
New Ferrocene-Based Metalloligand with Two Triazole Carboxamide Pendant Arms and Its Iron(II) Complex: Synthesis, Crystal Structure, 57Fe Mössbauer Spectroscopy, Magnetic Properties and Theoretical Calculations


Abstract
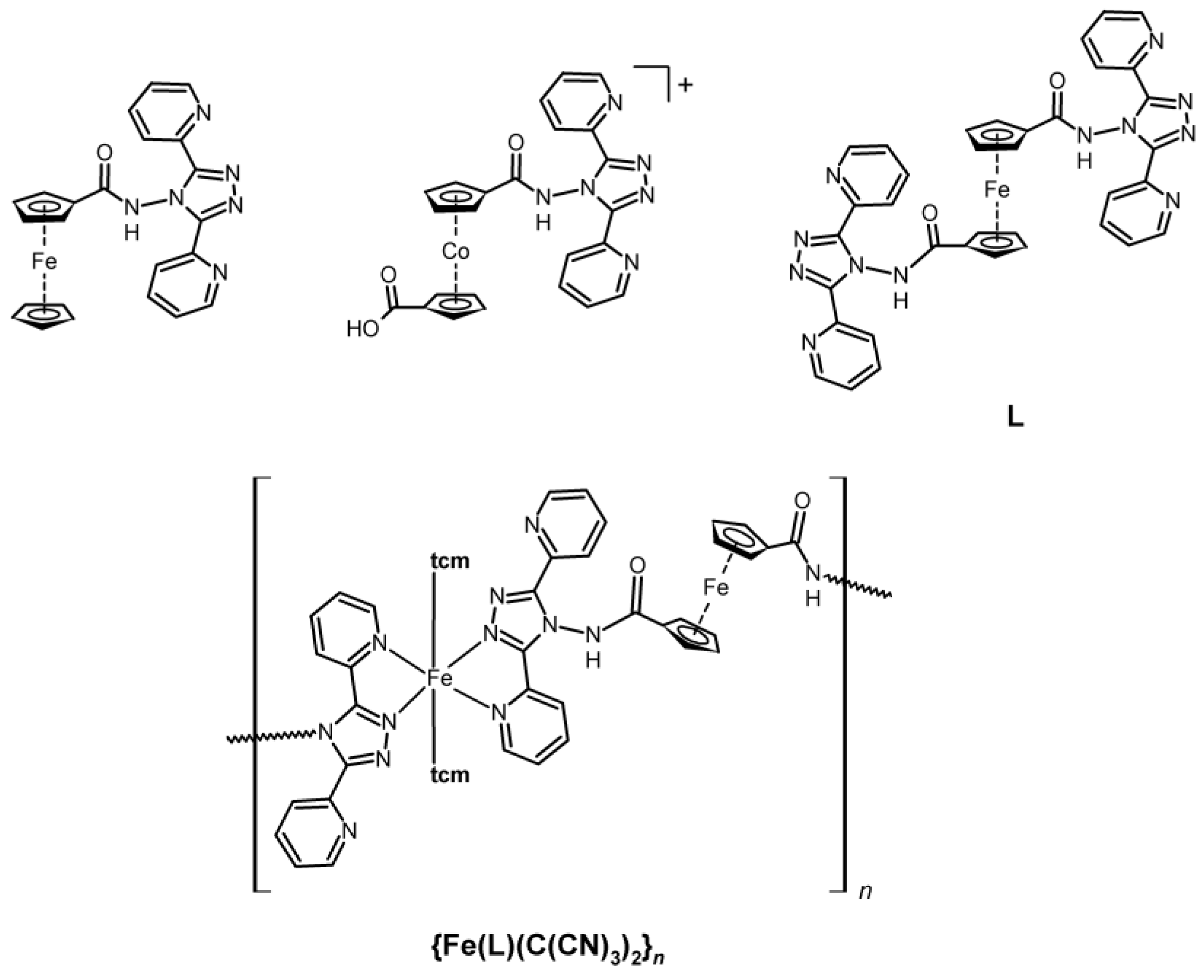
1. Introduction
2. Results and Discussion
2.1. Synthesis of Metalloligand L and FeII Complex
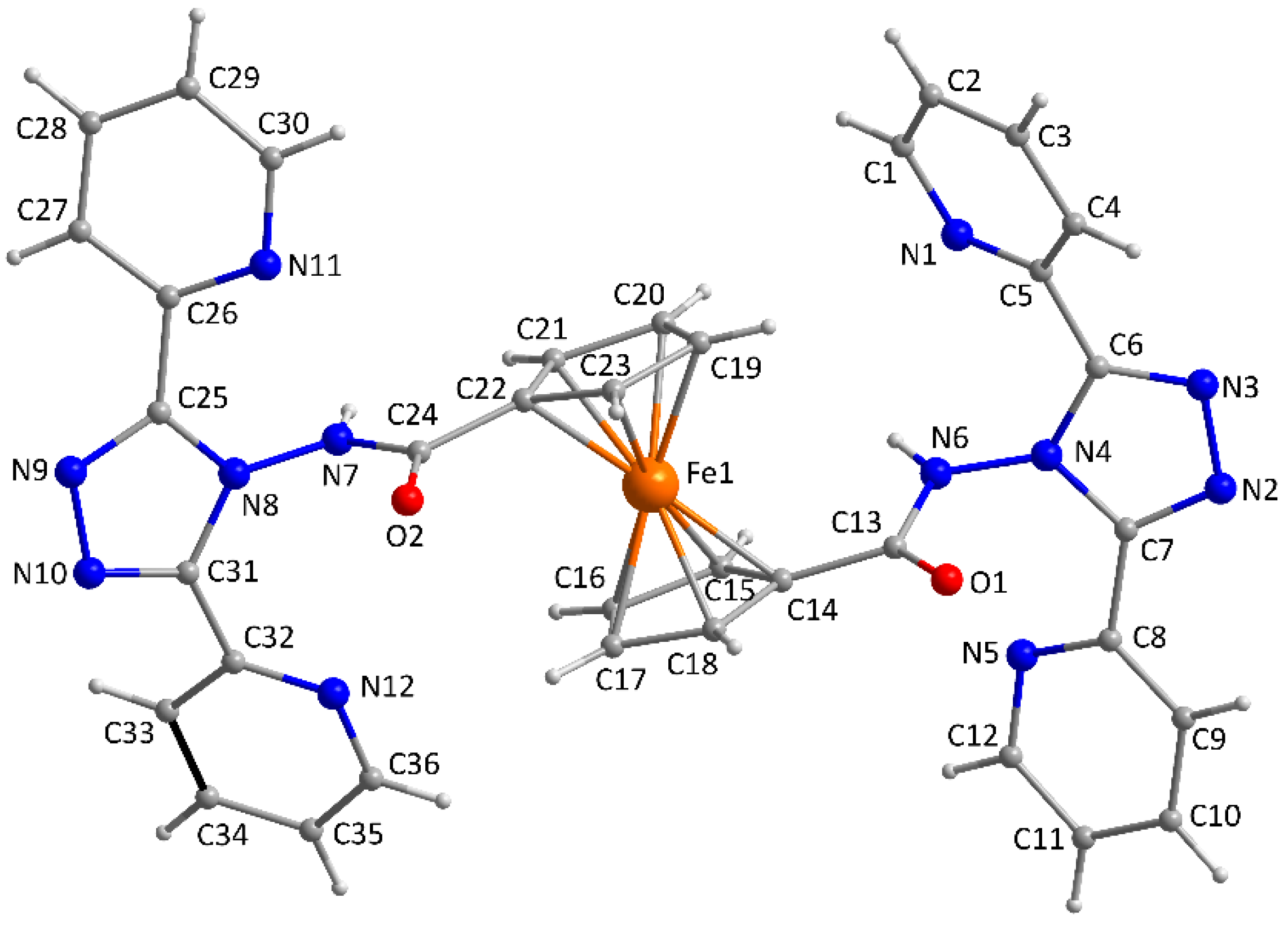
2.2. Description of the Crystal Structure of the Ligand (L)
2.3. UV-VIS Absorption Spectroscopic Studies
2.4. Electrochemical Properties
2.5. Magnetic Properties
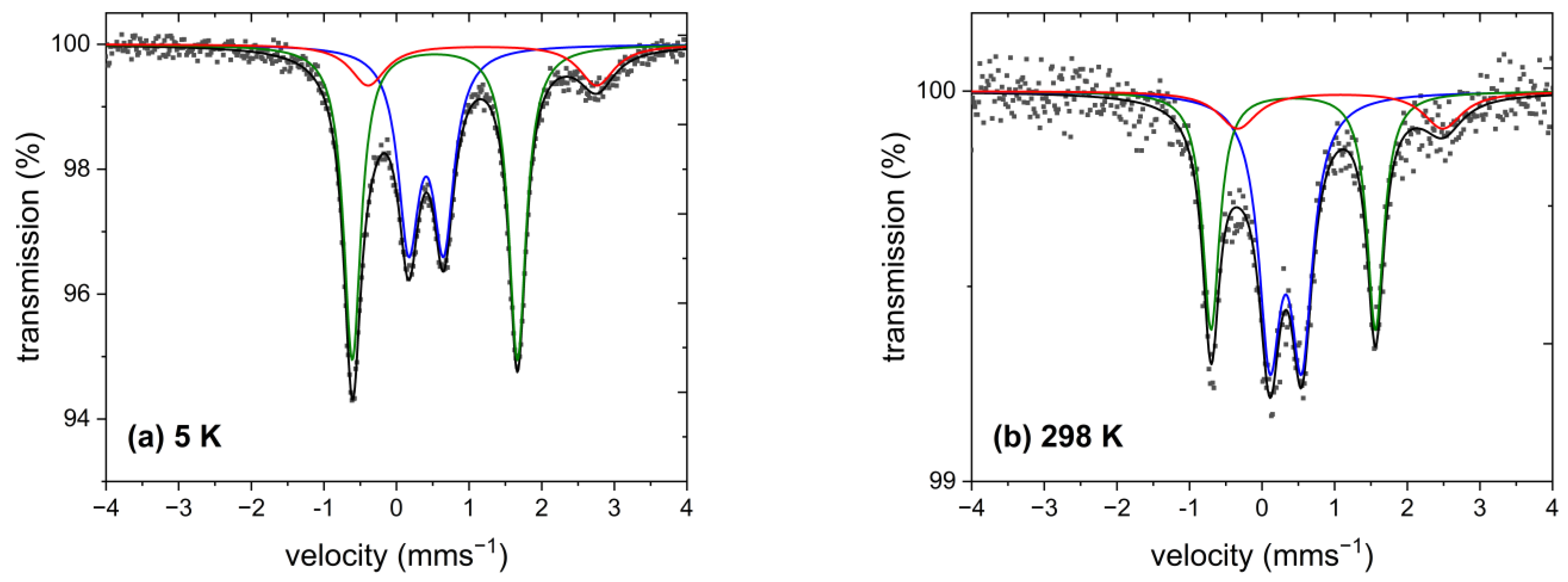
2.6. 57Fe Mössbauer Spectroscopy
2.7. Theoretical Calculations
3. Materials and Methods
3.1. Materials and Syntheses
3.1.1. Synthesis of the Ligand (L)
3.1.2. Synthesis of the Complex {Fe(L)(C(CN)3)2}n (1)
3.2. Analytical Methods
3.3. X-ray Crystallography
3.4. Theoretical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumar, G.; Gupta, R. Molecular designed architectures–the metalloligand way. Chem. Soc. Rev. 2013, 42, 9403–9453. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Kumar, G.; Gupta, R. Effect of pyridyl donors from organic ligands: Versus metalloligands on material design. Inorg. Chem. Front. 2021, 8, 1334–1373. [Google Scholar] [CrossRef]
- Gao, W.-X.; Zhang, H.-N.; Jin, G.-X. Supramolecular catalysis based on discrete heterometallic coordination-driven metallacycles and metallacages. Coord. Chem. Rev. 2019, 386, 69–84. [Google Scholar] [CrossRef]
- Cook, T.R.; Stang, P.J. Recent Development in the Preparation and Chemistry of Metallacycles and Metallacages via Coordination. Chem. Rev. 2015, 115, 7001–7045. [Google Scholar] [CrossRef] [PubMed]
- Horikoshi, R.; Mochida, T. Ferrocene-containing coordination polymers: Ligand design and assembled structure. Eur. J. Inorg. Chem. 2010, 2010, 5355–5371. [Google Scholar] [CrossRef]
