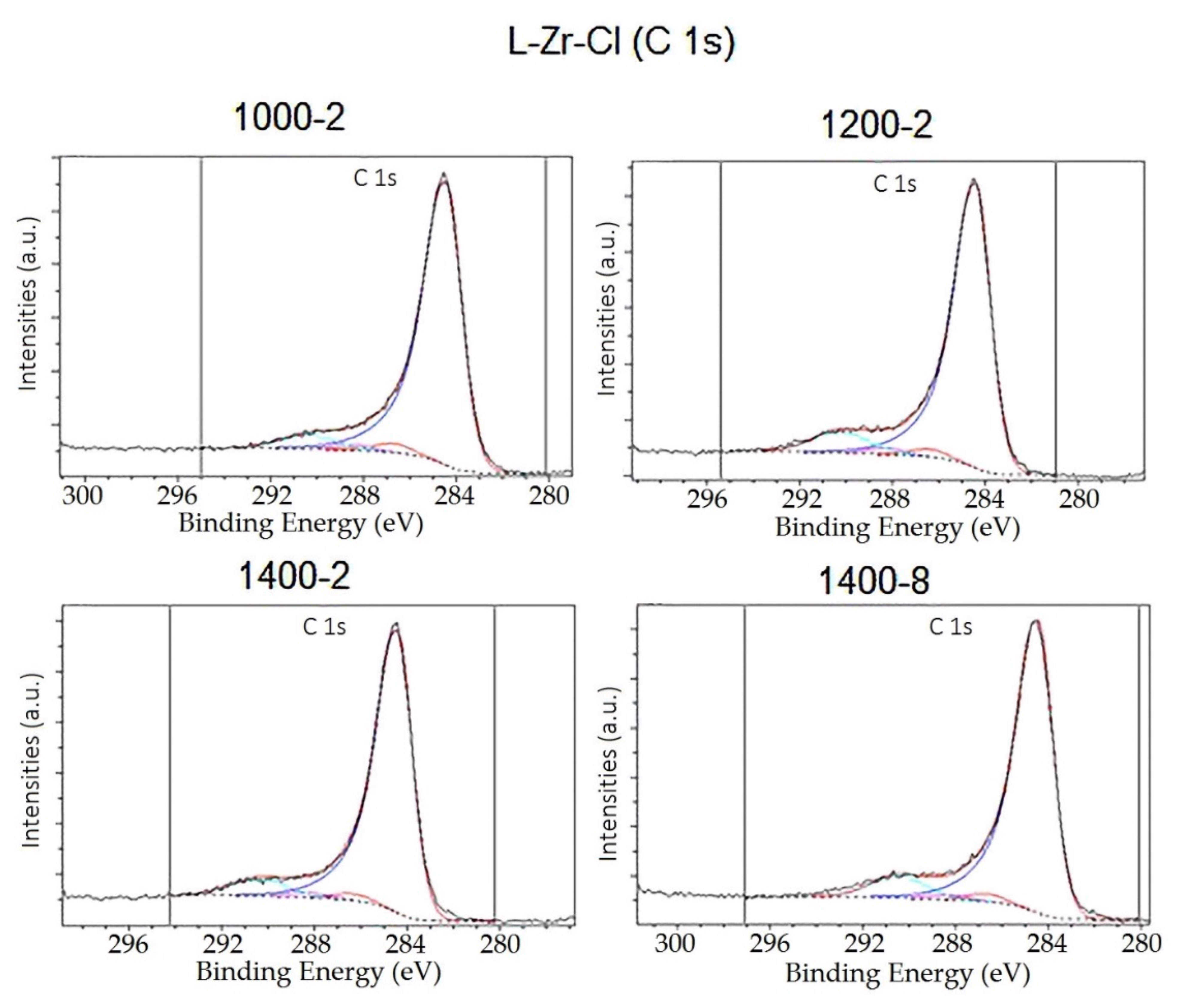
2.2. The Chemical Nature of Zirconium in the L-Zr-X Samples
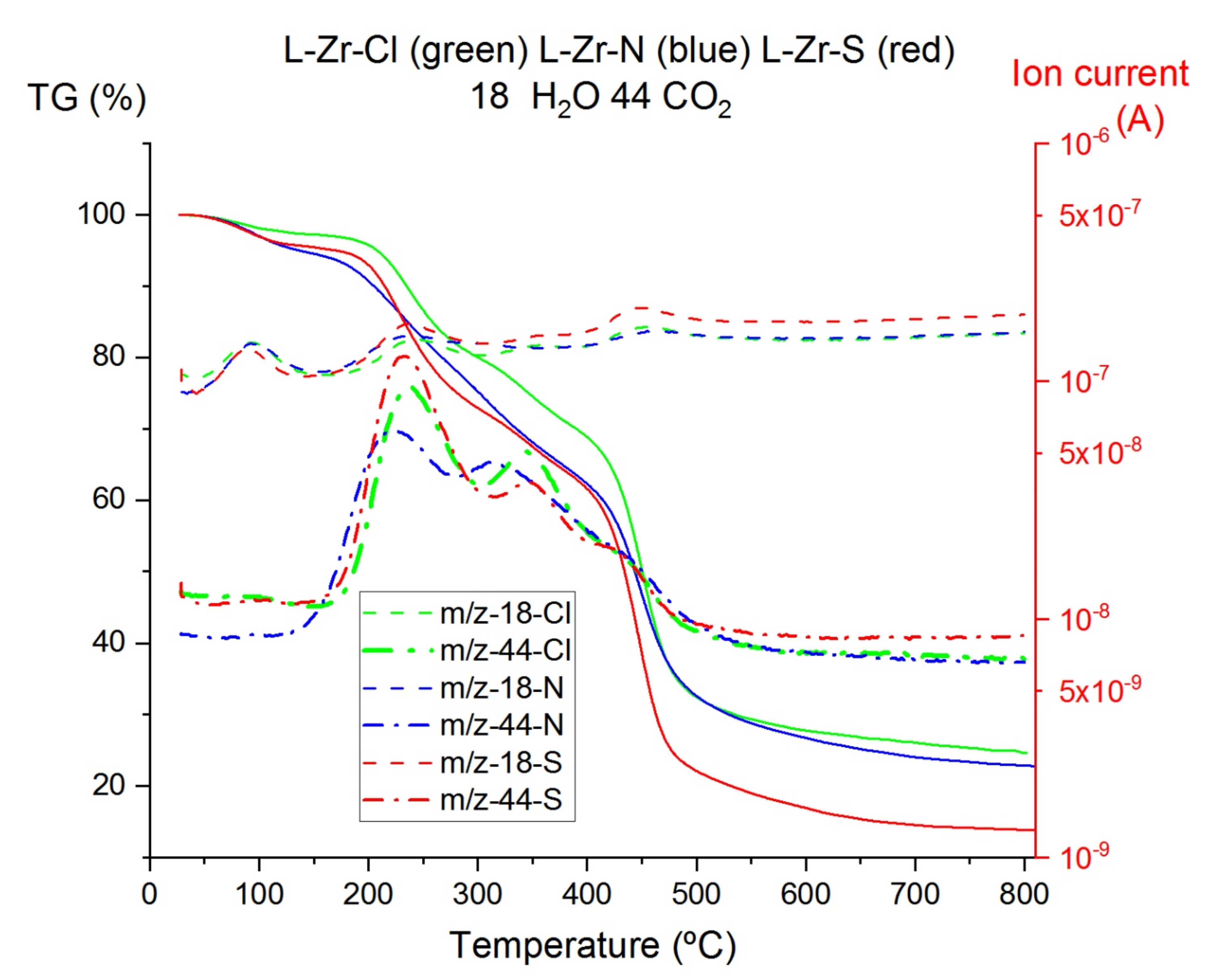
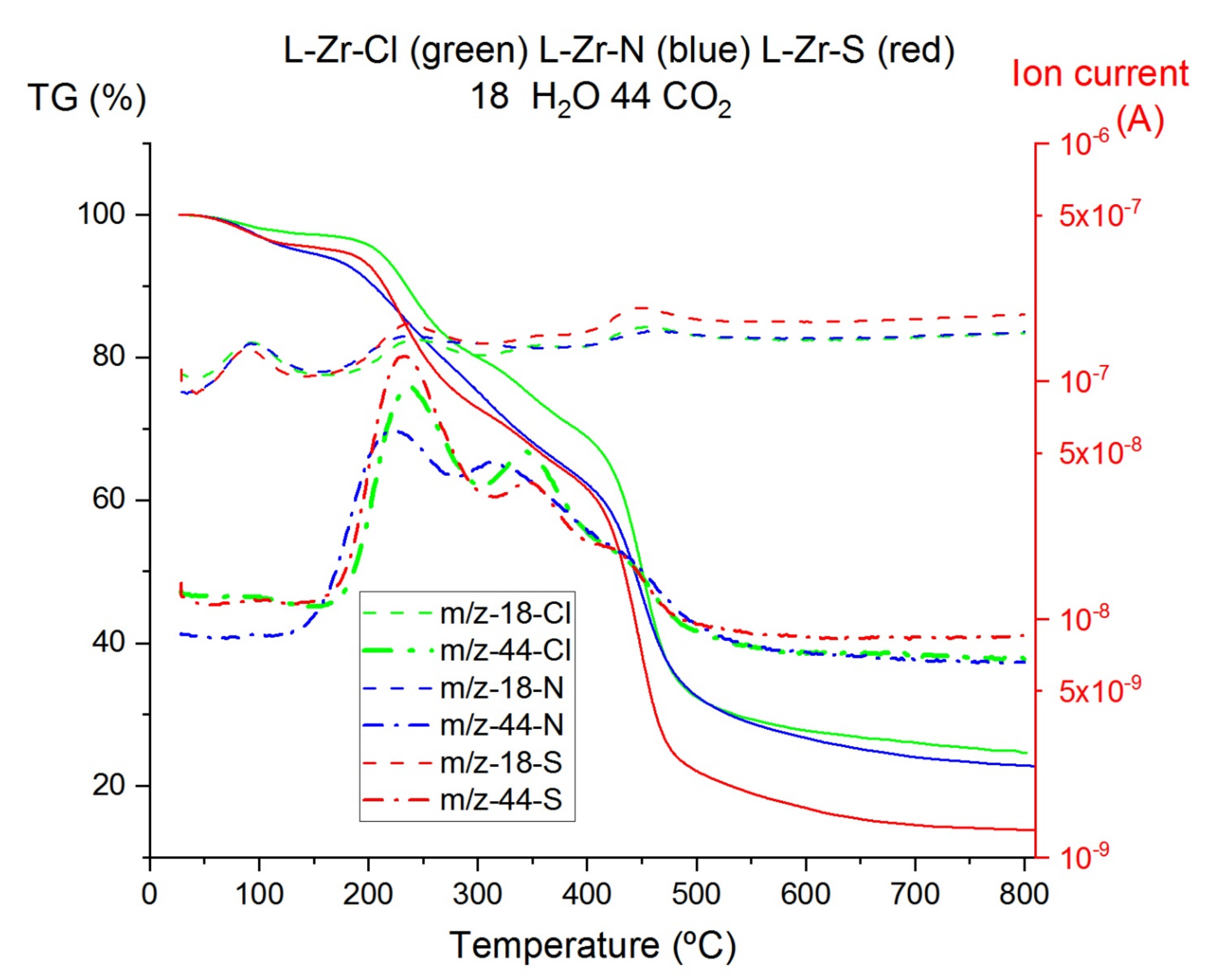
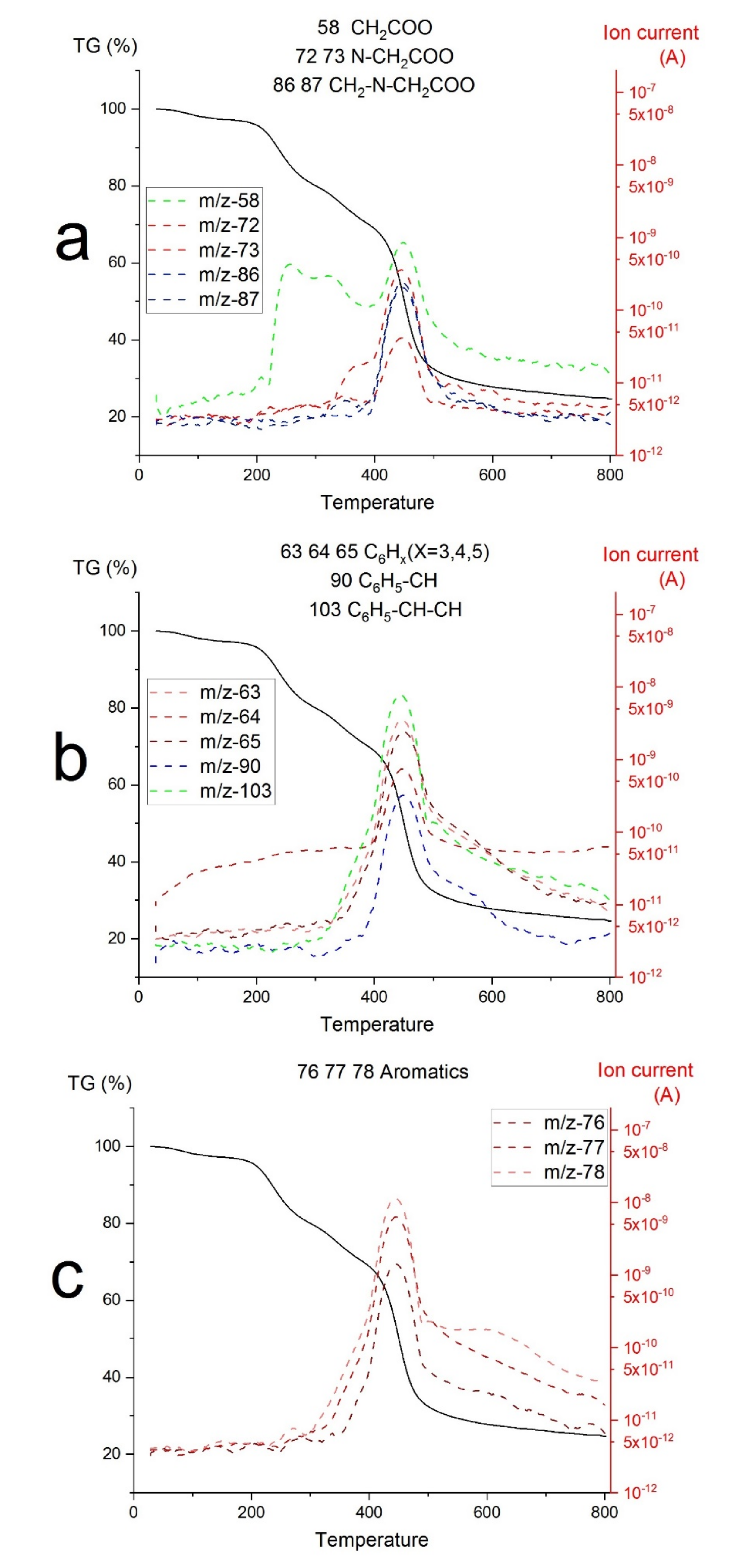
2.3. Thermal Studies on Compounds L-Zr-X
2.4. The Composition of Carbonization Products
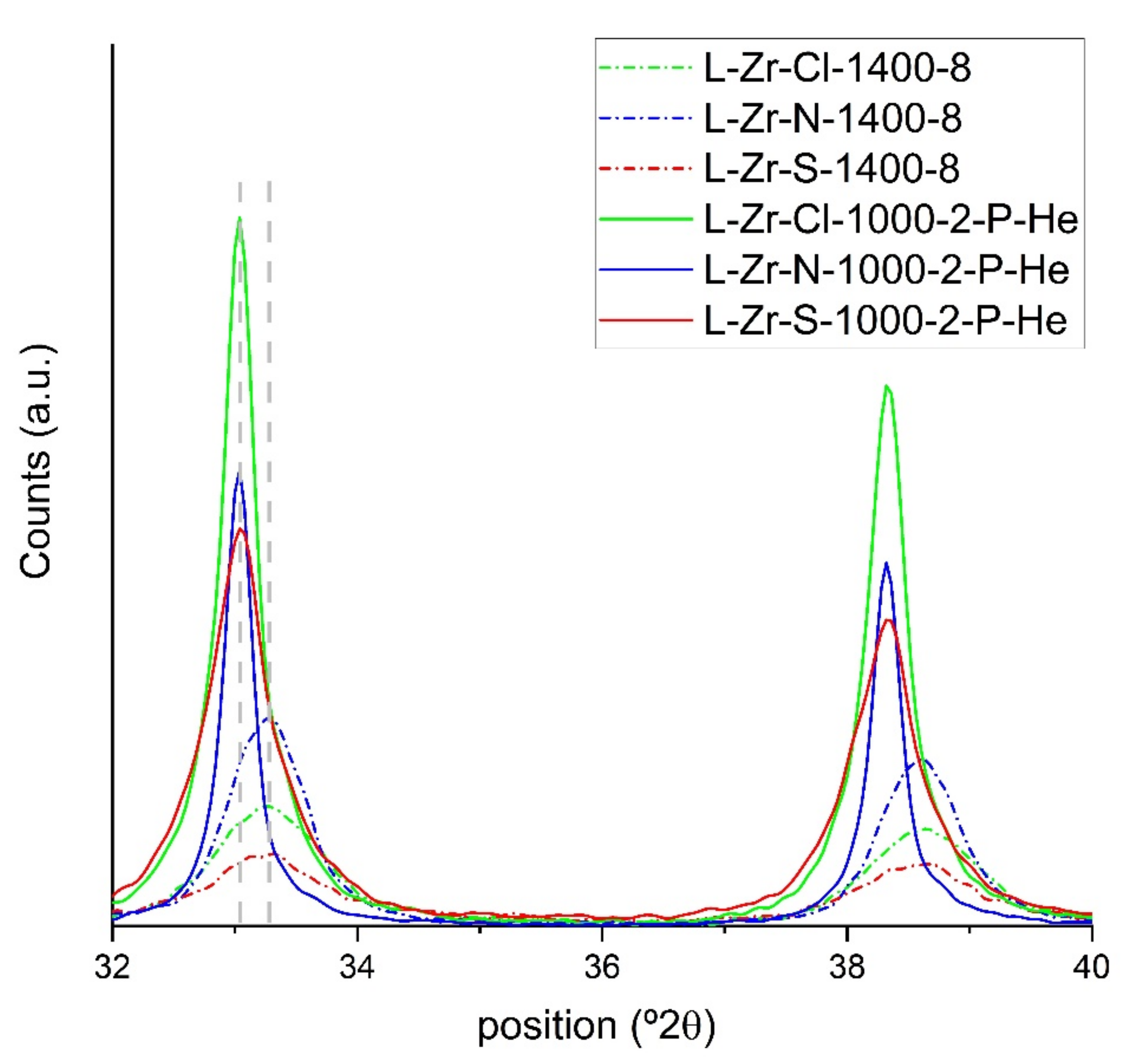
