
Bidimensional Polyiodide Netting Stabilized by a Cu(II) Macrocyclic Complex


Abstract
:1. Introduction
2. Results and Discussion
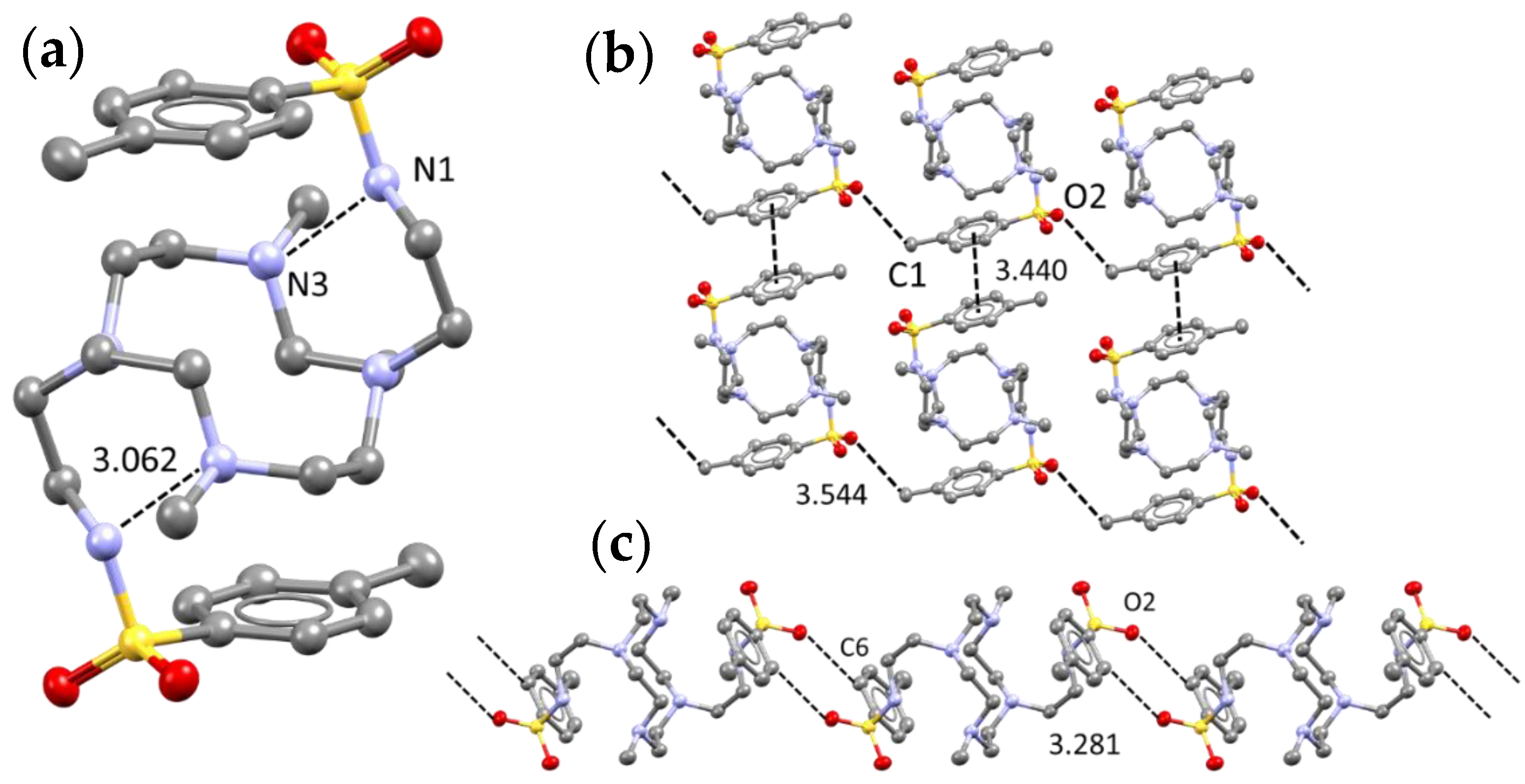
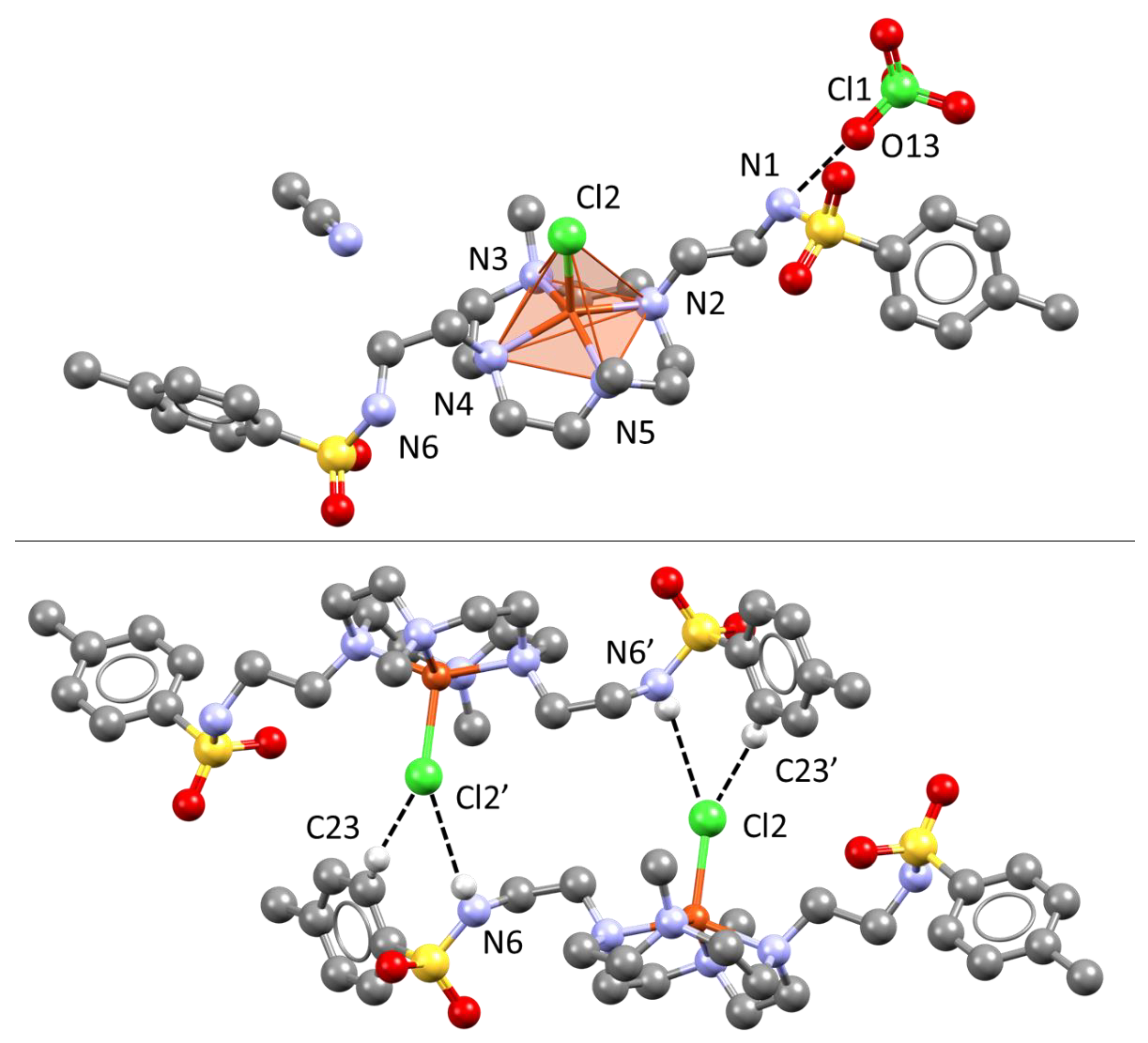
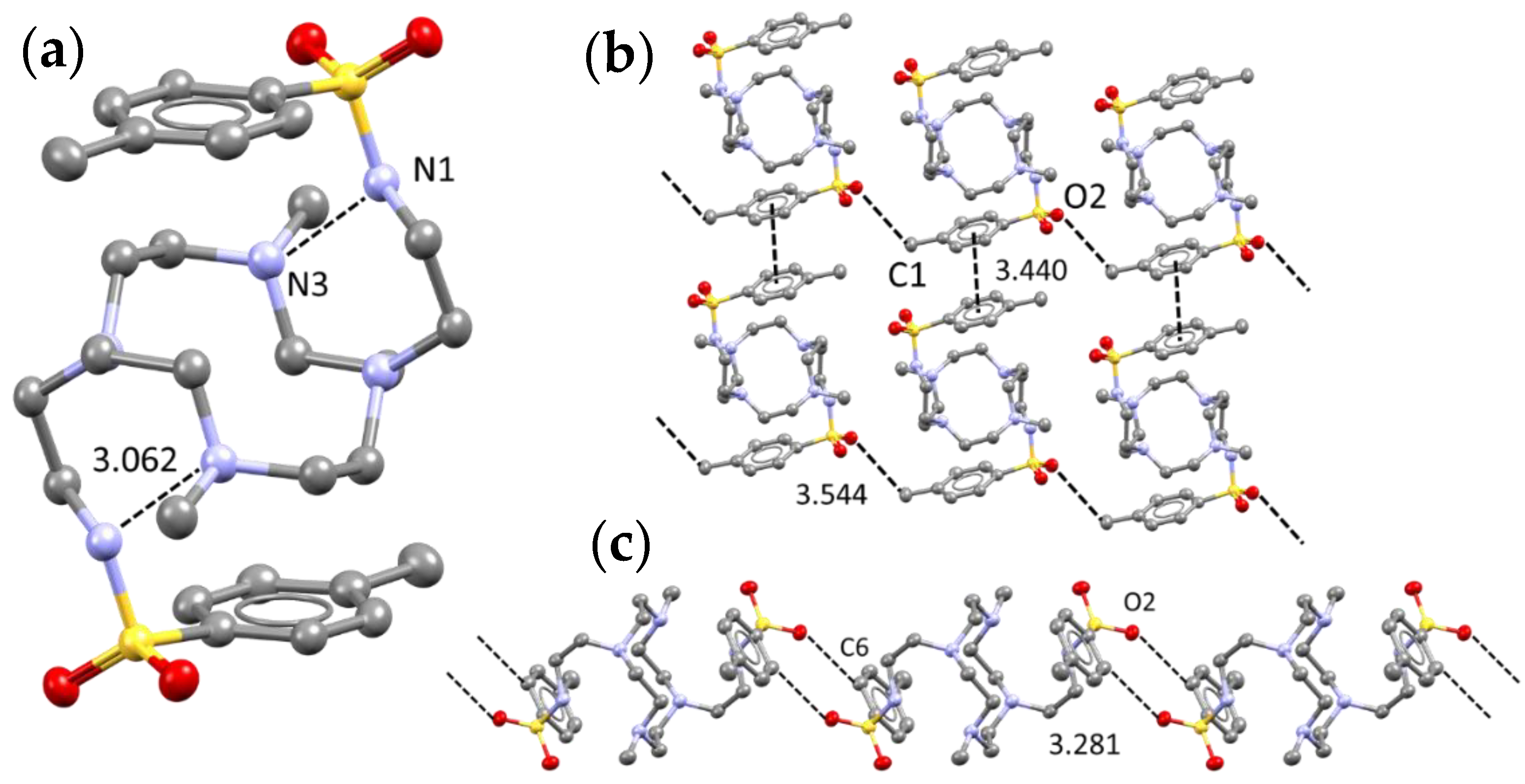
2.1. Crystal Structure of L
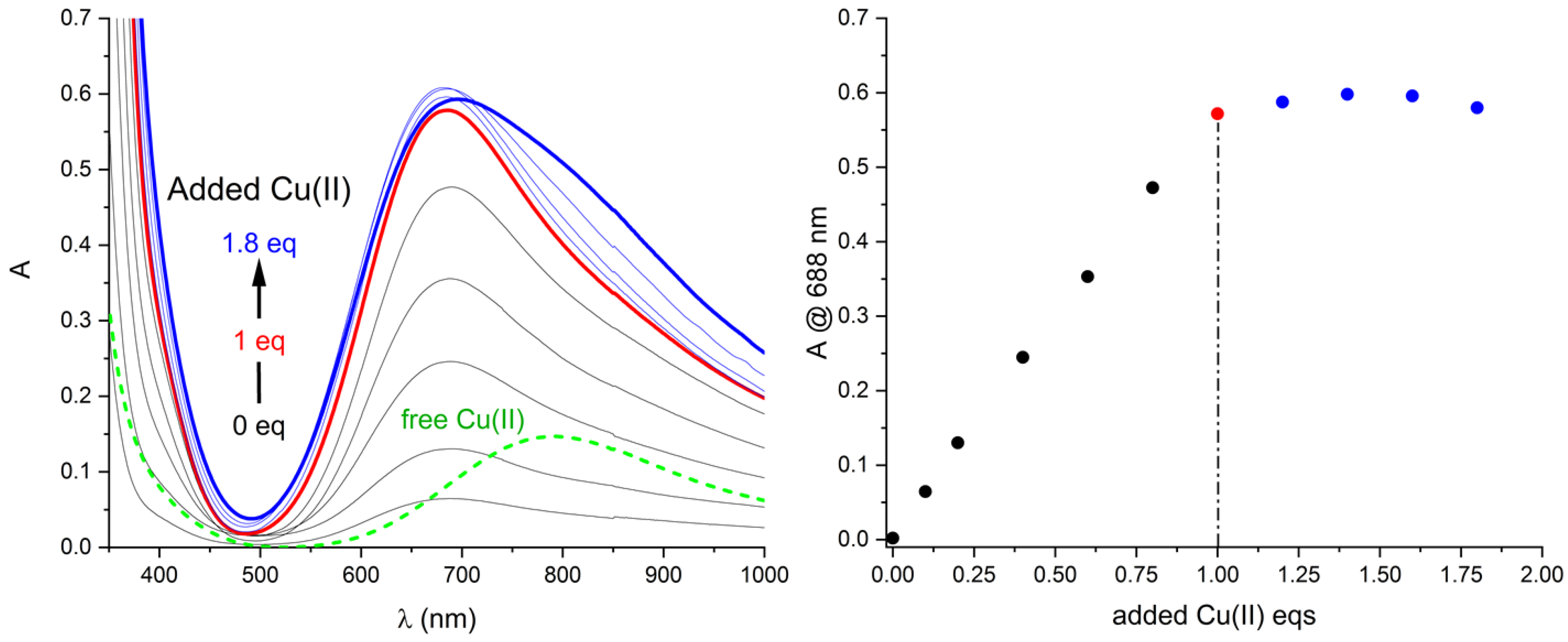
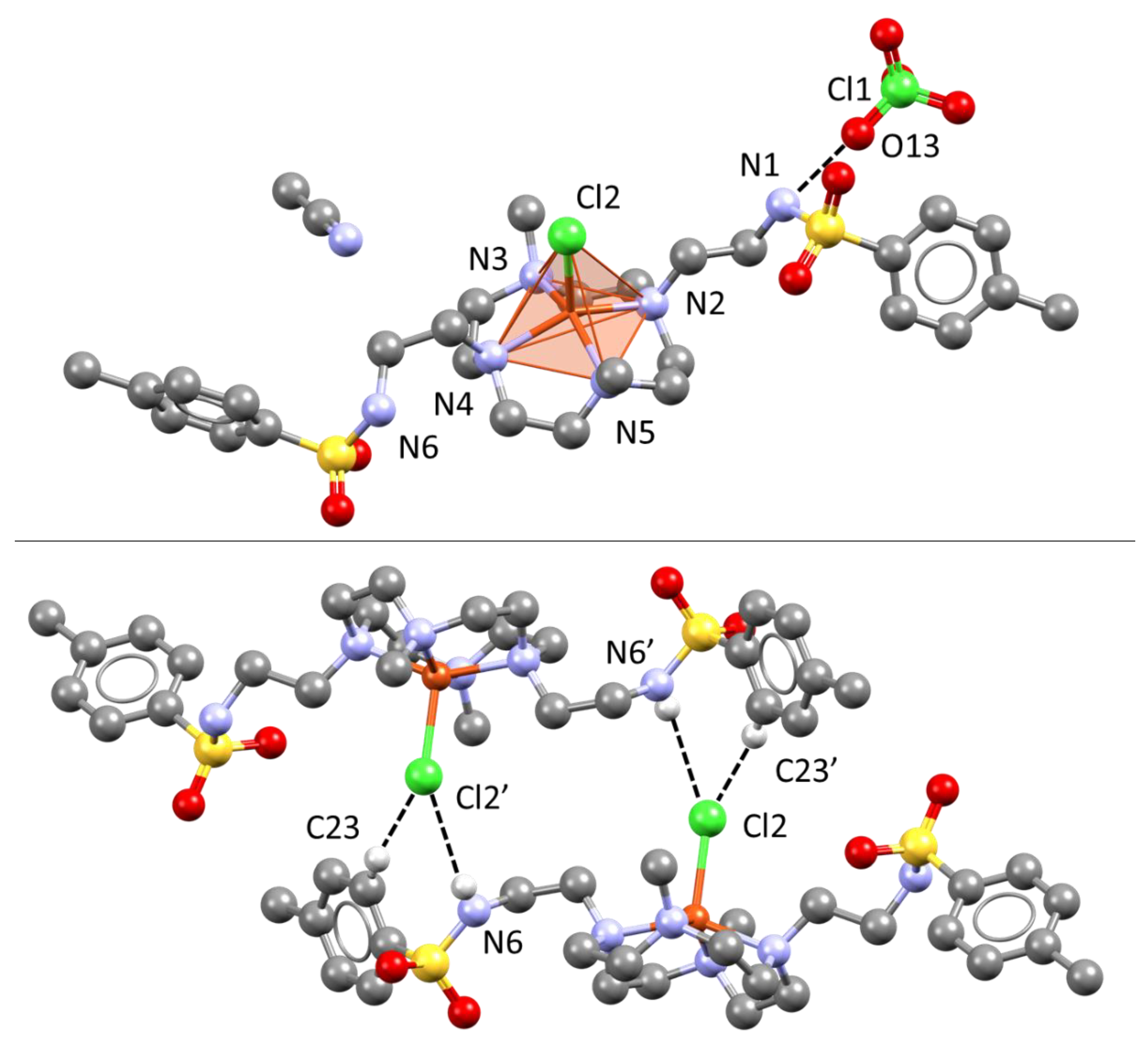
2.2. Cu(II) Complexes: Solution and Solid State
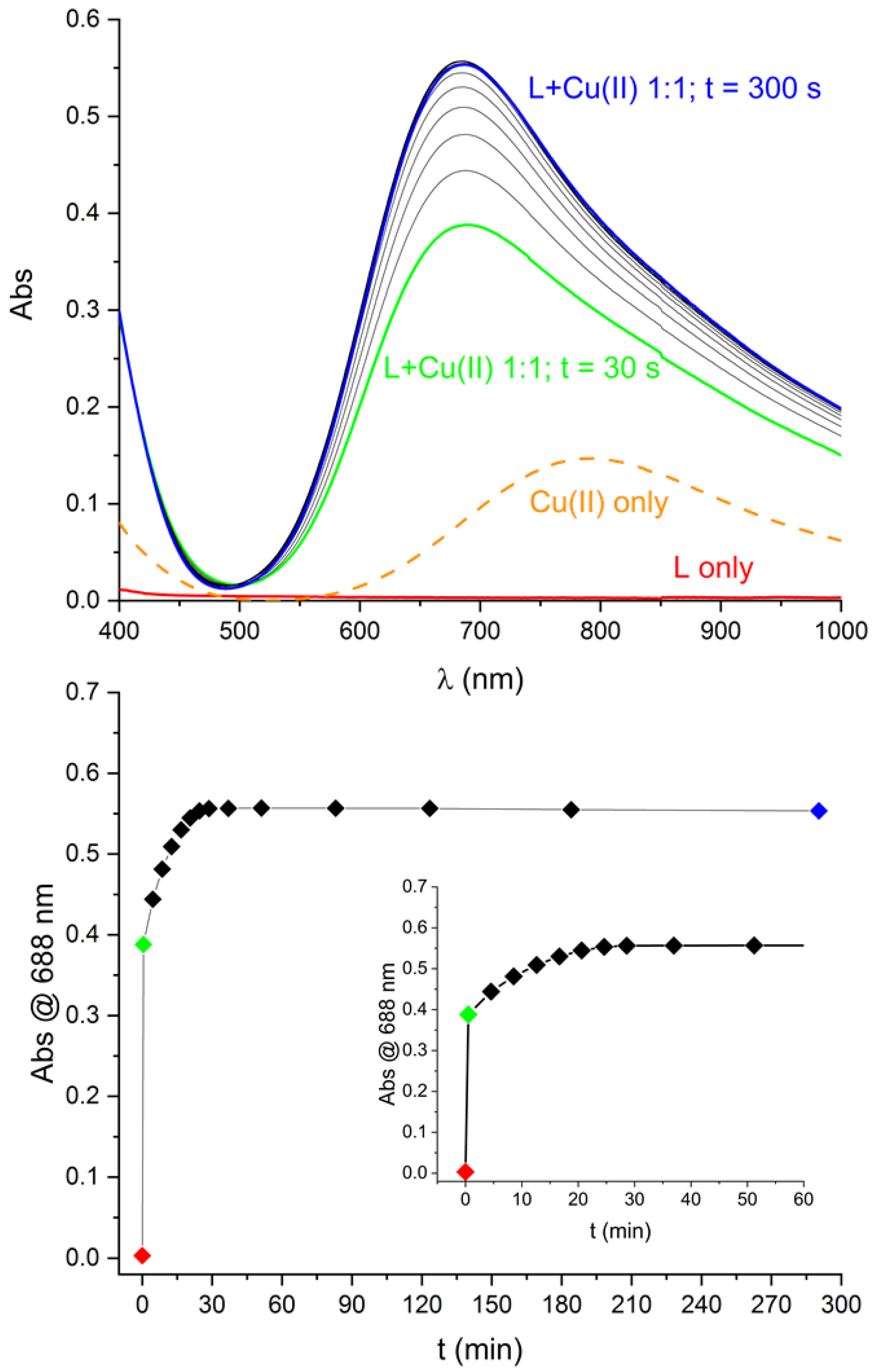
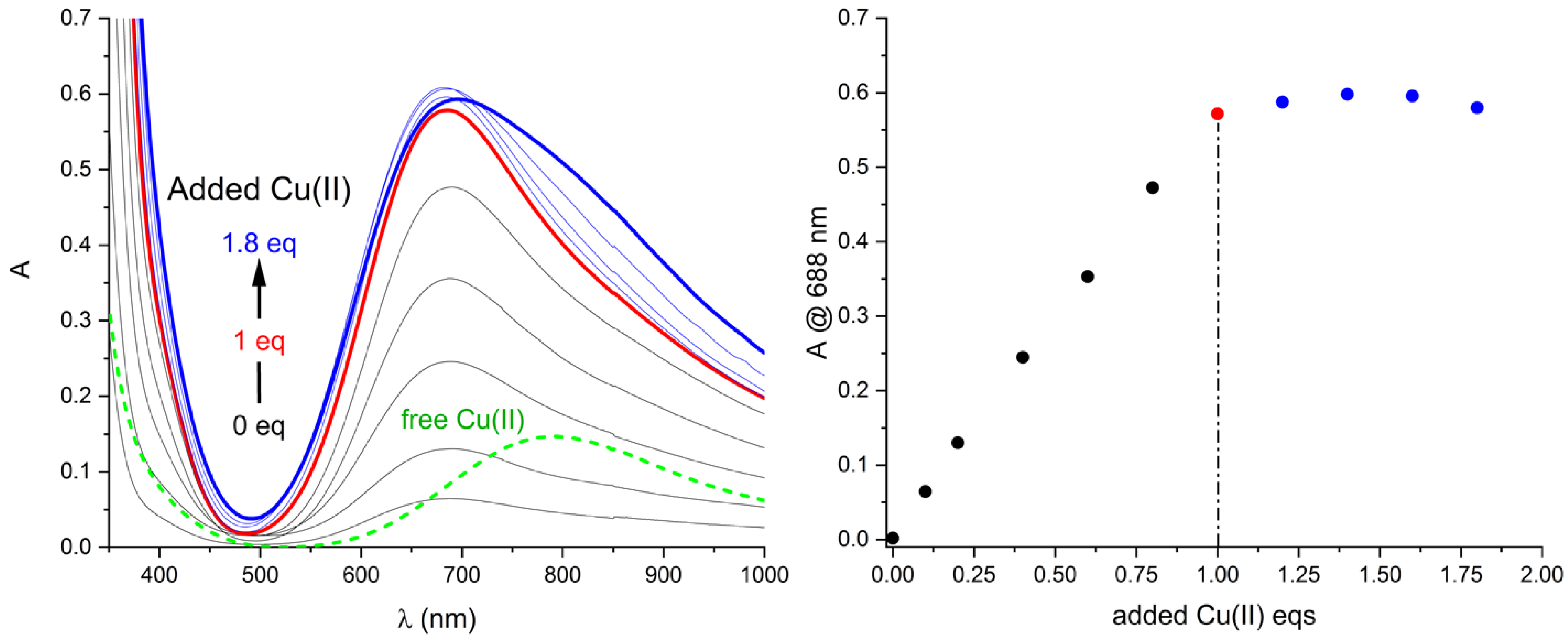
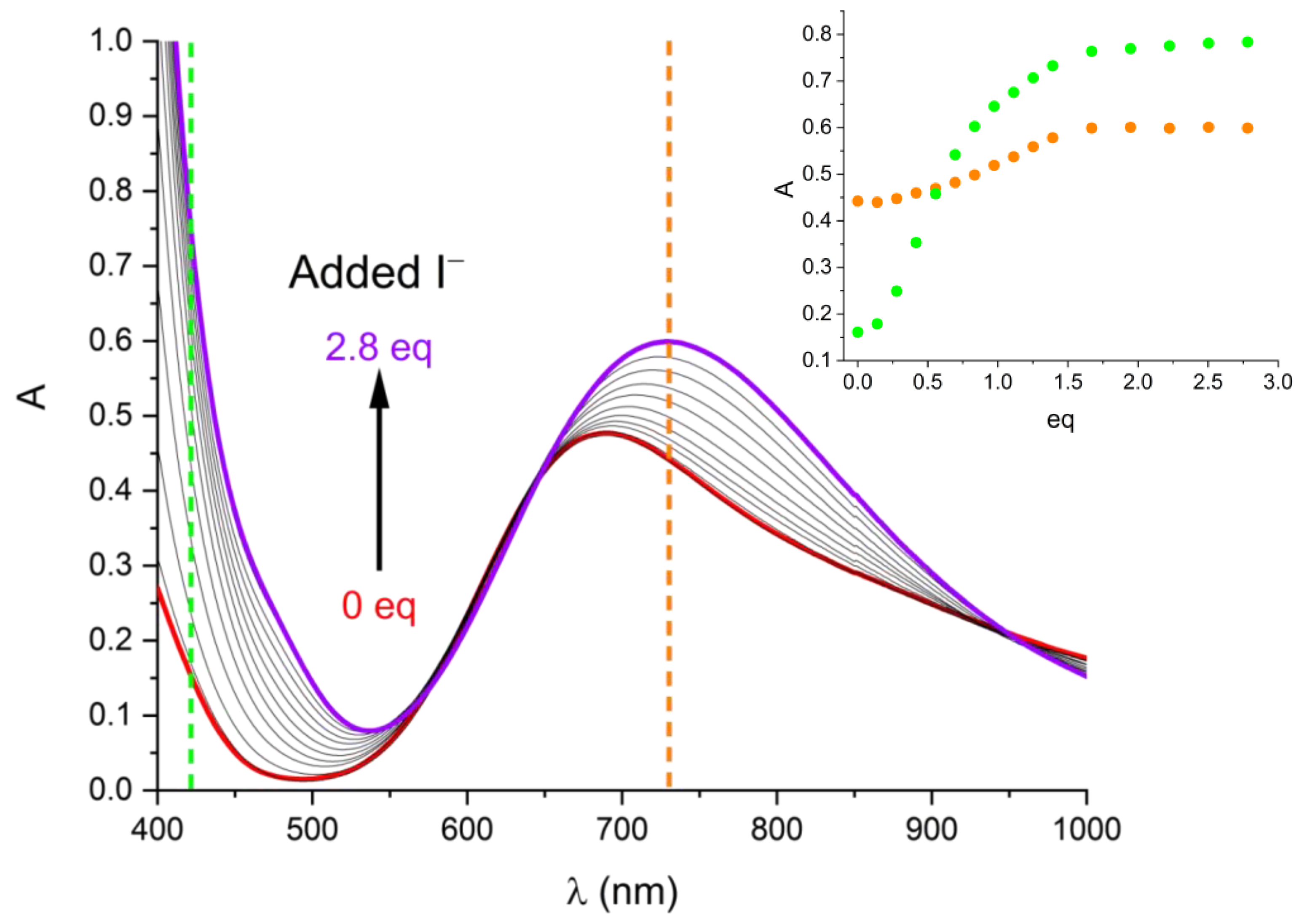
2.3. Solution Stability of [Cu(L)]2+ Complex in the Presence of Iodide
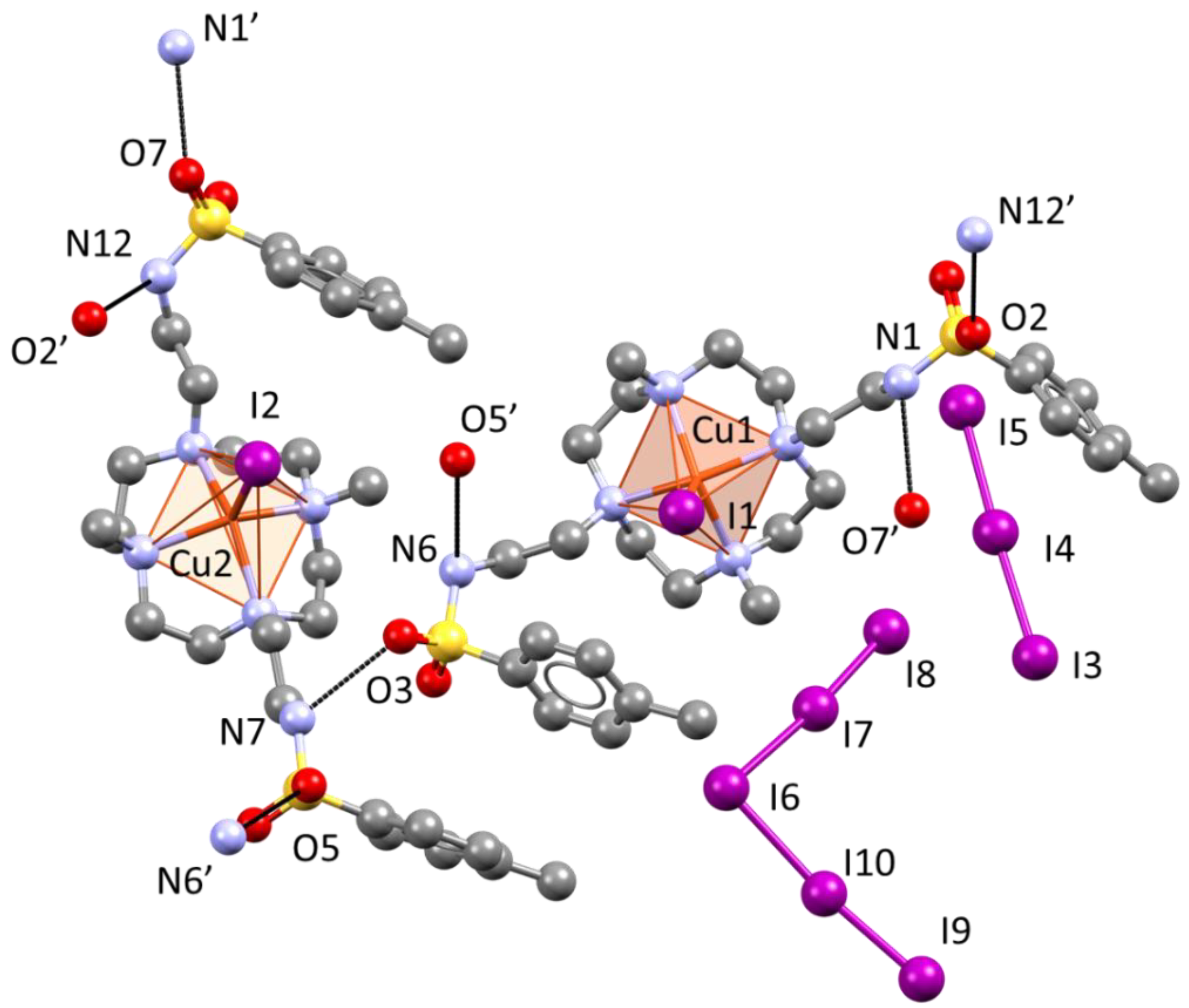
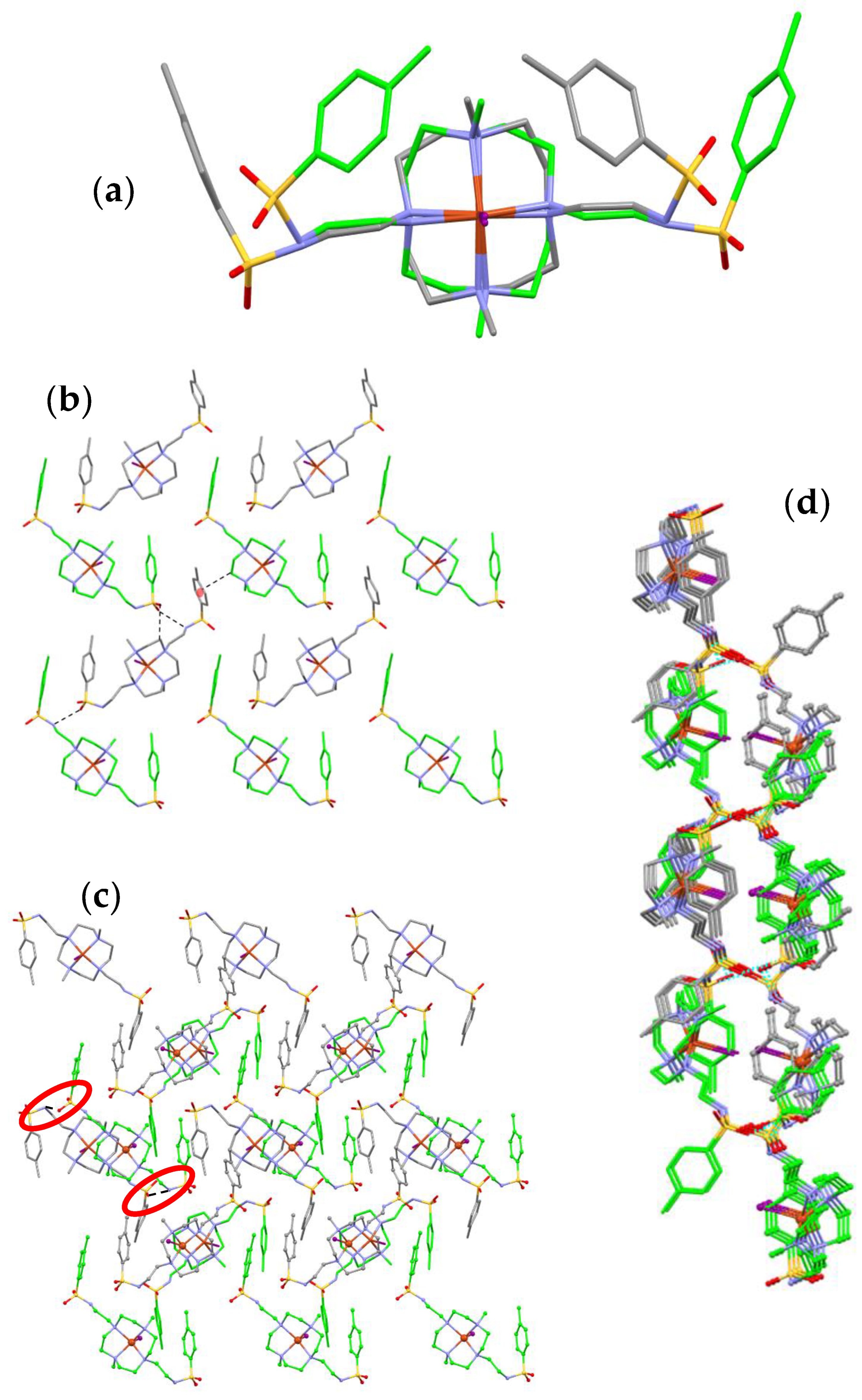
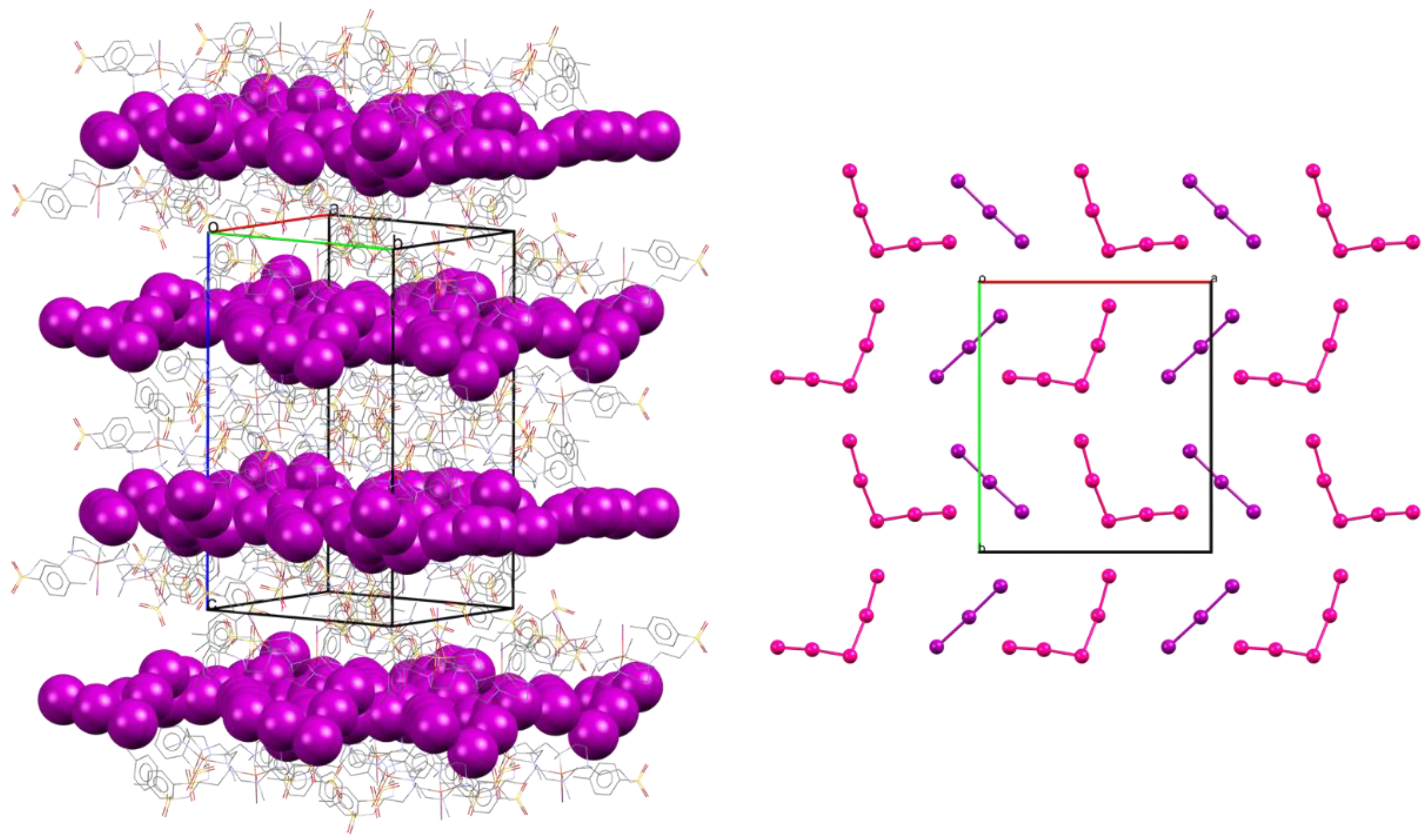
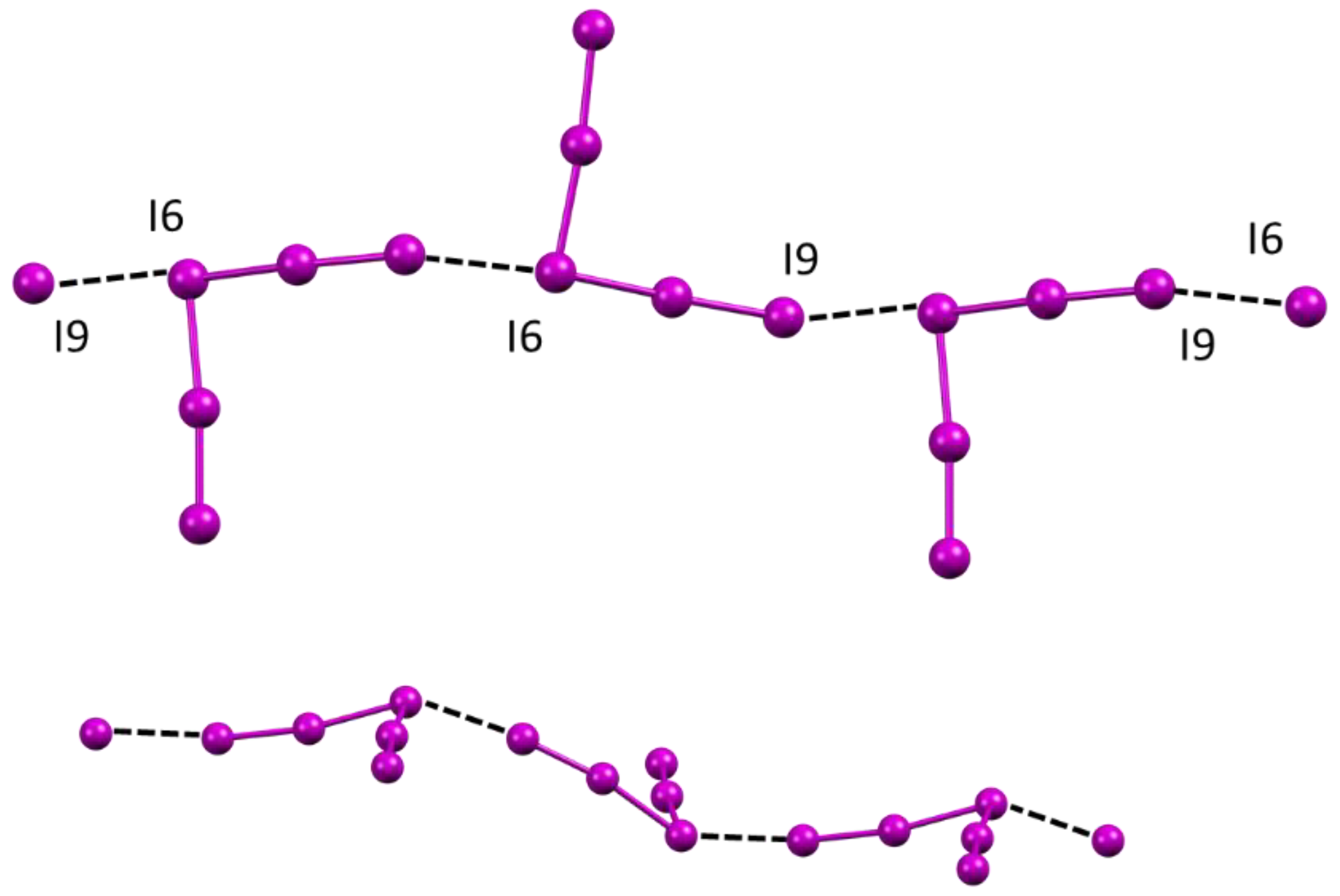
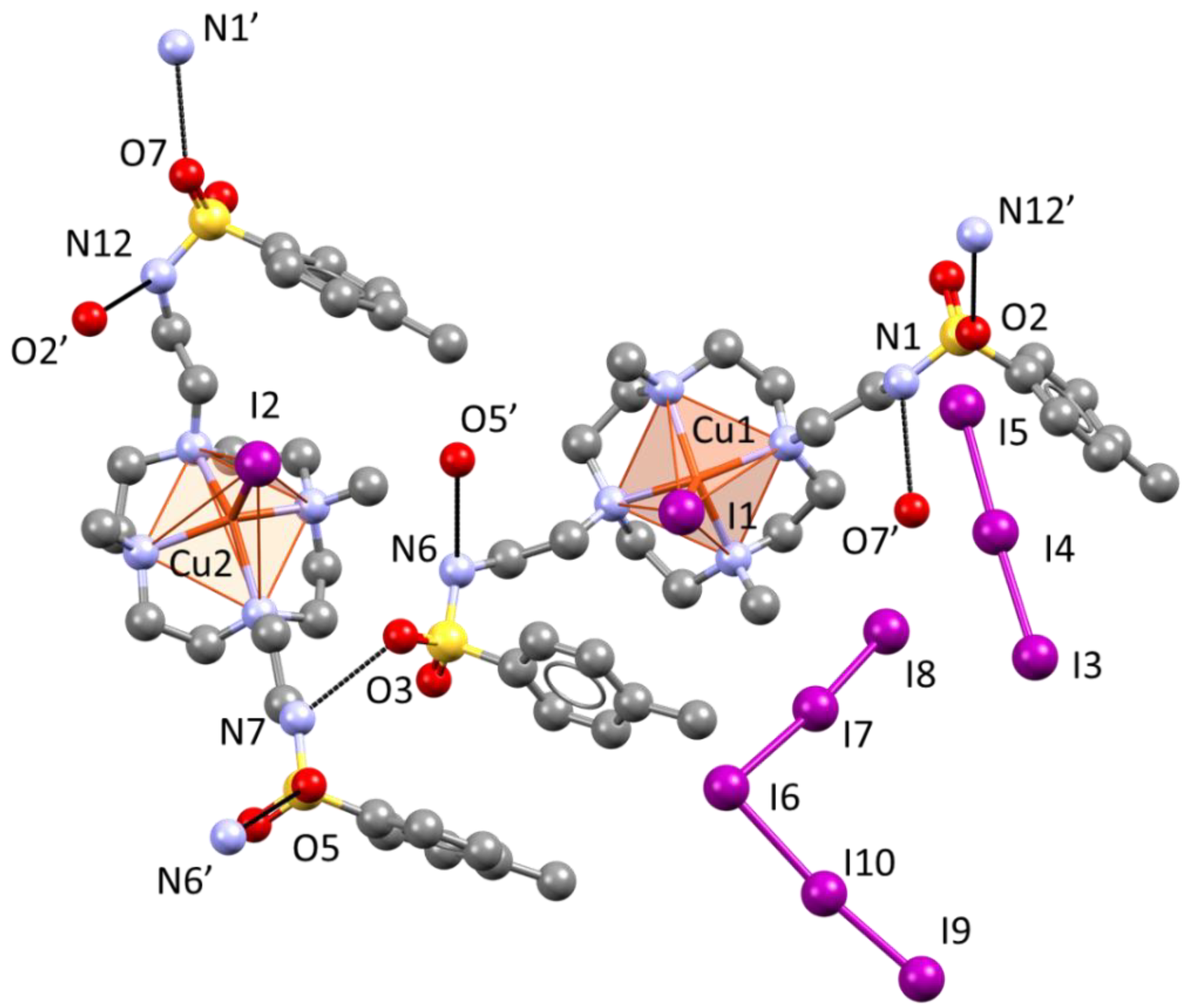
2.4. Crystal Structure of [Cu(L)I]2I3I5
3. Materials and Methods
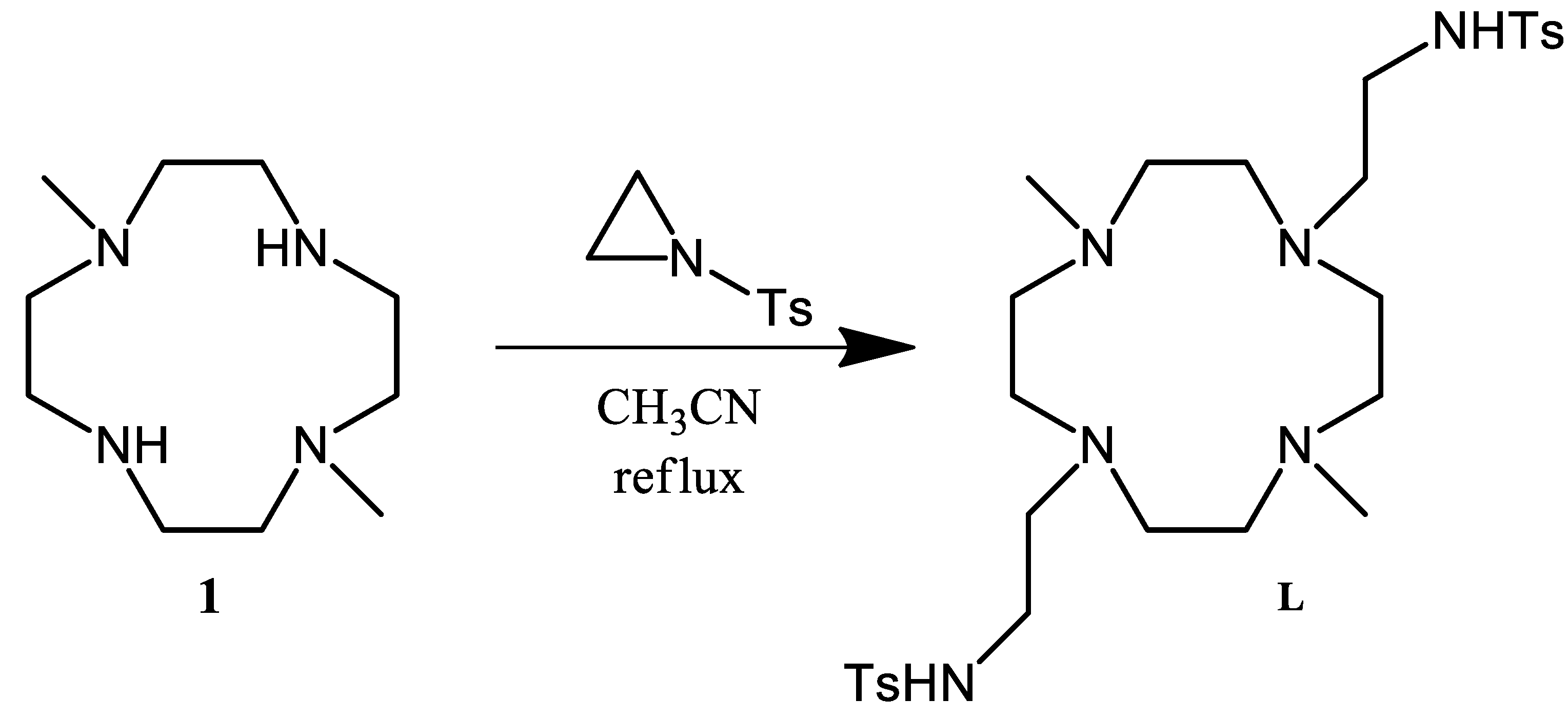
3.1. Synthesis of L
3.2. UV-Vis and NMR Spectroscopy
3.3. Crystals Preparation
3.4. XRD Data Collection and Structure Refinement
3.4.1. Crystal Data for L
3.4.2. Crystal Data for [Cu(L)Cl]ClO4∙CH3CN
3.4.3. Crystal Data for [Cu(L)I]2I3I5