- Jensen, T.; Pedersen, H.; Bang-Andersen, B.; Madsen, R.; Jørgensen, M. Palladium-catalysed aryl amination-heck cyclization cascade: A one-flask approach to 3-substituted indoles. Angew. Chem. Int. Ed. 2008, 47, 888–890. [Google Scholar] [CrossRef]
- Štěpnička, P. Phoshino-carboxamides: The inconspicuous gems. Chem. Soc. Rev. 2012, 41, 4273–4305. [Google Scholar] [CrossRef]
- Xie, Y.; Lin, R.-B.; Chen, B. Old Materials for New Functions: Recent Progress on Metal Cyanide Based Porous Materials. Adv. Sci. 2022, 9, 92104234. [Google Scholar] [CrossRef]
- Seredyuk, M.; Gaspar, A.B.; Ksenofontov, V.; Reiman, S.; Galyametdinov, Y.; Haase, W.; Rentschler, E.; Gütlich, P. Multifunctional materials exhibiting spin crossover and liquid-crystalline properties. Hyperfine Interact. 2005, 166, 385–390. [Google Scholar] [CrossRef]
- Javed, M.K.; Sulaiman, A.; Yamashita, M.; Li, Z.-Y. Shedding light on bifunctional luminescent spin crossover materials. Coord. Chem. Rev. 2022, 467, 214625. [Google Scholar] [CrossRef]
- Lacroix, P.G.; Malfant, I.; Real, J.-A.; Rodriguez, V. From magnetic to nonlinear optical switches in spin-crossover complexes. Eur. J. Inorg. Chem. 2013, 2013, 615–627. [Google Scholar] [CrossRef]
- Scott, H.S.; Gartshone, C.J.; Guo, S.-X.; Moubaraki, B.; Bond, A.M.; Batten, S.R.; Murray, K.S. Ferrocen-appended ligands for use in spin crossover-redox “hybrid” complexes of iron(ii) and cobalt (ii). Dalton Trans. 2014, 43, 15212–15220. [Google Scholar] [CrossRef] [PubMed]
- Moliner, N.; Gaspar, A.B.; Muñoz, M.C.; Niel, V.; Cano, J.; Real, J.A. Light- and thermal-induced spin crossover in {Fe(abpt)2[N(CN)2]2}. Synthesis, structure, magnetic properties, and high-spin–low-spin relaxation studies. Inorg. Chem. 2001, 40, 3986–3991. [Google Scholar] [CrossRef] [PubMed]