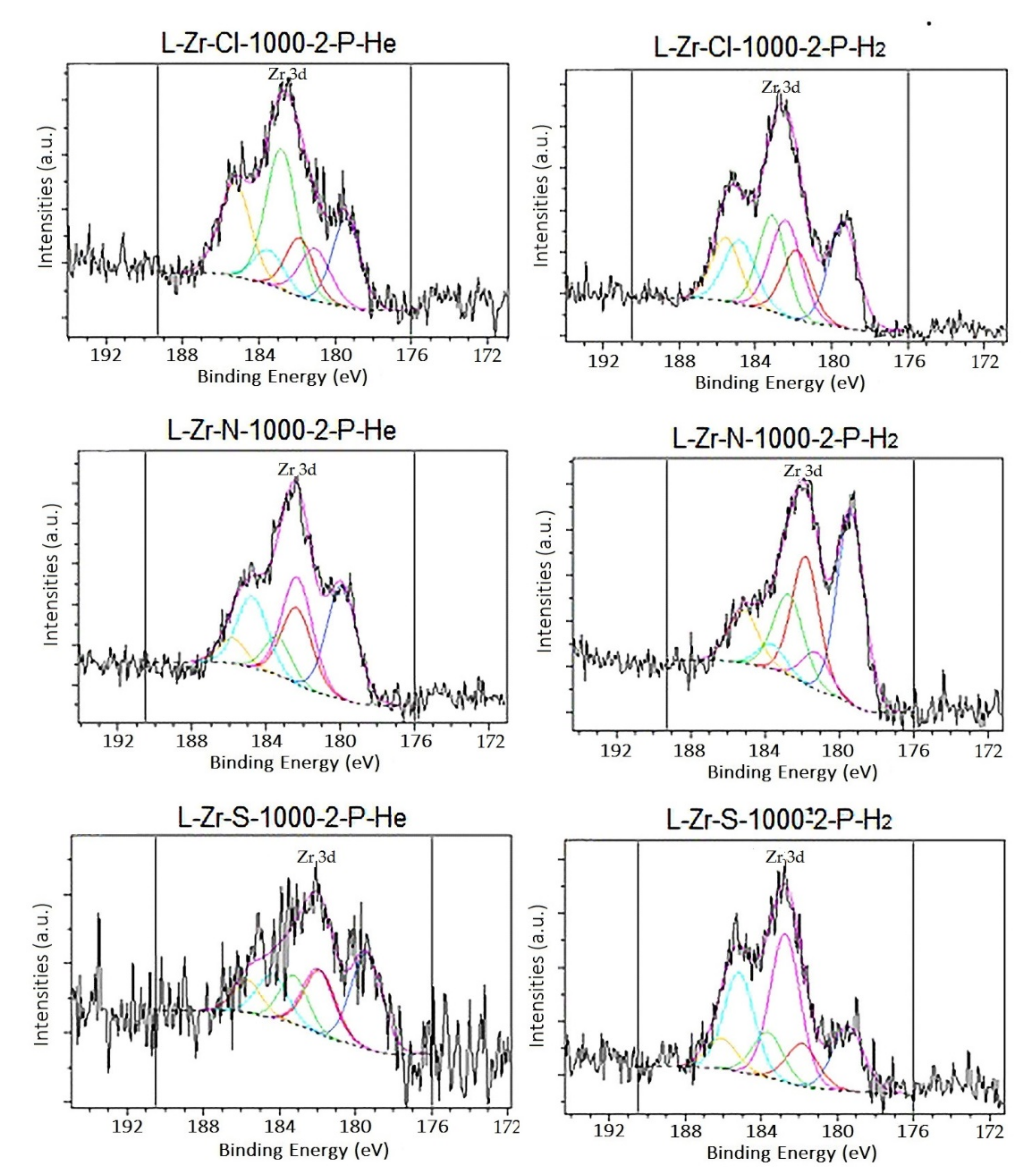
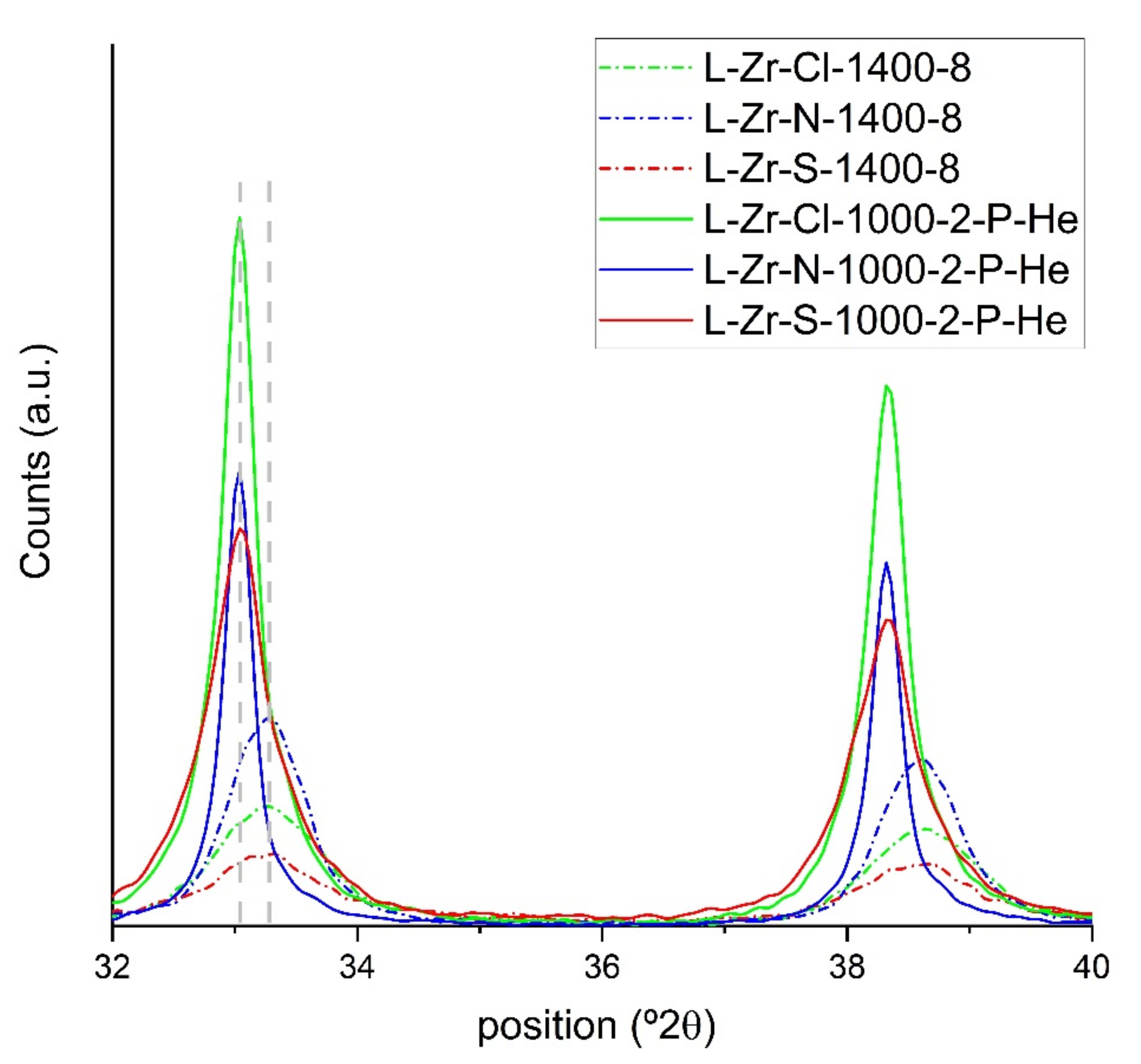
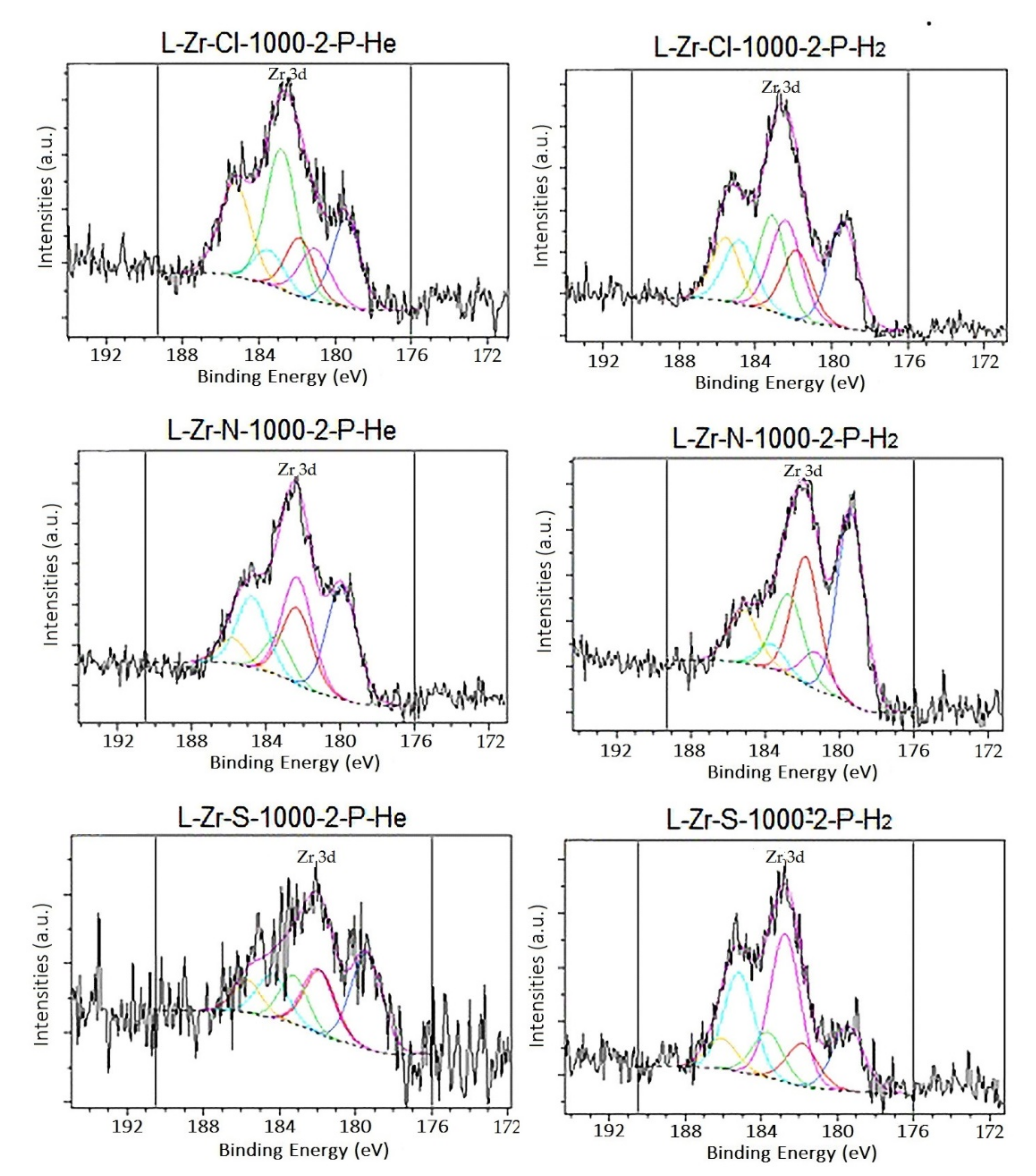
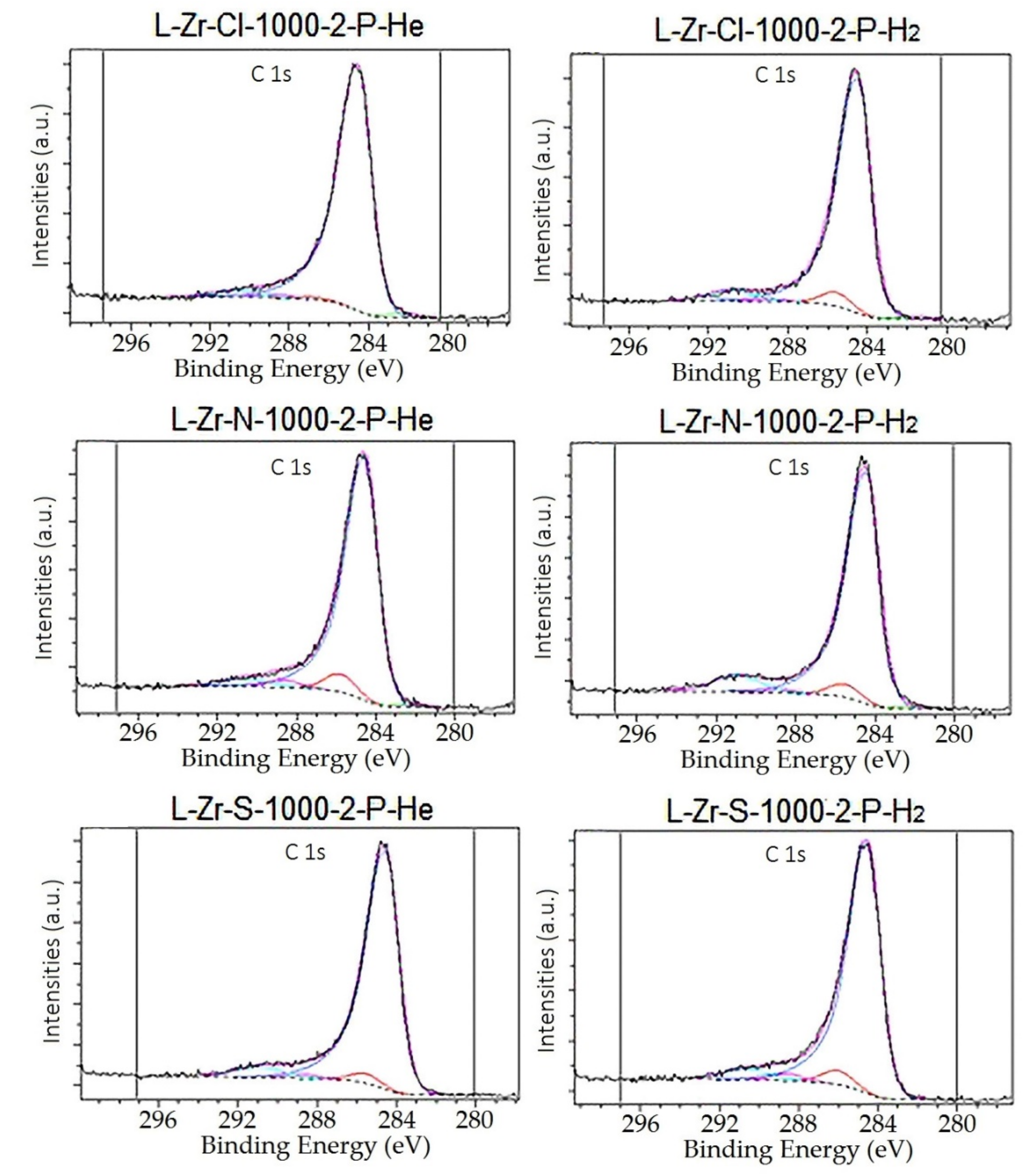
2.5. Surface Characterization of the Carbon Composites Containing Nano-ZrO2 and Nano-ZrC
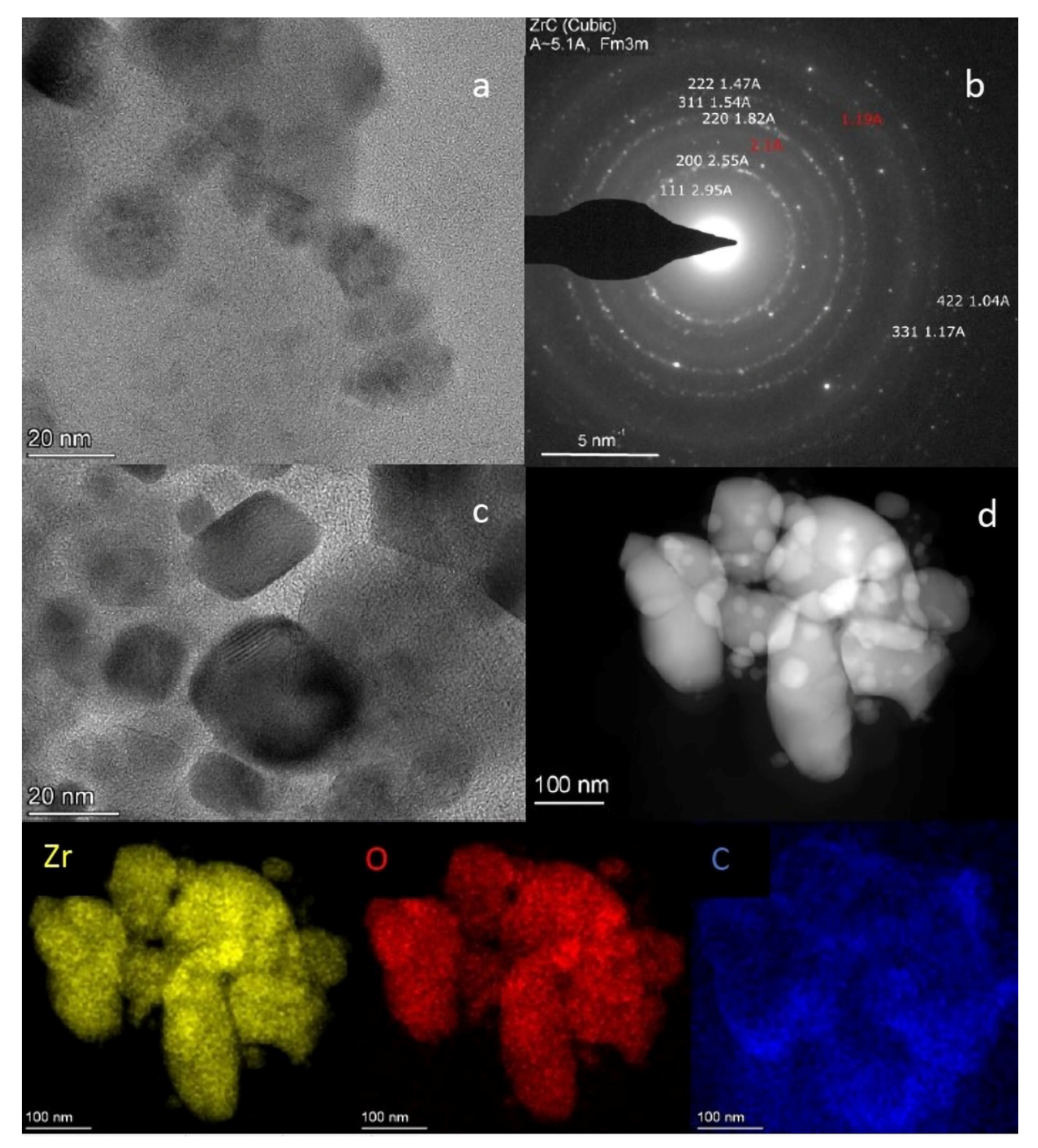
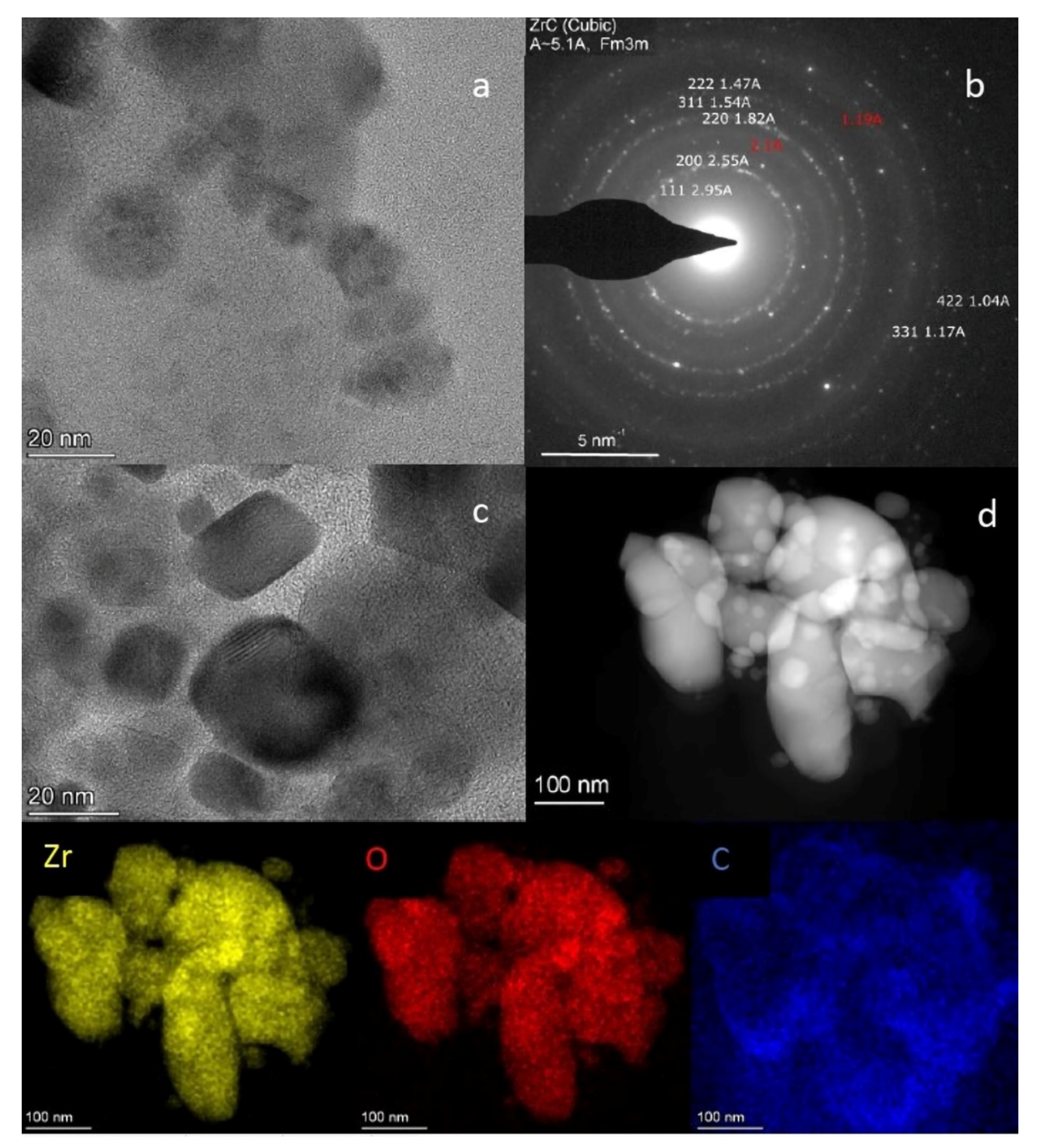
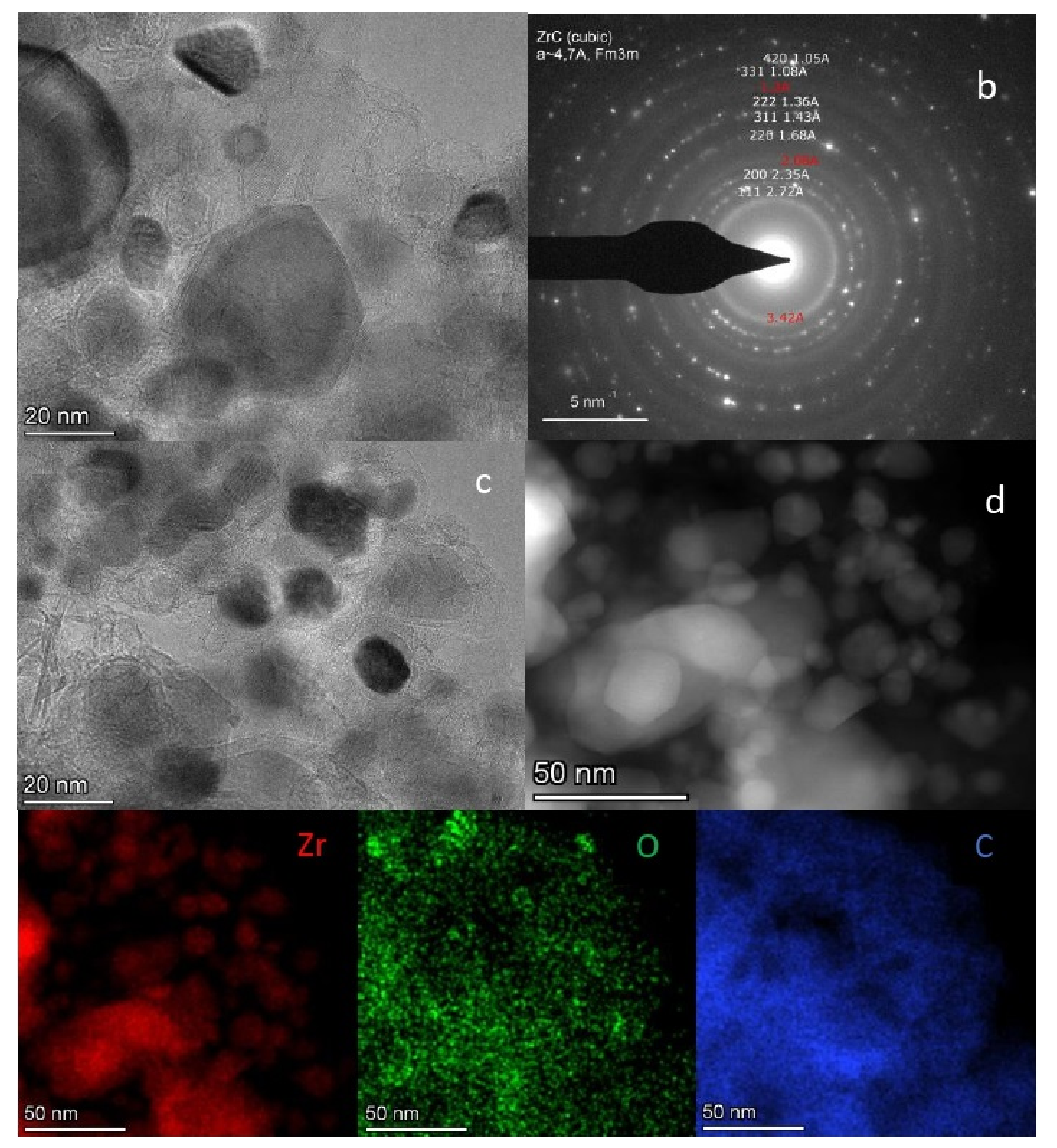