3.4.4. Data Presentation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Svensson, P.H.; Kloo, L. Synthesis, Structure, and Bonding in Polyiodide and Metal Iodide−Iodine Systems. Chem. Rev. 2003, 103, 1649–1684. [Google Scholar] [CrossRef] [PubMed]
- Savastano, M. Words in Supramolecular Chemistry: The Ineffable Advances of Polyiodide Chemistry. Dalton Trans. 2021, 50, 1142–1165. [Google Scholar] [CrossRef] [PubMed]
- Abbas, Q.; Fitzek, H.; Schröttner, H.; Dsoke, S.; Gollas, B. Immobilization of Polyiodide Redox Species in Porous Carbon for Battery-Like Electrodes in Eco-Friendly Hybrid Electrochemical Capacitors. Nanomaterials 2019, 9, 1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Z.; Tian, H.; Zhang, S.; Yan, X.; Ying, H.; He, W.; Liang, C.; Zhang, W.; Hou, X.; Han, W.-Q. Polyiodide-Shuttle Restricting Polymer Cathode for Rechargeable Lithium/Iodine Battery with Ultralong Cycle Life. ACS Appl. Mater. Interfaces 2018, 10, 17933–17941. [Google Scholar] [CrossRef] [PubMed]
- Bella, F.; Galliano, S.; Falco, M.; Viscardi, G.; Barolo, C.; Grätzel, M.; Gerbaldi, C. Unveiling Iodine-Based Electrolytes Chemistry in Aqueous Dye-Sensitized Solar Cells. Chem. Sci. 2016, 7, 4880–4890. [Google Scholar] [CrossRef] [Green Version]
