Probing Small Distances in Live Cell Imaging



Abstract
1. Introduction
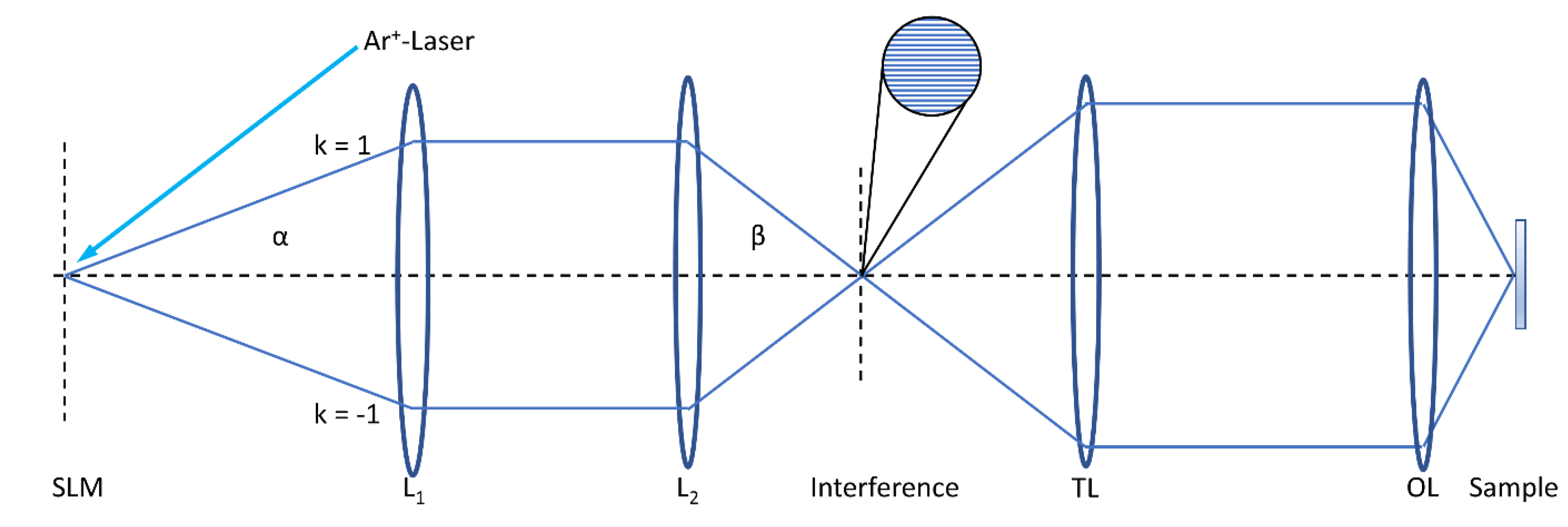
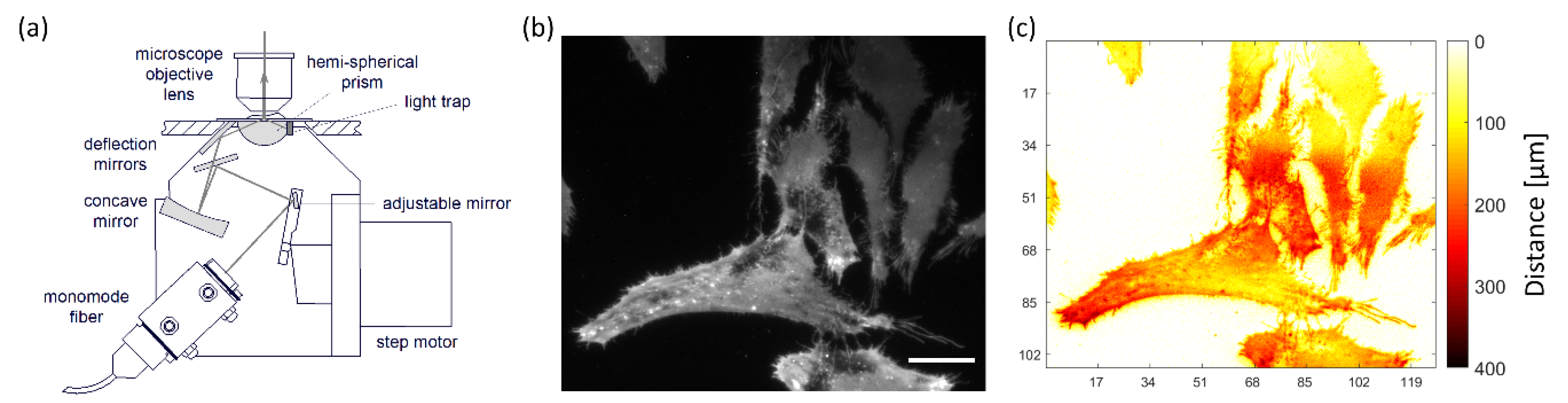
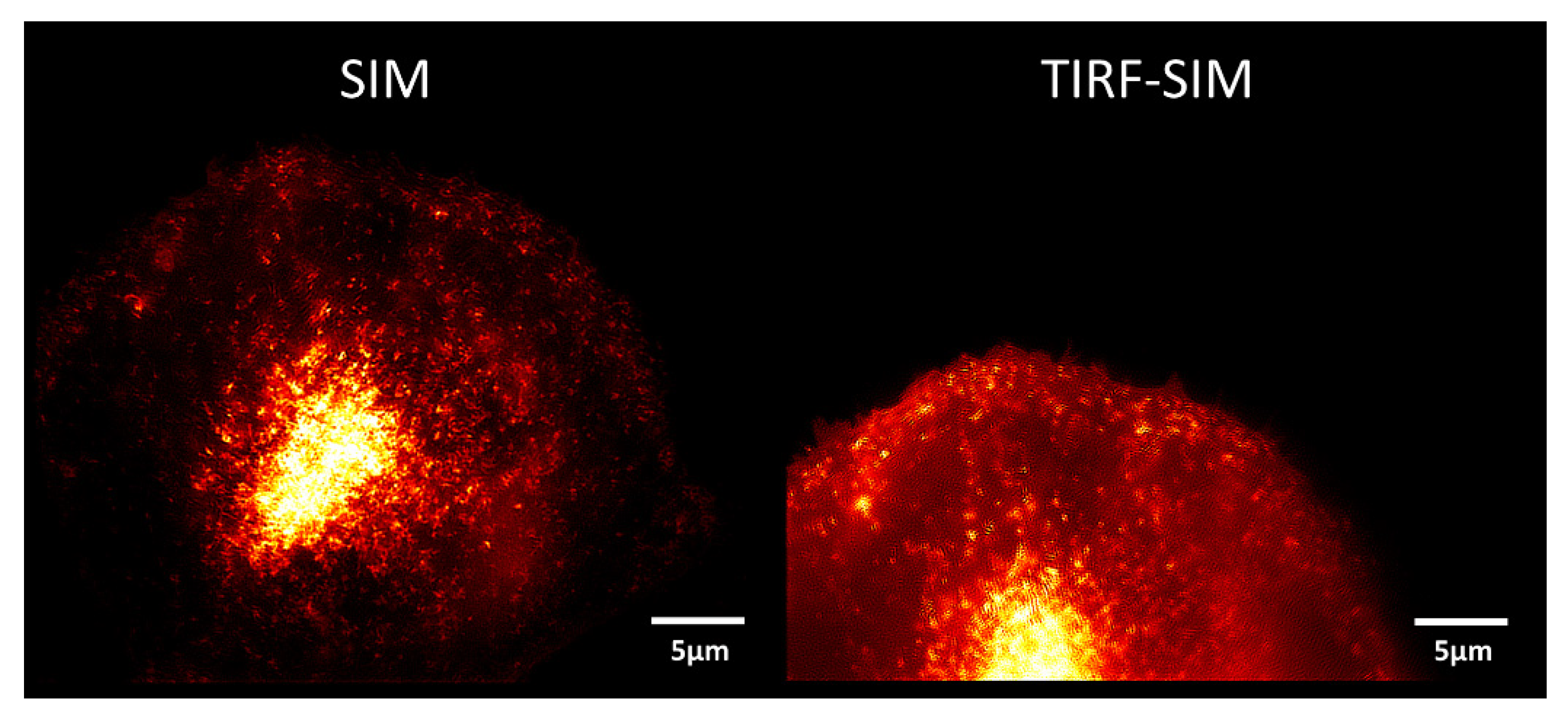
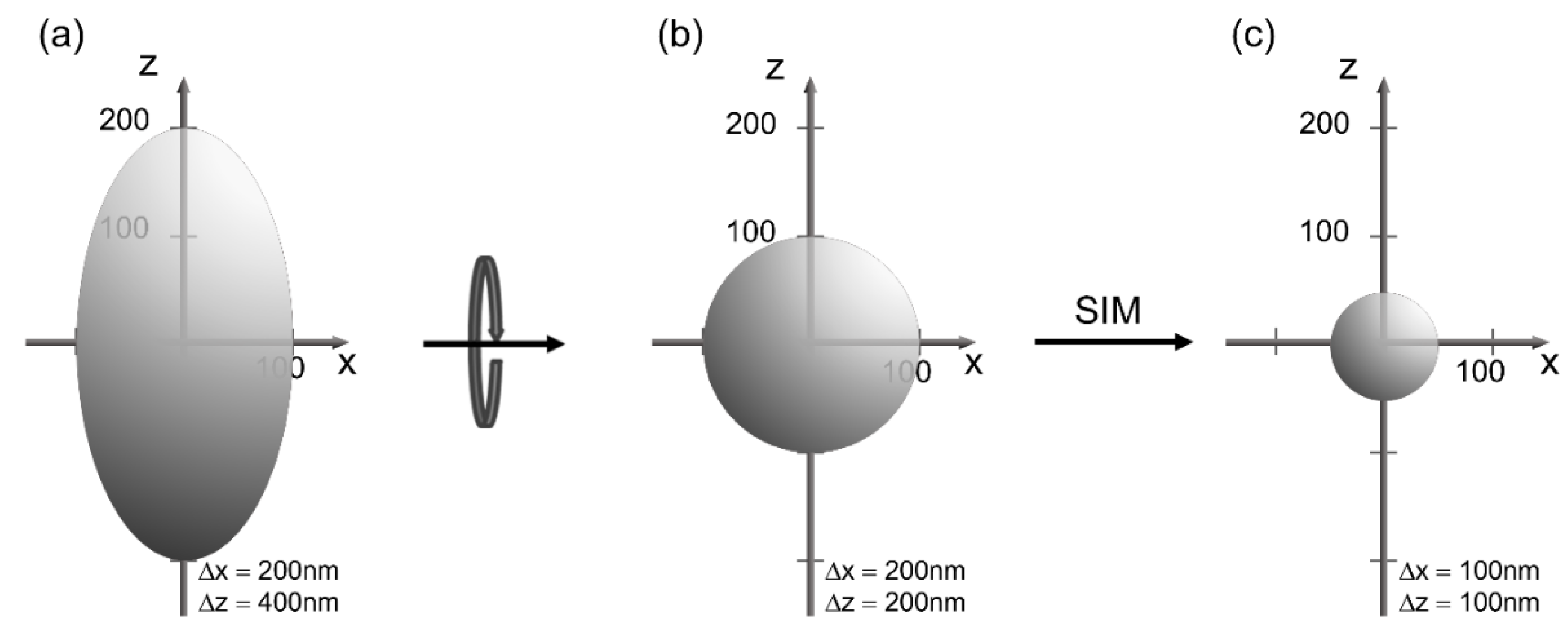
2. Microscopy
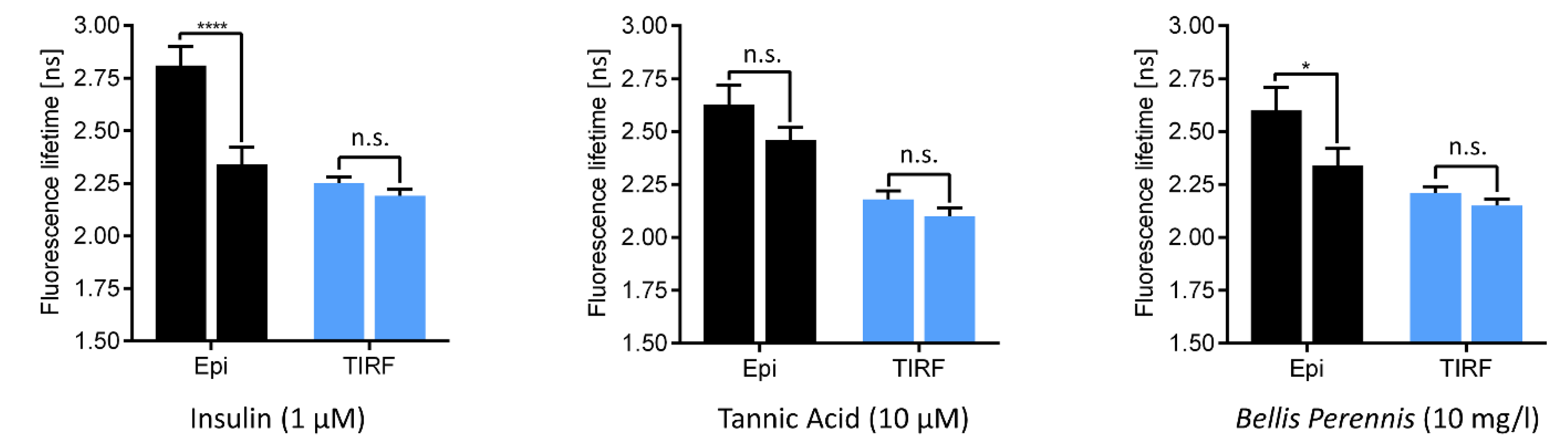
3. Sensing
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Axelrod, D. Cell-substrate contacts illuminated by total internal reflection fluorescence. J. Cell Biol. 1981, 89, 141–145. [Google Scholar] [CrossRef]
- Schneckenburger, H. Total internal reflection fluorescence microscopy: Technical innovations and novel applications. Curr. Opin. Biotechnol. 2005, 16, 13–18. [Google Scholar] [CrossRef]
- Förster, T. Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann. Phys. 1948, 437, 55–75. [Google Scholar] [CrossRef]
- Masters, B.R. Paths to Förster’s resonance energy transfer (FRET) theory. Eur. Phys. J. 2014, H39, 87–139. [Google Scholar] [CrossRef]
- Clegg, R.M.; Sener, M. From Foerster resonance energy transfer to coherent resonance energy transfer and back. In Optical Biopsy VII; Alfano, R.R., Ed.; SPIE Bios: San Francisco, CA, USA, 2010; pp. 59–79. [Google Scholar] [CrossRef]
- Schneckenburger, H. Förster resonance energy transfer–What can we learn and how can we use it? Methods Appl. Fluoresc. 2019, 8, 013001. [Google Scholar] [CrossRef]
- De Luca, G.M.R.; Breedijk, R.M.P.; Brandt, R.A.J.; Zeelenberg, C.H.C.; de Jong, B.E.; Timmermans, W.; Azar, L.N.; Hoebe, R.A.; Stallinga, S.; Manders, E.M.M. Re-scan confocal microscopy: Scanning twice for better resolution. Biomed. Opt. Express 2013, 4, 2644–2656. [Google Scholar] [CrossRef]
- Schneckenburger, H.; Richter, V. Laser Scanning versus Wide-Field−Choosing the Appropriate Microscope in Life Sciences. Appl. Sci. 2021, 11, 733. [Google Scholar] [CrossRef]