- Dupouy, G.; Marchivie, M.; Triki, S.; Sala-Pala, J.; Gómez-Garcia, C.J.; Pillet, S.; Lecomte, C.; Létard, J.-F. Photoinduced HS state in the first spin-crossover chain containing a cyanocarbanion as bridging ligand. Chem. Commun. 2009, 23, 3404–3406. [Google Scholar] [CrossRef]
- Gasser, G.; Carr, J.D.; Coles, S.J.; Green, S.J.; Hursthouse, M.B.; Cafferkey, S.M.; Stoeckli-Evans, H.; Tucker, J.H.R. Synthesis and complexation properties of novel triazoyl-based ferrocenyl ligands. J. Organomet. Chem. 2010, 695, 249–255. [Google Scholar] [CrossRef]
- Braga, D.; Polito, M.; Giaffreda, S.L.; Grepioni, F. Novel organometallic building blocks for molecular crystal engineering. Part 4, Synthsis and characterization of mono- And bis-amido derivatives of [CoIII(η5-C5H4COOH)2]+ and their utilization as ligands. Dalton Trans. 2005, 34, 2766–2773. [Google Scholar] [CrossRef]
- Meng, X.; Li, G.; Hou, H.; Han, H.; Fan, Y.; Zhu, Y.; Du, C. A series of metal-ferrocenedicarboxylate coordination polymers: Crystal structures, magnetic and luminescence properties. J. Organomet. Chem. 2003, 679, 153–161. [Google Scholar] [CrossRef]
- Kühnert, J.; Rüffer, T.; Ecorchard, P.; Bräuer, B.; Lan, Y.; Powell, A.K.; Lang, H. Reaction chemistry of 1,1′-ferrocenedicarboxylate towards M(ii) salts (M = Co, Ni, Cu): Solid-state structure and electrochemical, electronic and magnetic properties of bis- and tetrametallic complexes and coordination polymers. Dalton Trans. 2009, 38, 4499–4508. [Google Scholar] [CrossRef]
- Khirid, S.; Jangid, D.K.; Biswas, R.; Meena, S.; Sahoo, S.C.; Verma, V.P.; Nandi, C.; Haldar, K.K.; Dhayal, R.S. Ferrocene decorated homoleptic silver(I) clusters: Synthesis, structure, and their electrochemical behaviour. J. Organomet. Chem. 2021, 948, 121923. [Google Scholar] [CrossRef]