- Paulsson, H.; Berggrund, M.; Svantesson, E.; Hagfeldt, A.; Kloo, L. Molten and Solid Metal-Iodide-Doped Trialkylsulphonium Iodides and Polyiodides as Electrolytes in Dye-Sensitized Nanocrystalline Solar Cells. Sol. Energy Mater. Sol. Cells 2004, 82, 345–360. [Google Scholar] [CrossRef]
- Fei, Z.; Bobbink, F.D.; Păunescu, E.; Scopelliti, R.; Dyson, P.J. Influence of Elemental Iodine on Imidazolium-Based Ionic Liquids: Solution and Solid-State Effects. Inorg. Chem. 2015, 54, 10504–10512. [Google Scholar] [CrossRef]
- McDaniel, J.G.; Yethiraj, A. Grotthuss Transport of Iodide in EMIM/I3 Ionic Crystal. J. Phys. Chem. B 2018, 122, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Zhao, G.; Zhao, X.; He, C.; Pang, S.; Shreeve, J.M. New Promises from an Old Friend: Iodine-Rich Compounds as Prospective Energetic Biocidal Agents. Acc. Chem. Res. 2021, 54, 332–343. [Google Scholar] [CrossRef]
- Savastano, M.; Bazzicalupi, C.; García, C.; Gellini, C.; López de la Torre, M.D.; Mariani, P.; Pichierri, F.; Bianchi, A.; Melguizo, M. Iodide and Triiodide Anion Complexes Involving Anion–π Interactions with a Tetrazine-Based Receptor. Dalton Trans. 2017, 46, 4518–4529. [Google Scholar] [CrossRef] [PubMed]