- Heintzmann, R.; Cremer, C. Laterally modulated excitation microscopy: Improvement of resolution by using a diffraction grating. In Optical Biopsies and Microscopic Techniques III; In Proceedings of the SPIE 3568, Stockholm, Sweden, 8–12 September 1998; SPIE: Bellingham, WA, USA, 1999; pp. 185–196. [Google Scholar] [CrossRef]
- Gustafsson, M.G.L.; Shao, L.; Carlton, P.M.; Wang, C.J.R.; Golubovskaya, I.N.; Cande, W.Z.; Agard, D.A.; Sedat, J.W. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys. J. 2008, 94, 4957–4970. [Google Scholar] [CrossRef]
- Hirvonen, L.M.; Wicker, K.; Mandula, O.; Heintzmann, R. Structured illumination microscopy of a living cell. Eur. Biophys. J. 2009, 38, 807–812. [Google Scholar] [CrossRef]
- Rego, E.H.; Shao, L.; Macklin, J.J.; Winoto, L.; Johansson, G.A.; Kamps-Hughes, N.; Davidson, M.W.; Gustafsson, M.G. Nonlinear structured-illumination microscopy with a photoswitchable protein reveals cellular structures at 50-nm resolution. Proc. Natl. Acad. Sci. USA 2012, 109, E135–E143. [Google Scholar] [CrossRef]
- Bi-Chang, C.; Legant, W.R.; Wang, K.; Shao, L.; Milkie, D.E.; Davidson, M.W.; Janetopoulos, C.; Wu, X.S.; Hammer, J.A., 3rd; Liu, Z. Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science 2014, 346, 1257998. [Google Scholar] [CrossRef]
- O’Shaughnessy, E.C.; Stone, O.J.; LaFosse, P.K.; Azoitei, M.L.; Tsygankov, L.; Heddleston, J.M.; Legant, W.R.; Wittchen, E.S.; Burridge, K.; Elston, T.C. Software for lattice light-sheet imaging of FRET biosensors, illustrated with a new Rap1 biosensor. J. Cell Biol. 2019, 218, 3153–3160. [Google Scholar] [CrossRef] [PubMed]