2.6. TEM Analysis of the L-Zr-S-1400-8 and L-Zr-N-1000-P-H2 Samples
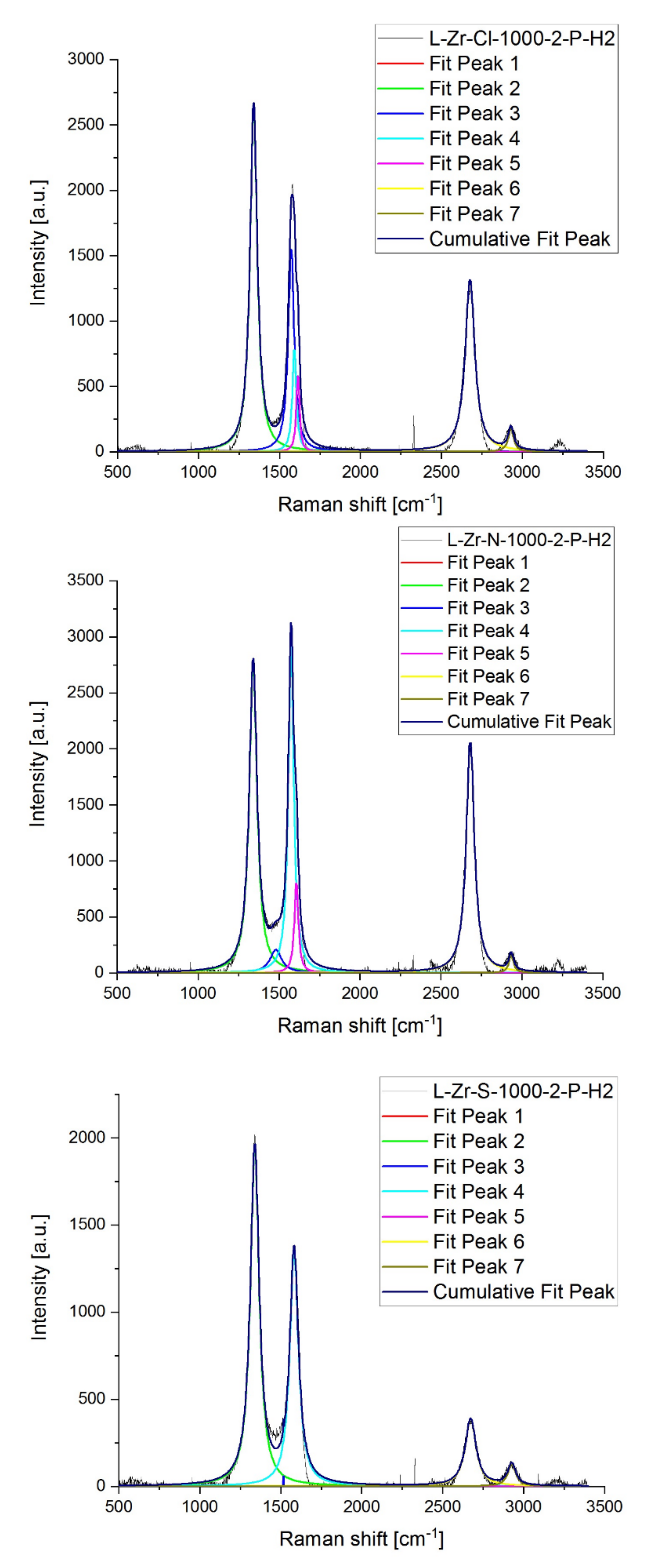
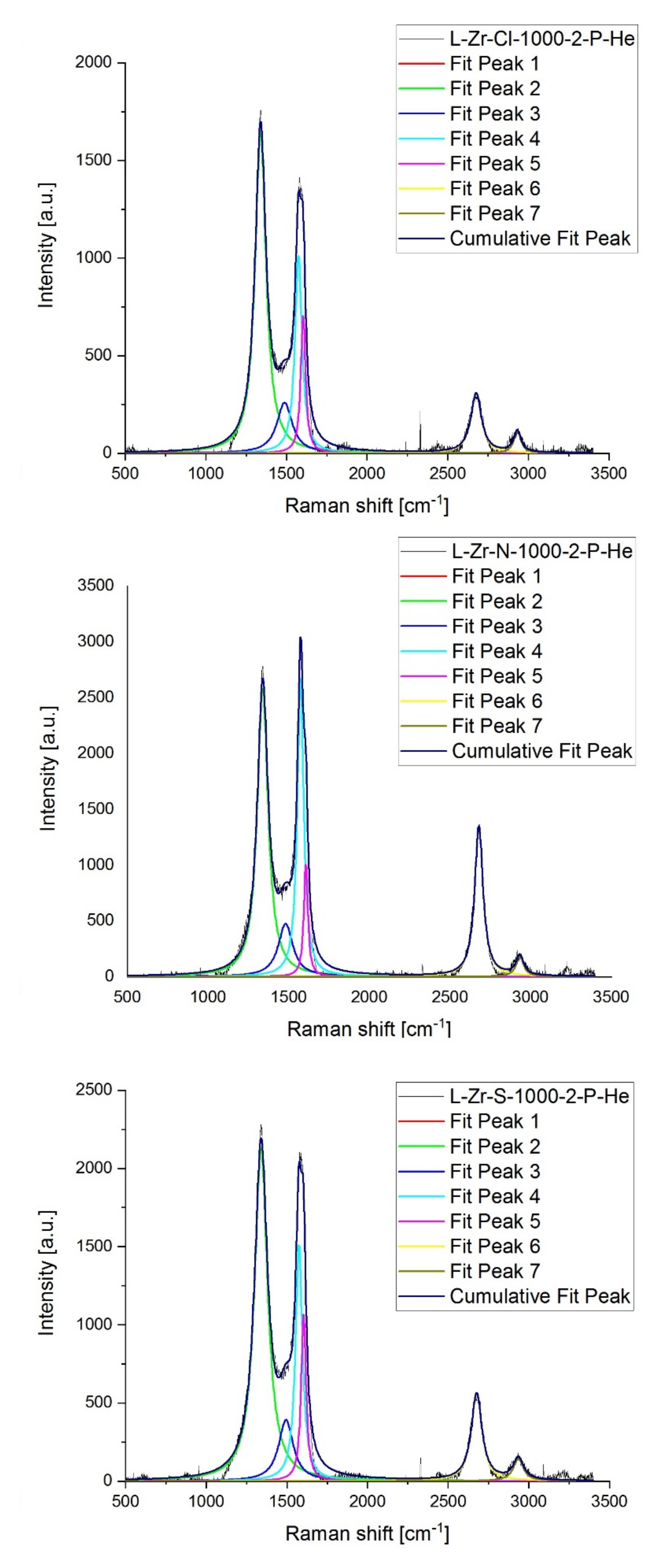
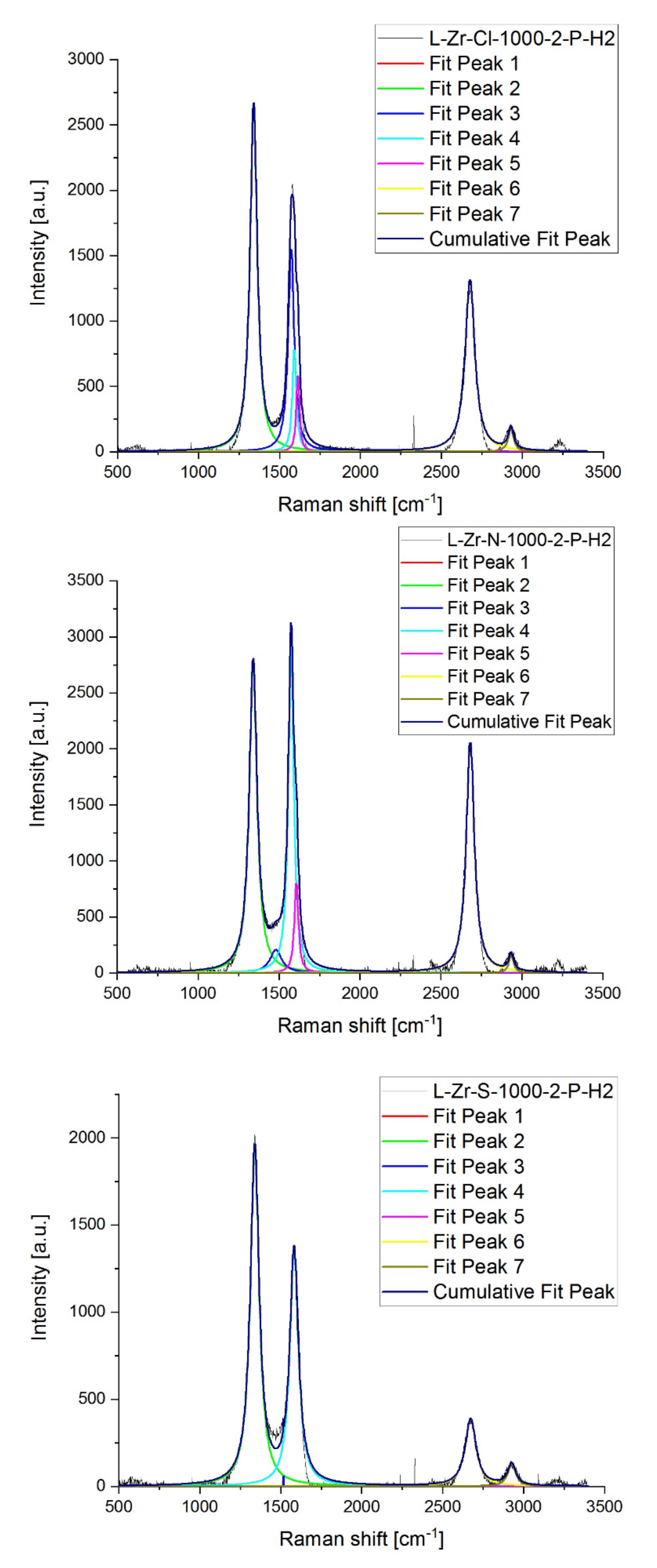
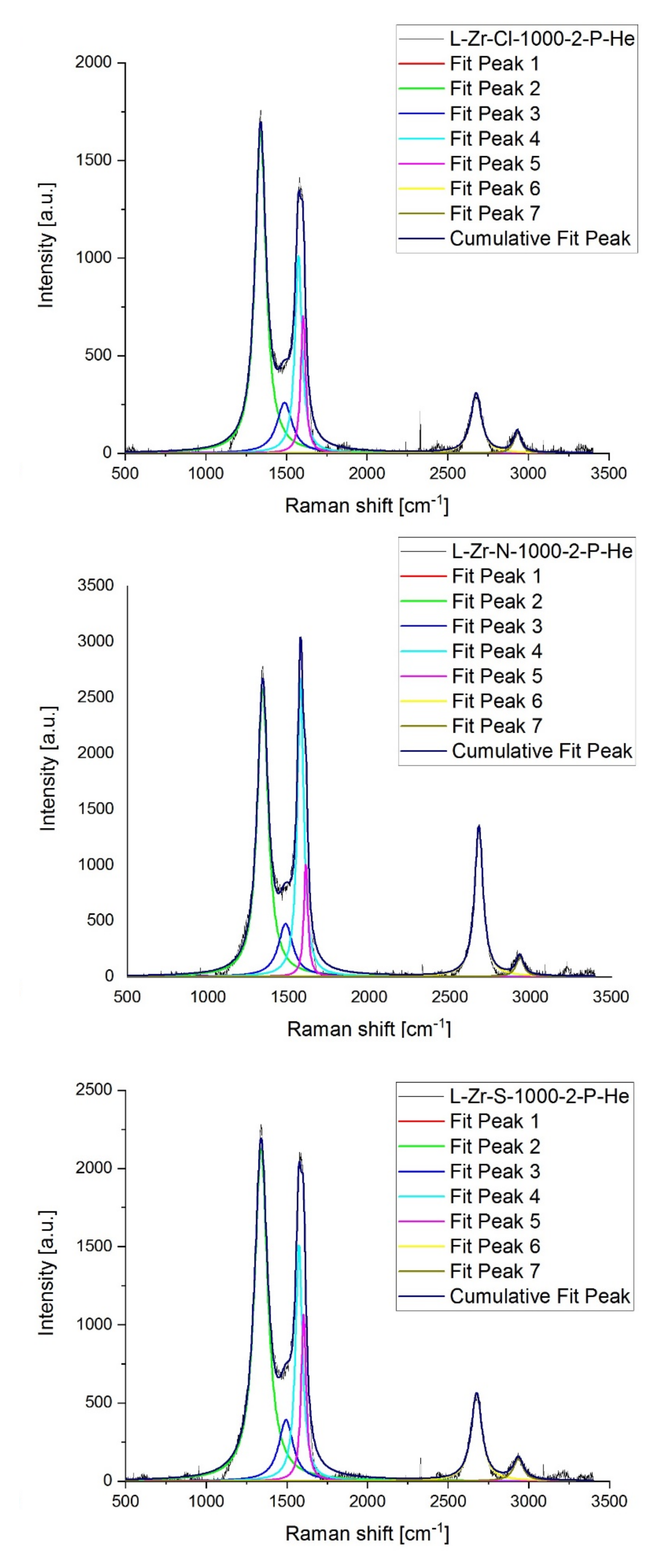
2.7. Raman Studies on ZrO2@C, ZrC@C and (ZrC,ZrO2)@C Composites
3. Materials and Methods
3.1. Experimental
3.1.1. Preparation of L-X-Zr Samples
3.1.2. Carbonization Experiments
3.1.3. RF Plasma Processing
3.1.4. Elemental Analysis
3.1.5. Specific Surface Area Measurements
3.1.6. Transmission Electron Microscopy
3.1.7. Thermal Studies
3.1.8. Powder X-ray Diffraction
3.1.9. Vibrational Spectroscopy
3.1.10. X-ray Photoelectron Spectroscopy
3.1.11. Solid-State NMR Measurements
4. Conclusions
- (1)
- Due to the low (2%) divinylbenzene content of the Zr-loaded iminodiacetate functional groups containing styrene-divinylbenzene (L-Zr-X) copolymers samples, a significant loss of the organic volatiles was observed during the carbonization reaction of the L-Zr-X samples, which resulted in high ZrO2/C and ZrC/C ratios in the precursor and product samples, respectively.
- (2)
- Changing the anion in the zirconium salt used to load the chelate-forming resin resulted in ZrO2@C precursors with varying reactivity levels towards carbonization and alterable Zr content.
- (3)
- ZrC0.54@C samples can be prepared with or without ZrO2 in a tubular furnace, with nanosize ZrC and ZrO2 content.