- Wei, K.-J.; Ni, J.; Liu, Y. Heterobimetallic Metal-Complex Assemblies Constructed from the Flexible Arm-Like Ligand 1,1′-Bis[(3-pyridylamino)carbonyl]ferrocene: Structural Versatility in the Solid State. Inorg. Chem. 2010, 49, 1834–1848. [Google Scholar] [CrossRef]
- Cao, C.-Y.; Wei, K.-J.; Ni, J.; Liu, Y. Solvent-induced two heterometallic coordination polymers based on a flexible ferrocenyl ligand. Inorg. Chem. Commun. 2010, 13, 19–21. [Google Scholar] [CrossRef]
- Djaković, S.; Maračić, S.; Lapić, J.; Kovalski, E.; Hildebrandt, A.; Lang, H.; Vrček, V.; Raić-Malić, S.; Cetina, M. Triazole-tethered ferrocene-quinoline conjugates: Solis-state structure analysis, electrochemistry and theoretical calculations. Struc. Chem. 2021, 32, 2291–2301. [Google Scholar] [CrossRef]
- Navrátil, M.; Císařová, I.; Štěpnička, P. Synthesis, Coordination and Electrochemistry of a Ferrocenyl-Tagged Aminobisphosphane Ligand. Eur. J. Inorg. Chem. 2021, 2021, 3781–3792. [Google Scholar] [CrossRef]
- Okabe, T.; Nakazaki, K.; Igaue, T.; Nakamura, N.; Donnio, D.; Guillon, D.; Gallani, J.-L. Synthesis and physical properties of ferrocene derivatives. XXI Crystal structure of a liquid crystalline ferrocene derivative, 1,1′-bis [3-[4-(4-methoxyphenoxycarbonyl)phenoxy]propyloxycarbonyl]ferrocene. J. Appl. Cryst. 2009, 42, 63–68. [Google Scholar] [CrossRef]
- Kowalski, K.; Szczupak, Ł.; Skiba, J.; Abbel-Rahman, O.S.; Winter, R.F.; Czerwieniec, R.; Therrien, B. Synthesis, structure, and spectroelectrochemistry of ferrocenyl-meldrum’s acid donor-acceptor systems. Organometallics 2014, 33, 4697–4705. [Google Scholar] [CrossRef]
- Labulo, A.H.; Omondi, B.; Nyamori, V. Synthesis, crystal structures and electrochemical properties of ferrocenyl imidazole derivatives. Heliyon 2019, 5, e02580. [Google Scholar] [CrossRef]