- Richter, V.; Lanzerstorfer, P.; Weghuber, J.; Schneckenburger, H. Super-resolution live cell microscopy of membrane-proximal fluorophores. Int. J. Mol. Sci. 2020, 21, 7099. [Google Scholar] [CrossRef] [PubMed]
- Stock, K.; Sailer, R.; Strauss, W.S.L.; Lyttek, M.; Steiner, R.; Schneckenburger, H. Variable-angle total internal reflection fluorescence microscopy (VA-TIRFM): Realization and application of a compact illumination device. J. Microsc. 2003, 211, 19–29. [Google Scholar] [CrossRef]
- Axelrod, D. Selective imaging of surface fluorescence with very high aperture microscope objectives. J. Biomed. Opt. 2001, 6, 6–13. [Google Scholar] [CrossRef]
- Van’t Hoff, M.; de Sars, V.; Oheim, M. A programmable light engine for quantitative single molecule TIRF and HILO imaging. Opt. Express 2008, 16, 18495–18504. [Google Scholar] [CrossRef]
- Brunstein, M.; Wicker, K.; Hérault, K.; Heintzmann, R.; Oheim, M. Full-field dual-color 100-nm super-resolution imaging reveals organization and dynamics of mitochondrial and ER networks. Opt. Express 2013, 21, 26162–26173. [Google Scholar] [CrossRef]
- Young, L.J.; Ströhl, F.; Kaminski, C.F. A Guide to Structured Illumination TIRF Microscopy at high speed with multiple colors. J. Vis. Exp. 2016, 111, e53988. [Google Scholar] [CrossRef]
- Guo, M.; Chandris, P.; Giannini, J.P.; Trexler, A.J.; Fischer, R.; Chen, J.; Vishwasrao, H.D.; Rey-Suarez, I.; Wu, Y.; Wu, X.; et al. Single-shot super-resolution total internal reflection fluorescence microscopy. Nat. Methods 2018, 15, 425–428. [Google Scholar] [CrossRef]
- Lanzerstorfer, P.; Stadlbauer, V.; Chtcheglova, L.A.; Haselgrübler, R.; Borgmann, D.; Wruss, J.; Hinterdorfer, P.; Schröder, K.; Winkler, S.M.; Höglinger, O.; et al. Identification of novel insulin mimetic drugs by quantitative total internal reflection fluorescence (TIRF) microscopy. Br. J. Pharmacol. 2014, 171, 5237–5251. [Google Scholar] [CrossRef]