- (4)
- The RF plasma treatment of ZrO2@C samples led to ZrC0.94@C samples with high (75–95%) nanosized ZrC0.94 content and a pore volume of 0.20–0.26 cm3/g, with or without graphite content.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pierson, H.O. Handbook of Refractory Carbides and Nitrides: Properties, Characteristics, Processing and Applications, 1st ed.; Noyes Publication: Westwood, NJ, USA, 1997. [Google Scholar]
- Liu, G.; Cheng, L.; Li, K.; Chen, Z.; Xiong, X.; Luan, X. Damage behavior of atomic oxygen on zirconium carbide coating modified carbon/carbon composite. Ceram. Int. 2020, 3, 3324–3331. [Google Scholar] [CrossRef]
- Scales, N.; Chen, J.; Aughterson, R.D.; Karatchevtseva, I.; Stopic, A.; Lumpkin, G.R.; Luca, V. Porous ZrC-carbon microspheres as potential insoluble target matrices for production of 188W/188Re. J. Radioanal. Nucl. Chem. 2018, 318, 835–847. [Google Scholar] [CrossRef]
- Scales, N.; Chen, J.; Hanley, T.L.; Riley, D.P.; Lumpkin, G.R.; Luca, V. Hierarchically porous carbon–zirconium carbide spheres as potentially reusable transmutation targets. Microporous Mesoporous Mater. 2015, 212, 100–109. [Google Scholar] [CrossRef]
- Martiz, A.; Károly, Z.; Bódis, E.; Fazekas, P.; Mohai, M.; Bertóti, I.; Keszler, A.M. In-flight Synthesis of Nanosized ZrC Particles from Various Precursors in RF-Thermal Plasma. Period. Polytech.-Chem. 2021, 65, 331–342. [Google Scholar] [CrossRef]
- Cheng, W.; Campolongo, M.J.; Tan, S.J.; Luo, D. Freestanding ultrathin nano-membranes via self-assembly. Nano Today. 2009, 4, 482–493. [Google Scholar] [CrossRef]
- Byrappa, K.; Ohara, S.; Adschiri, T. Nanoparticles synthesis using supercritical fluid technology towards biomedical applications. Adv. Drug Deliv. Rev. 2008, 60, 32714. [Google Scholar] [CrossRef]
- Hussain, I.; Jalil, A.A.; Hamid, M.Y.S.; Hassan, N.S. Recent advances in catalytic systems in the prism of physicochemical properties to remediate toxic CO pollutants: A state-of-the-art review. Chemosphere 2021, 277, 130285. [Google Scholar] [CrossRef]
- Kocsis, T.; May, Z.; Czégény, Z.; Sreedhar, B.; Pawar, R.P.; Kótai, L. Perspectives of magnetic and nanosized metal-containing amorphous carbon composite chemosorbents and catalysts. Nano Stud. 2016, 14, 7–35. [Google Scholar]
- Dou, B.; Wang, C.; Song, Y.; Chen, H.; Jiang, B.; Yang, M.; Xu, Y. Solid sorbents for in-situ CO2 removal during sorption-enhanced steam reforming process: A review. Renew. Sust. Energy Rev. 2016, 53, 536–546. [Google Scholar] [CrossRef]
- Ciddor, L.; Bennett, J.A.; Hunns, J.A.; Wilson, K.; Lee, A.F. Catalytic upgrading of bio-oils by esterification. J. Chem. Technol. Biotechnol. 2015, 90, 780–795. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Cheng, R.; Liao, L.; Duan, X. High-performance thin-film electronics based on inorganic nanostructures and composites. Nano Today 2013, 8, 514–530. [Google Scholar] [CrossRef]
- Basumatary, S. Transesterification with heterogeneous catalyst in production of biodiesel: A review. J. Chem. Pharm. 2013, 5, 1–7. [Google Scholar]
- Fedorov, P.P.; Yarotskaya, E.G. Zirconium dioxide. Review. Kondens. Sredy Mezhfaznye Granitsy Condens. Matter Interphases 2021, 23, 169–187. [Google Scholar] [CrossRef]
- Dunlap, C.J.; Mcneff, C.V.; Stoll, D.; Carr, P.W. Zirconia stationary phases for extreme separations. Anal. Chem. 2001, 73, 599A–607A. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camps, M.; Chatzopoulos, M.; Camps, J.M.; Montheards, J.P. Chloromethylation of Polystyrenes and Styrene Copolymers. Applications. JMS-Rev. Macromol. Chem. Phys. 1987, 27, 505–557. [Google Scholar]
- Yuchi, A.; Yoshida, N. Adsorption of tetravalent metal ions chelating resins containing iminodiacetic acid groups. Bull. Chem. Soc. Jpn. 2000, 73, 1841–1842. [Google Scholar] [CrossRef]
- Auer, B.M.; Skinner, J.L. IR and Raman spectra of liquid water: Theory and interpretation. J. Chem. Phys. 2008, 128, 224511. [Google Scholar] [CrossRef]
- Ikawa, K.; Iwamoto, K. Coating microspheres with zirconium carbide-carbon alloy. J. Nucl. Mater. 1974, 52, 128–130. [Google Scholar] [CrossRef]
- Ikawa, K. Vapor deposition of zirconium carbide-carbon composites by the chloride process. JCOMA 1972, 29, 233–239. [Google Scholar] [CrossRef]
- González Vílchez, M.C.; Puerta Vizcaíno, M.F.; Gargallo, E. Thermal behavior of iminodiacetic acid and its disodium salt. Thermochim. Acta 1980, 42, 295–303. [Google Scholar] [CrossRef]
- Li, F.; Huang, X.; Zhang, G.J. Scalable foaming assisted synthesis of ZrC nanopowder by carbothermal reduction. Ceram Int. 2015, 41, 3335–3338. [Google Scholar] [CrossRef]
- Sacks, M.D.; Wang, C.A.; Yang, Z.; Jain, A. Carbothermal reduction synthesis of nanocrystalline zirconium carbide and hafnium carbide powders using solution-derived precursors. J. Mat. Sci. 2004, 39, 6057–6066. [Google Scholar] [CrossRef]
- Byun, H.S.; Burford, R.P.; Fane, A.G. Sulfonation of crosslinked asymmetric membranes based on polystyrene and divinylbenzene. J Appl. Polym. Sci. 1994, 52, 825–835. [Google Scholar] [CrossRef]
- Dobson, K.D.; McQuillan, A.J. An Infrared Spectroscopic Study of Carbonate Adsorption to Zirconium Dioxide Sol-Gel Films from Aqueous Solutions. Langmuir 1997, 13, 3392–3396. [Google Scholar] [CrossRef]
- Jiang, J.; May, I.; Sarsfield, M.J.; Ogden, M.; Fox, D.O.; Jones, C.J.; Mayhew, P. Spectroscopic Study of the Dissolution of Cesium Phosphomolybdate and Zirconium Molybdate by Ammonium Carbamate. J. Sol. Chem. 2005, 34, 443–468. [Google Scholar] [CrossRef]
- Fitzgerald, J.J.; Weiss, R.A. Cation-Anion and Cation-Cation Interactions in Sulfonated Polystyrene Ionomers Spectroscopic Studies of the Effects of Solvents. ACS 1986, 302, 35–53. [Google Scholar]
- El-NahhalI, I.M.; Zaggout, F.R.; Nassar, M.A.; El-Ashgar, N.M.; Maquet, J.; Babonneau, F.; Chehimi, M. Synthesis, Characterization and Applications of Immobilized Iminodiacetic Acid-Modified Silica. J Sol-Gel Sci. Technol. 2003, 28, 255–265. [Google Scholar] [CrossRef]
- Jiang, J.; Renshaw, J.C.; Sarsfield, M.J.; Livens, F.R.; Collison, D.; Charnock, J.M.; Eccles, H. Solution Chemistry of Uranyl Ion with Iminodiacetate and Oxydiacetate: A Combined NMR/EXAFS and Potentiometry/Calorimetry Study. Inorg. Chem. 2003, 42, 1233–1240. [Google Scholar] [CrossRef]
- Busca, G.; Lorenzelli, V. Infrared spectroscopic identification of species arising from reactive adsorption of carbon oxides on metal oxide surfaces. Mater. Chem. 1982, 7, 89–126. [Google Scholar] [CrossRef]
- Kawashiro, K.; Morimoto, S.; Yoshida, H. Mass spectra of butyl esters and N-trifluoroacetyl butyl esters of some iminodicarboxylic acids upon electron impact. Bull. Chem. Soc. Jpn. 1984, 57, 1097–1103. [Google Scholar] [CrossRef] [Green Version]
- Fazekas, P.; Czégény, Z.; Mink, J.; Bódis, E.; Klébert, S.; Németh, C.; Keszler, A.M.; Károly, Z.; Szépvölgyi, J. Decomposition of poly(vinyl chloride) in inductively coupled radiofrequency thermal plasma. Chem. Eng. J. 2016, 302, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Komarov, V.S.; Yatsevskaya, M.I.; Sycheva, O.A. Properties of activated carbon produced from spent ion-exchange resins. Dokl. Akad. Nauk. BSSR 1985, 29, 1010–1023. [Google Scholar]
- Chinthaka Silva, G.W.; Kercher, A.A.; Hunn, J.D.; Martin, R.C.; Jellison, G.E.; Meyer, H.M. Characterization of zirconium carbides using electron microscopy; optical anisotropy; Auger depth profiles, X-ray diffraction and electron density calculated by charge flipping method. J. Solid State Chem. 2012, 194, 91–99. [Google Scholar] [CrossRef]
- Gusev, A.I. Structural stability boundaries for nonstoichiometric compounds. Phys. Status Solidi A 1989, 111, 443–450. [Google Scholar] [CrossRef]