- Savastano, M.; Martínez-Camarena, Á.; Bazzicalupi, C.; Delgado-Pinar, E.; Llinares, J.M.; Mariani, P.; Verdejo, B.; García-España, E.; Bianchi, A. Stabilization of Supramolecular Networks of Polyiodides with Protonated Small Tetra-Azacyclophanes. Inorganics 2019, 7, 48. [Google Scholar] [CrossRef] [Green Version]
- Savastano, M.; Bazzicalupi, C.; Gellini, C.; Bianchi, A. Genesis of Complex Polyiodide Networks: Insights on the Blue Box/I−/I2 Ternary System. Crystals 2020, 10, 387. [Google Scholar] [CrossRef]
- Savastano, M.; Bazzicalupi, C.; Gellini, C.; Bianchi, A. Infinite Supramolecular Pseudo-Polyrotaxane with Poly[3]Catenane Axle: Assembling Nanosized Rings from Mono- and Diatomic I− and I2 Tectons. Chem. Commun. 2020, 56, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Camarena, Á.; Savastano, M.; Llinares, J.M.; Verdejo, B.; Bianchi, A.; García-España, E.; Bazzicalupi, C. Stabilization of Polyiodide Networks with Cu(II) Complexes of Small Methylated Polyazacyclophanes: Shifting Directional Control from H-Bonds to I⋯I Interactions. Inorg. Chem. Front. 2020, 7, 4239–4255. [Google Scholar] [CrossRef]