- Ruckstuhl, T.; Verdes, D. Supercritical angle fluorescence (SAF) microscopy. Opt. Express 2004, 12, 4246–4254. [Google Scholar] [CrossRef] [PubMed]
- Barroca, T.; Balaa, K.; Delahaye, J.; Lévêque-Fort, S.; Fort, E. Full-field supercritical angle fluorescence microscopy for live cell imaging. Opt Lett. 2011, 36, 3051–3053. [Google Scholar] [CrossRef] [PubMed]
- Wildanger, D.; Medda, R.; Kastrup, L.; Hell, S.W. A compact STED microscope providing 3D nanoscale resolution. J. Microsc. 2009, 236, 35–43. [Google Scholar] [CrossRef]
- Balzarotti, F.; Eilers, Y.; Gwosch1, K.C.; Gynnå, A.H.; Westphal, V.; Stefani, F.D.; Elf, J.; Hell, S.W. Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes. Science 2017, 355, 606–612. [Google Scholar] [CrossRef]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–796. [Google Scholar] [CrossRef]
- Hess, S.T.; Girirajan, T.P.; Mason, M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006, 91, 4258–4272. [Google Scholar] [CrossRef]
- Cox, S.; Rosten, E.; Monypenny, J.; Jovanovic-Talisman, T.; Burnette, D.T.; Lippincott-Schwartz, J.; Jones, G.E.; Heintzmann, R. Bayesian localization microscopy reveals nanoscale podosome dynamics. Nat. Methods 2011, 9, 195–200. [Google Scholar] [CrossRef]
- Richter, V.; Bruns, S.; Bruns, T.; Weber, P.; Wagner, M.; Cremer, C.; Schneckenburger, H. Axial Tomography in Live Cell Laser Microscopy. J. Biomed. Opt. 2017, 22, 91505. [Google Scholar] [CrossRef]
- Bruns, T.; Schickinger, S.; Schneckenburger, H. Sample holder for axial rotation of specimens in 3D Microscopy. J. Microsc. 2015, 260, 30–36. [Google Scholar] [CrossRef]