- Zhang, E.; Hou, H.; Meng, X.; Liu, Y.; Liu, Y.; Fan, Y. Ferrocenyl functional coordination polymers based on mono-, bi., and heterotrinuclear organometallic building blocks: Syntheses, structures, and properties. Cryst. Growth. Des. 2009, 9, 903–913. [Google Scholar] [CrossRef]
- Reynes, O.; Maillard, F.; Moutet, J.-C.; Royal, G.; Saint-Aman, E.; Stanciu, G.; Dutasta, J.-P.; Gosse, I.; Mulatier, J.-C. Complexation and electrochemical sensing of anions by amide-substituted ferrocenyl ligands. J. Organomet. Chem. 2001, 637–639, 356–363. [Google Scholar] [CrossRef]
- Magalhães, C.I.R.; Gomes, A.C.; Lopes, A.D.; Gonçalves, I.S.; Pillinger, M.; Jin, E.; Kim, I.; Ko, Y.H.; Kim, K.; Nowik, I.; et al. Ferrocene and ferrocenium inclusion compounds with cucurbiturils: A study of metal atom dynamics probed by Mössbauer spectroscopy. Phys. Chem. Chem. Phys. 2017, 19, 21548–21555. [Google Scholar] [CrossRef]
- Siebler, D.; Linseis, M.; Gasi, T.; Carrella, L.M.; Winter, R.F.; Förster, C.; Heinze, K. Oligonuclear ferrocene amides: Mixed-valent peptides and potential redox-switchable foldamers. Chem. Eur. J. 2011, 17, 4540–4551. [Google Scholar] [CrossRef]
- Gütlich, P.; Ensling, J. Inorganic Electronic Structure and Spectroscopy; Solomon, E.I., Lever, A.B.P., Eds.; Wiley: New York, NY, USA, 1999; Volume I, p. 161. [Google Scholar]
- Greenwood, N.N.; Gibb, T.C. Mössbauer Spectroscopy; Chapman and Hall Ltd.: London, UK, 1971. [Google Scholar]
- Matouzenko, G.S.; Bousseksou, A.; Borshch, S.A.; Perrin, M.; Zein, S.; Salmon, L.; Molnar, G.; Lecocq, S. Cooperative Spin Crossover and Order-Disorder Phenomena in a Mononuclear Compound [Fe(DAPP)(abpt)](ClO4)2,[DAPP = [Bis(3-aminopropyl)(2-pyridylmethyl)amine], abpt = 4-Amino-3,5-bis(pyridin-2-yl)-1,2,4-triazole]. Inorg. Chem. 2004, 43, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Herchel, R.; Trávníček, Z.; Zbořil, R. Tuning of the critical temperature in iron(II) spin-crossover materials based on bridging polycyanidometallates: Pentacyanidonitrosylferrate(II) and hexacyanidoplatinate(IV). Inorg. Chem. 2011, 50, 12390–12392. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, X.; Lu, T.; Yuan, A.; Yan, X. Potential optical molecular switch: Lithium@cyclo[18]carbon complex transforming betweeen two stable configurations. Carbon 2022, 187, 78–85. [Google Scholar] [CrossRef]
- Gallenkamp, C.; Kramm, U.I.; Proppe, J.; Krewald, V. Calibration of computation Mössbauer spectroscopy to unravel active sites in FeNC catalysts for the oxygen reduction reaction. Int. J. Quantum Chem. 2021, 121, e26394. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C. Purification of Laboratory Chemicals, 6th ed.; Butterworth-Heinemann, Elsevier: Oxford, UK, 2009. [Google Scholar]
- Miyaji, H.; Dudic, M.; Gasser, G.; Green, S.J.; Moran, N.; Prokeš, I.; Labat, G.; Stoeckli-Evans, H.; Strawbridge, S.M.; Tucker, J.H.R. A heterodifunctionalised ferrocene derivatives that self-assembles in solution through complementary hydrogen-bonding interactions. Dalton Trans. 2004, 33, 2831–2832. [Google Scholar] [CrossRef]
- Petrov, A.R.; Jess, K.; Freytag, M.; Jones, P.G.; Tamm, M. Large-scale preparation of 1,1′-ferrocenedicarboxylic acids, a key compoound for the synthesis of 1,1′-disubstituted ferrocene derivatives. Organometallics 2013, 32, 5946–5954. [Google Scholar] [CrossRef]
- Pavlishchuk, V.V.; Addison, A.W. Conversion constants for redox potentials measured versus different reference electrodes in acetonitrile solutions at 25 °C. Inorg. Chim. Acta 2000, 298, 97–102. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment–Olex2, dissected. Acta Crystallogr. Sect. A 2015, 71, 59–75. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction (2020) CrysAlisPro 1.171.40.82a. Available online: http://www.rigaku.com/ (accessed on 12 September 2020).