- Martínez-Camarena, Á.; Savastano, M.; Blasco, S.; Delgado-Pinar, E.; Giorgi, C.; Bianchi, A.; García-España, E.; Bazzicalupi, C. Assembly of Polyiodide Networks with Cu(II) Complexes of Pyridinol-Based Tetraaza Macrocycles. Inorg. Chem. 2022, 61, 368–383. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Englert, U. N-(6-Methyl-2-Pyridyl)Mesitylenesulfonamide: An Efficient Template for Polyiodides. Cryst. Growth Des. 2014, 14, 1057–1066. [Google Scholar] [CrossRef]
- Martínez-Camarena, Á.; Savastano, M.; Bazzicalupi, C.; Bianchi, A.; García-España, E. Stabilisation of Exotic Tribromide (Br3−) Anions via Supramolecular Interaction with A Tosylated Macrocyclic Pyridinophane. A Serendipitous Case. Molecules 2020, 25, 3155. [Google Scholar] [CrossRef]
- Ciampolini, M.; Micheloni, M.; Nardi, N.; Paoletti, P.; Dapporto, P.; Zanobini, F. Synthesis and Characterisation of 1,7-Dimethyl-1,4,7,10-Tetra-Azacyclododecane: Crystal Structure of the Nickel(II) Bromide Monohydrate Complex of This Macrocycle; Thermodynamic Studies of Protonation and Metal Complex Formation. J. Chem. Soc. Dalton Trans. 