- Verdaasdonk, J.S.; Stephens, A.D.; Haase, J.; Bloom, K. Bending the rules: Widefield microscopy and the Abbe limit of resolution. J. Cell. Physiol. 2014, 229, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Swedlow, J.R. Quantitative fluorescence microscopy and image deconvolution. Methods Cell Biol. 2013, 114, 407–426. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Rueden, C.T.; White, J.G.; Eliceiri, K.W. Applications of combined spectral lifetime microscopy for biology. Biotechniques 2006, 41, 249–257. [Google Scholar] [CrossRef]
- Zhu, H.; Fan, J.; Du, J.; Peng, X. Fluorescent Probes for Sensing and Imaging within Specific Cellular Organelles. Acc. Chem. Res. 2016, 49, 2115–2126. [Google Scholar] [CrossRef]
- Tregidgo, C.; Levitt, J.A.; Suhling, K. Effect of refractive index on the fluorescence lifetime of green fluorescent protein. J. Biomed. Opt. 2008, 13, 031218. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.; Zhang, J. Dynamic visualization of calcium-dependent signaling in cellular microdomains. Cell Calcium 2015, 58, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Schneckenburger, H.; Weber, P.; Wagner, M.; Enderle, S.; Kalthof, B.; Schneider, L.; Herzog, C.; Weghuber, J.; Lanzerstorfer, P. Combining TIR and FRET in molecular test systems. Int. J. Mol. Sci. 2019, 20, 648. [Google Scholar] [CrossRef] [PubMed]
- Heim, R.; Tsien, R.Y. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr. Biol. 1996, 6, 178–182. [Google Scholar] [CrossRef]
- Sorkin, A.; McClure, M.; Huang, F.; Carter, R. Interaction of EGF receptor and Grb2 in living cells visualized by fluorescence resonance energy transfer (FRET) microscopy. Curr. Biol. 2000, 10, 1395–1398. [Google Scholar] [CrossRef]