- Brandenburg, K.; Putz, H. Diamond—Crystal and Molecular Structure Visualization, ver. 2.1.b; Crystal Impact GbR: Bonn, Germany, 1999. [Google Scholar]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system–Version 5.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Neese, F. An Improvement of the Resolution of the Identity Approximation for the Formation of the Cuolomb Matrix. J. Comp. Chem. 2003, 24, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94, elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvatation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Perdew, J.P.; Kurth, S.; Zupan, A.; Blaha, P. Accurate density functional with correct formal properties: A step beyond the generalized gradient approximation. Phys. Rev. Lett. 1999, 82, 2544–2547. [Google Scholar] [CrossRef]
- Perdew, J.P.; Tao, J.; Staroverov, V.N.; Scuseria, G.E. Meta-generalized gradient approximation: Explanation of a realistic nonempirical density functional. J. Chem. Phys. 2004, 120, 6898–6911. [Google Scholar] [PubMed]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree–Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree–Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Izsák, R.; Neese, F. An overlap fitted chain of spheres exchange method. J. Chem. Phys. 2011, 135, 144105. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fe1-C14 | 2.026(7) | Fe1-C19 | 2.029(7) | |
| Fe1-C15 | 2.025(6) | Fe1-C20 | 2.038(7) | |
| Fe1-C16 | 2.051(7) | Fe1-C21 | 2.046(7) | |
| Fe1-C17 | 2.034(7) | Fe1-C22 | 2.023(7) | |
| Fe1-C18 | 2.042(8) | Fe1-C23 | 2.019(6) | |
| C19-C20-C21 | 109.1(7) | C14-C15-C16 | 108.4(7) | |
| C20-C21-C22 | 108.3(7) | C15-C16-C17 | 106.8(7) | |
| C21-C22-C23 | 107.0(7) | C16-C17-C18 | 110.4(7) | |
| C22-C23-C19 | 108.8(6) | C17-C18-C14 | 106.4(7) | |
| C23-C19-C20 | 106.3(7) | C18-C14-C15 | 107.9(8) |
| {Fe(abpt)}LS | {Fe(abpt)}HS | {Fe(Cp)} | ||||
|---|---|---|---|---|---|---|
| Experimental data b | δ | ΔEQ | δ | ΔEQ | δ | ΔEQ |
| 1 (5 K) | 0.41(1) | 0.48(1) | 1.18(1) | 3.15(2) | 0.53(1) | 2.28(1) |
| 1 (298 K) | 0.33(1) | 0.44(1) | 1.08(4) | 2.83(1) | 0.43(1) | 2.27(1) |
| [Fe(DAPP)(abpt)](ClO4)2 (80 K) [33] | 0.570(1) | 0.419(1) | ||||
| [Fe(DAPP)(abpt)](ClO4)2 (211 K) [33] | 1.022(1) | 1.385(2) | ||||
| [Fe(abpt)2(µ-Fe(CN)5(NO))]n (25 K) [34] | 0.517 | 0.478 | 1.065 | 3.736 | ||
| DFT calculated data | δ | ΔEQ | δ | ΔEQ | δ | ΔEQ |
| 1’(LS) | 0.54 | 0.63 | 0.60 0.60 | 2.24 2.25 | ||
| 1’(HS) | 1.14 | 3.28 | 0.60 0.60 | 2.24 2.25 | ||
| [Fe(DAPP)(abpt)]2+ (LS) | 0.61 | 0.25 | ||||
| [Fe(DAPP)(abpt)]2+ (HS) | 1.06 | 3.41 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antal, P.; Nemec, I.; Pechoušek, J.; Herchel, R. New Ferrocene-Based Metalloligand with Two Triazole Carboxamide Pendant Arms and Its Iron(II) Complex: Synthesis, Crystal Structure, 57Fe Mössbauer Spectroscopy, Magnetic Properties and Theoretical Calculations. Inorganics 2022, 10, 199. https://doi.org/10.3390/inorganics10110199
Antal P, Nemec I, Pechoušek J, Herchel R. New Ferrocene-Based Metalloligand with Two Triazole Carboxamide Pendant Arms and Its Iron(II) Complex: Synthesis, Crystal Structure, 57Fe Mössbauer Spectroscopy, Magnetic Properties and Theoretical Calculations. Inorganics. 2022; 10(11):199. https://doi.org/10.3390/inorganics10110199
Chicago/Turabian StyleAntal, Peter, Ivan Nemec, Jiří Pechoušek, and Radovan Herchel. 2022. "New Ferrocene-Based Metalloligand with Two Triazole Carboxamide Pendant Arms and Its Iron(II) Complex: Synthesis, Crystal Structure, 57Fe Mössbauer Spectroscopy, Magnetic Properties and Theoretical Calculations" Inorganics 10, no. 11: 199. https://doi.org/10.3390/inorganics10110199
APA StyleAntal, P., Nemec, I., Pechoušek, J., & Herchel, R. (2022). New Ferrocene-Based Metalloligand with Two Triazole Carboxamide Pendant Arms and Its Iron(II) Complex: Synthesis, Crystal Structure, 57Fe Mössbauer Spectroscopy, Magnetic Properties and Theoretical Calculations. Inorganics, 10(11), 199. https://doi.org/10.3390/inorganics10110199