1984, 1357–1362. [Google Scholar] [CrossRef]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of Equilibria in Solution. Determination of Equilibrium Constants with the HYPERQUAD Suite of Programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Pidcock, E. Achiral Molecules in Non-Centrosymmetric Space Groups. Chem. Commun. 2005, 3457–3459. [Google Scholar] [CrossRef]
- Baldauf, C.; Günther, R.; Hofmann, H.-J. Conformational Properties of Sulfonamido Peptides. J. Mol. Struct.-THEOCHEM 2004, 675, 19–28. [Google Scholar] [CrossRef]
- Ambrosi, G.; Formica, M.; Fusi, V.; Giorgi, L.; Guerri, A.; Micheloni, M.; Paoli, P.; Pontellini, R.; Rossi, P. A New Macrocyclic Cryptand with Squaramide Moieties: An Overstructured CuII Complex That Selectively Binds Halides: Synthesis, Acid/Base- and Ligational Behavior, and Crystal Structures. Chem. Eur. J. 2007, 13, 702–712. [Google Scholar] [CrossRef]
- Yong, L.; Guo-Ping, X.; Cheng-Tai, W. Synthesis of Novel N-Aminoethyl Cyclic Polyamines. Chin. J. Chem. 1998, 16, 448–451. [Google Scholar] [CrossRef]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of