- Verveer, P.J.; Wouters, F.S.; Reynolds, A.R.; Bastiaens, P.I. Quantitative imaging of lateral ErbB1 receptor signal propagation in the plasma membrane. Science 2000, 290, 1567–1570. [Google Scholar] [CrossRef]
- Mahajan, N.P.; Harrison-Shostak, D.C.; Michaux, J.; Herman, B. Novel mutant green fluorescent protein protease substrates reveal the activation of specific caspases during apoptosis. Chem. Biol. 1999, 6, 401–409. [Google Scholar] [CrossRef]
- Angres, B.; Steuer, H.; Weber, P.; Wagner, M.; Schneckenburger, H. A membrane-bound FRET-based caspase sensor for detection of apoptosis using fluorescence lifetime and total internal reflection microscopy. Cytometry A 2009, 75, 420–427. [Google Scholar] [CrossRef]
- Kiyokawa, E.; Hara, S.; Nakamura, T.; Matsuda, M. Fluorescence (Förster) resonance energy transfer imaging of oncogene activity in living cells. Cancer Sci. 2006, 97, 8–15. [Google Scholar] [CrossRef]
- Von Arnim, C.A.; von Einem, B.; Weber, P.; Wagner, M.; Schwanzar, D.; Spoelgen, R.; Strauss, W.L.; Schneckenburger, H. Impact of cholesterol level upon APP and BACE proximity and APP cleavage. Biochem. Biophys. Res. Commun. 2008, 370, 207–212. [Google Scholar] [CrossRef]
- Levitt, J.A.; Matthews, D.R.; Ameer-Beg, S.M.; Suhling, K. Fluorescence lifetime and polarization-resolved imaging in cell biology. Curr. Opin. Biotechnol. 2009, 20, 28–36. [Google Scholar] [CrossRef]
- Grewal, T.; Enrich, C. Annexins-modulators of EGF receptor signalling and trafficking. Cell Signal. 2009, 21, 847–858. [Google Scholar] [CrossRef]
- Schneckenburger, H.; Weber, P.; Wagner, M.; Enderle, S.; Weghuber, J.; Lanzerstorfer, P. Combining TIR and FRET: From fluorescence microscopy to a multi-well reader system. Adv. Microsc. Imaging Proc. SPIE-OSA 2019, 11076, 110761C. [Google Scholar] [CrossRef]