- Senczyk, D. The lattice parameters of MeC-type carbides; nitrides and carbonitrides of transition metals of IV and V groups of the periodic system. In Proceedings of the 11th Conference of Applied Crystallography, Kozubnik, Poland; 1984; Volume 1, pp. 234–240. [Google Scholar]
- Martiz, A.; Károly, Z.; Trif, L.; Mohai, M.; Bereczki, L.; Németh, P.; Molnár, Z.; Menyhárd, A.; Pawar, R.P.; Tekale, S.; et al. Plasma-assisted preparation of nano-(ZrC; ZrO2)@carbon composites from Zr-loaded sulfonated styrene–divinylbenzene copolymers. J. Therm. Anal. Calorim. 2022, 1–13, in press. [Google Scholar] [CrossRef]
- Pechishcheva, N.V.; Shunyaev, K.Y.; Melchakova, O.V. Zirconium in modern analytical chemistry. Rev. Anal. Chem. 2018, 37, 1–26. [Google Scholar] [CrossRef]
- Reilman, R.F.; Msezane, A.; Manson, S.T. Relative intensities in photoelectron spectroscopy of atoms and molecules. J. Electron. Spectr. Relat. Phenom. 1976, 8, 389–394. [Google Scholar] [CrossRef]
- Martiz, A.; Farkas, A.; Karoly, Z.; Franguelli, F.P.; Samaniego, S.K.; Menyhard, A.; Kotai, L. Raman studies on carbon-containing phases in nanosized-ZrO2/C and nanosized-(ZrC; ZrO2)/C composites. Nano Stud. 2022, 10, 21–22. [Google Scholar]
- Devia, D.H.; Sykes, A.G. Aqueous solution chemistry of zirconium(IV). 1. Kinetic studies on hydrogen ion and general acid (HX) induced dissociations of the tetrameric ion [Zr4(OH)8(H2O)16]8+. Inorg. Chem. 1981, 20, 910–913. [Google Scholar] [CrossRef]
- Lokshin, E.P.; Tareeva, O.A. Sorption of Zirconium from Nitrate and Sulfate Solutions. Theor. Found. Chem. Eng. 2019, 53, 688–692. [Google Scholar] [CrossRef]
- Clearfield, A. Structural aspects of zirconium chemistry. Rev. Pure Appl. Chem. 1964, 14, 91–108. [Google Scholar]
- Fathy, M.; Moghny, T.A.; Awadallah, A.E.; El-Bellihi, A.H. Nano Composites of Polystyrene Divinyl Benzene Resin Based on Oxidized Multi-Walled Carbon Nanotubes. Int. J. Mod. Org. Chem. 2013, 2, 67–80. [Google Scholar]
- Shishlov, N.M.; Khurasan, S.L. Effect of ion interactions on the IR spectrum of benzenesulfonate ion. Restoration of sulfonate ion symmetry in sodium benzenesulfonate dimer. J. Mol. Struct. 2016, 1123, 360–366. [Google Scholar]
- De, R.; Lee, H.; Das, B. Exploring the interactions in binary mixtures of polyelectrolytes: Influence of mixture composition; concentration; and temperature on counter-ion condensation. J. Mol. Liq. 2018, 251, 94–99. [Google Scholar] [CrossRef]
- Claramunt, S.; Varea, A.; Lopez-Diaz, D.; Velazquez, M.; Cornet, A.; Cirera, A. The Importance of Interbands on the Interpretation of the Raman Spectrum of Graphene Oxide. J. Phys. Chem. C 2015, 119, 10123–10129. [Google Scholar] [CrossRef]
- Vollebregt, S.; Ishihara, R.; Tichelaar, F.D.; Hou, Y.; Beenakker, C.I.M. Influence of the growth temperature on the first and secondorder Raman band ratios and widths of carbon nanotubes and fibers. Carbon 2012, 50, 3542–3554. [Google Scholar] [CrossRef]
- Cuesta, A.; Dhamelincourt, P.; Laureyns, J. Raman Microprobe studies on carbon materials. Carbon 1994, 8, 1523–1532. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Label | Description of Sample |
|---|---|
| L-X | H-form of iminodiacetate-functionalized STY-DVB copolymers containing 2 mass% DVB. |
| L-X-Zr | Zirconium-loaded iminodiacetate-functionalized STY-DVB copolymer samples containing 2 mass% DVB with ZrOX2 (X=Cl NO3) and Zr(SO4)2 (X=S) salts. |
| L-X-Zr-T-t | Samples obtained by carbonizing L-X-Zr resins at the given T temperature and for a carbonization time of t. |
| L-Zr-X-1000-2-P-H2/He | Plasma-treated samples from L-Zr-X-1000-2 samples containing ZrO2@C in a H2 or He atmosphere. |
| Composite | ZrC, (wt%) (Size in nm) | ZrO2 (wt%) (Size in nm) | Carbon (wt%) | BET Surface Area (m2/g) |
|---|---|---|---|---|
| L-Zr-Cl-1000-2 | - | 65 (5) Ortho/tetra = 1.36 | 35 | 140 |
| L-Zr-Cl-1200-2 | 75 (10) | 15 (11) tetra | 10 | 311 |
| L-Zr-Cl-1400-2 | 80 (10) | 10 (12) tetra | 10 | 310 |
| L-Zr-Cl-1400-8 | 85 (14) | 5 (26) tetra/mono = 1.92 | 10 | 245 |
| L-Zr-N-1000-2 | - | 80(5) ortho/tetra = 0.77 | 20 | 278 |
| L-Zr-N-1200-2 | 65 (10) | 25 (7) tetra | 10 | 447 |
| L-Zr-N-1400-2 | 95 (17) | - | 5 | 430 |
| L-Zr-N-1400-8 | 95 (18) | - | 5 | 438 |
| L-Zr-S-1000-2 | - | 10 (8) | 90 | 33 |
| L-Zr-S-1200-2 | 50 (8) | 10 (8) ortho/tetra = 0.26 | 40 | 41 |
| L-Zr-S-1400-2 | 30(13) | 15 (8) tetra | 55 | 54 |
| L-Zr-S-1400-8 | 50 (56) | 5 (31) mono/cubic = 1.05 | 55 | 35 |
| Sample | ZrC, (wt%) (Size in nm) | Carbon (wt%) | BET Surface Area (m2/g) | Total Pore Volume (cm3/g) |
|---|---|---|---|---|
| L-Cl-1000-2-P-He | 95 (49) | 5 | 50 | 0.21 |
| L-Cl-1000-2-P-H2 | 90 (40) | 10 | 62 | 0.26 |
| L-N-1000-2-P-He | 95 (62) | 5 | 52 | 0.22 |
| L-N-1000-2-P-H2 | 90 (35) | 15 | 82 | 0.22 |
| L-S-1000-2-P-He | 80 (40) | 20 | 52 | 0.20 |
| L-S-1000-2-P-H2 | 75 (42) | 20 | 58 | 0.22 |
| Sample | D * | D | D” | G | D’ | 2D | D + D’ | ID/IG | I2D/IG | |
|---|---|---|---|---|---|---|---|---|---|---|
| L-Zr-Cl-1000-2-P-H2 | R shift [cm−1] | 1100 | 1340 | 1572 | 1592 | 1614 | 2676 | 2929 | ||
| Intensity | 0 | 2650 | 1552 | 784 | 585 | 1313 | 173 | 3.38 | 1.68 | |
| FWHM | 0 | 53.53 | 44.38 | 29.40 | 23.47 | 71.72 | 40.98 | |||
| L-Zr-Cl-1000-2-P-He | R shift [cm−1] | 1100 | 1339 | 1488 | 1574 | 1603 | 2675 | 2930 | ||
| Intensity | 0 | 1647 | 259 | 1012 | 705 | 305 | 110 | 1.63 | 0.30 | |
| FWHM | 0 | 81.88 | 115.5 | 52.17 | 35.59 | 79.04 | 54.11 | |||
| L-Zr-N-1000-2-P-H2 | R shift [cm−1] | 1100 | 1340 | 1480 | 1574 | 1606 | 2680 | 2933 | ||
| Intensity | 0 | 2762 | 206 | 2907 | 802 | 2070 | 157 | 0.95 | 0.71 | |
| FWHM | 0 | 57.92 | 88.54 | 40.51 | 29.73 | 60.06 | 35.56 | |||
| L-Zr-N-1000-2-P-He | R shift [cm−1] | 1100 | 1339 | 1481 | 1574 | 1607 | 2679 | 2933 | ||
| Intensity | 0 | 2581 | 475 | 2670 | 1004 | 1343 | 178 | 0.97 | 0.50 | |
| FWHM | 0 | 80.57 | 109.4 | 46.94 | 30.00 | 62.47 | 57.14 | |||
| L-Zr-S-1000-2-P-H2 | R shift [cm−1] | 1100 | 1339 | 1517 | 1582 | 2078 | 2671 | 2925 | ||
| Intensity | 0 | 1943 | 60.55 | 1349 | 0 | 387 | 127 | 1.44 | 0.29 | |
| FWHM | 0 | 65.45 | 0.113 | 64.27 | 0 | 79,9 | 59.33 | |||
| L-Zr-S-1000-2-P-He | R shift [cm−1] | 1100 | 1340 | 1494 | 1575 | 1604 | 2676 | 2934 | ||
| Intensity | 0 | 2128 | 392 | 1511 | 1068 | 558 | 143 | 1.41 | 0.37 | |
| FWHM | 0 | 97.33 | 109.5 | 51.36 | 35.97 | 80.13 | 75.83 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martiz, A.; Károly, Z.; Domján, A.; Mohai, M.; Bereczki, L.; Trif, L.; Farkas, A.; László, K.; Menyhárd, A.; Kótai, L. Nano-ZrO2@C, Nano-(ZrC, ZrO2)@C and Nano-ZrC@C Composites Prepared by Plasma-Assisted Carbonization of Zr-Loaded Iminodiacetate-Functionalized Styrene-Divinylbenzene Copolymers. Inorganics 2022, 10, 77. https://doi.org/10.3390/inorganics10060077
Martiz A, Károly Z, Domján A, Mohai M, Bereczki L, Trif L, Farkas A, László K, Menyhárd A, Kótai L. Nano-ZrO2@C, Nano-(ZrC, ZrO2)@C and Nano-ZrC@C Composites Prepared by Plasma-Assisted Carbonization of Zr-Loaded Iminodiacetate-Functionalized Styrene-Divinylbenzene Copolymers. Inorganics. 2022; 10(6):77. https://doi.org/10.3390/inorganics10060077
Chicago/Turabian StyleMartiz, Alejandro, Zoltán Károly, Attila Domján, Miklós Mohai, Laura Bereczki, László Trif, Attila Farkas, Krisztina László, Alfréd Menyhárd, and László Kótai. 2022. "Nano-ZrO2@C, Nano-(ZrC, ZrO2)@C and Nano-ZrC@C Composites Prepared by Plasma-Assisted Carbonization of Zr-Loaded Iminodiacetate-Functionalized Styrene-Divinylbenzene Copolymers" Inorganics 10, no. 6: 77. https://doi.org/10.3390/inorganics10060077
APA StyleMartiz, A., Károly, Z., Domján, A., Mohai, M., Bereczki, L., Trif, L., Farkas, A., László, K., Menyhárd, A., & Kótai, L. (2022). Nano-ZrO2@C, Nano-(ZrC, ZrO2)@C and Nano-ZrC@C Composites Prepared by Plasma-Assisted Carbonization of Zr-Loaded Iminodiacetate-Functionalized Styrene-Divinylbenzene Copolymers. Inorganics, 10(6), 77. https://doi.org/10.3390/inorganics10060077