Silver and Molybdenum Microfocus X-Ray Sources for Single-Crystal Structure Determination. J Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A Short History of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Distances (Å) | Bond Angles (°) | ||
|---|---|---|---|
| Cu1-N2 | 2.071(4) | Cl2 -Cu1 -N4 | 105.3(1) |
| Cu1-N3 | 2.047(4) | Cl2 -Cu1 -N2 | 106.7(1) |
| Cu1-N4 | 2.081(4) | Cl2 -Cu1 -N5 | 103.6(1) |
| Cu1-N5 | 2.041(4) | Cl2 -Cu1 -N3 | 106.5(1) |
| Cu1-Cl2 | 2.372(1) | N4 -Cu1 -N2 | 148.0(2) |
| N4 -Cu1 -N5 | 86.1(2) | ||
| N4 -Cu1 -N3 | 86.1(2) | ||
| N2 -Cu1 -N5 | 86.2(2) | ||
| N2 -Cu1 -N3 | 85.2(2) | ||
| N5 -Cu1 -N3 | 149.8(2) | ||
| Bond Distances (Å) | Bond Angles (°) | ||||
|---|---|---|---|---|---|
| Cu1-N2 | 2.08(1) | I1-Cu1-N2 | 107.7(3) | I2-Cu2-N8 | 107.3(3) |
| Cu1-N3 | 2.06(1) | I1-Cu1-N3 | 108.4(3) | I2-Cu2-N9 | 103.7(4) |
| Cu1-N4 | 2.13(1) | I1-Cu1-N4 | 106.4(3) | I2-Cu2-N10 | 106.9(3) |
| Cu1-N5 | 2.07(1) | I1-Cu1-N5 | 103.6(4) | I2-Cu2-N11 | 109.3(4) |
| Cu1-I1 | 2.670(2) | N2-Cu1-N3 | 86.3(5) | N8-Cu2-N9 | 85.8(5) |
| Cu2-N8 | 2.13(1) | N2-Cu1-N4 | 145.9(5) | N8-Cu2-N10 | 145.8(5) |