- Bruns, T.; Strauss, W.S.L.; Sailer, R.; Wagner, M.; Schneckenburger, H. Total internal reflectance fluorescence reader for selective investigations of cell membranes. J. Biomed. Opt. 2006, 11, 34011. [Google Scholar] [CrossRef]
- Stadlbauer, V.; Lanzerstorfer, P.; Neuhauser, C.; Weber, F.; Stübl, F.; Weber, P.; Wagner, M.; Plochberger, B.; Wieser, S.; Schneckenburger, H.; et al. Fluorescence Microscopy-Based Quantitation of GLUT4 Translocation: High Throughput or High Content? Int. J. Mol. Sci. 2020, 21, 7964. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organelle | Diameter (typ.) | Technique |
|---|---|---|
| nucleus | 6–12 µm | SIM 1 |
| mitochondrium | 0.5–1.5 µm | |
| lysosome | 0.1–1.2 µm | |
| microtubule | ~25 nm | STED/MINFLUX/SMLM 1 |
| Actin filament | ~7 nm | |
| membrane | 4–5 nm | TIRFM |
| protein | 3–6 nm |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richter, V.; Lanzerstorfer, P.; Weghuber, J.; Schneckenburger, H. Probing Small Distances in Live Cell Imaging. Photonics 2021, 8, 176. https://doi.org/10.3390/photonics8060176
Richter V, Lanzerstorfer P, Weghuber J, Schneckenburger H. Probing Small Distances in Live Cell Imaging. Photonics. 2021; 8(6):176. https://doi.org/10.3390/photonics8060176
Chicago/Turabian StyleRichter, Verena, Peter Lanzerstorfer, Julian Weghuber, and Herbert Schneckenburger. 2021. "Probing Small Distances in Live Cell Imaging" Photonics 8, no. 6: 176. https://doi.org/10.3390/photonics8060176
APA StyleRichter, V., Lanzerstorfer, P., Weghuber, J., & Schneckenburger, H. (2021). Probing Small Distances in Live Cell Imaging. Photonics, 8(6), 176. https://doi.org/10.3390/photonics8060176