| Cu2-N9 | 2.05(1) | N2-Cu1-N5 | 84.6(5) | N8-Cu2-N11 | 85.5(5) |
| Cu2-N10 | 2.12(1) | N3-Cu1-N4 | 84.8(4) | N9-Cu2-N10 | 85.1(5) |
| Cu2-N11 | 2.08(1) | N3-Cu1-N5 | 147.9(5) | N9-Cu2-N11 | 147.0(5) |
| Cu2-I2 | 2.662(2) | N4-Cu1-N5 | 85.6(5) | N10-Cu2-N11 | 84.4(5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savastano, M.; Monini, V.; Bazzicalupi, C.; Bianchi, A. Bidimensional Polyiodide Netting Stabilized by a Cu(II) Macrocyclic Complex. Inorganics 2022, 10, 12. https://doi.org/10.3390/inorganics10010012
Savastano M, Monini V, Bazzicalupi C, Bianchi A. Bidimensional Polyiodide Netting Stabilized by a Cu(II) Macrocyclic Complex. Inorganics. 2022; 10(1):12. https://doi.org/10.3390/inorganics10010012
Chicago/Turabian StyleSavastano, Matteo, Valeria Monini, Carla Bazzicalupi, and Antonio Bianchi. 2022. "Bidimensional Polyiodide Netting Stabilized by a Cu(II) Macrocyclic Complex" Inorganics 10, no. 1: 12. https://doi.org/10.3390/inorganics10010012
APA StyleSavastano, M., Monini, V., Bazzicalupi, C., & Bianchi, A. (2022). Bidimensional Polyiodide Netting Stabilized by a Cu(II) Macrocyclic Complex. Inorganics, 10(1), 12. https://doi.org/10.3390/inorganics10010012