
Optimization of an Analytical Method for Indoxacarb Residues in Fourteen Medicinal Herbs Using GC–μECD, GC–MS/MS and LC–MS/MS

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemical and Reagents
2.2. Medicinal Herbs
2.3. Sample Preparation
2.4. SPE Clean-Up
2.5. Instrumentation
2.5.1. GC–μECD Analysis
2.5.2. GC–MS/MS Analysis
2.5.3. LC–MS/MS Analysis
2.6. Method Validation
2.7. Matrix Effect
3. Results and Discussion
3.1. Selection of Extraction Method
3.2. Optimization of SPE Condition
3.3. Method Validation
3.3.1. GC–μECD Analysis
3.3.2. GC–MS/MS Analysis
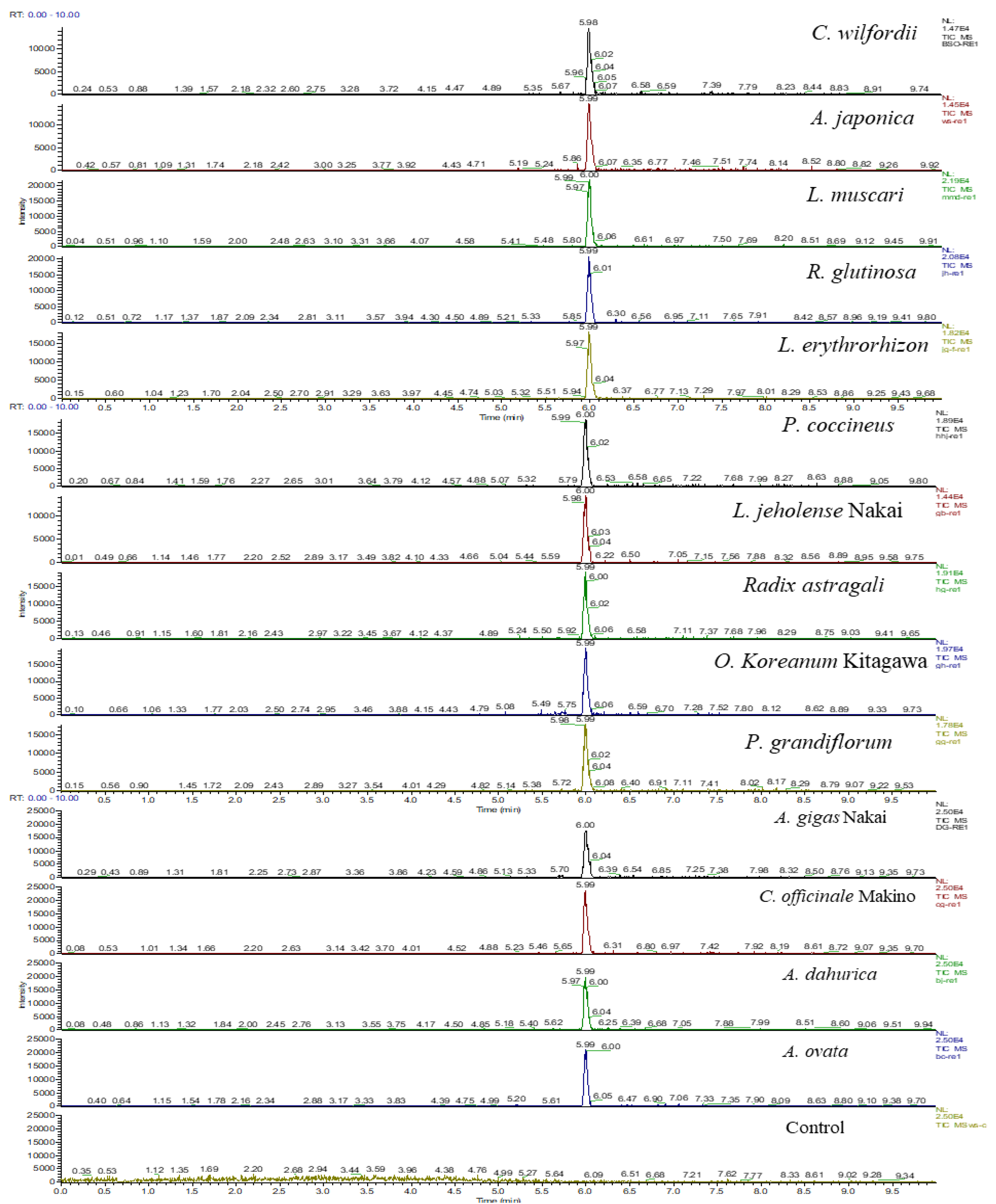
3.3.3. LC–MS/MS Analysis
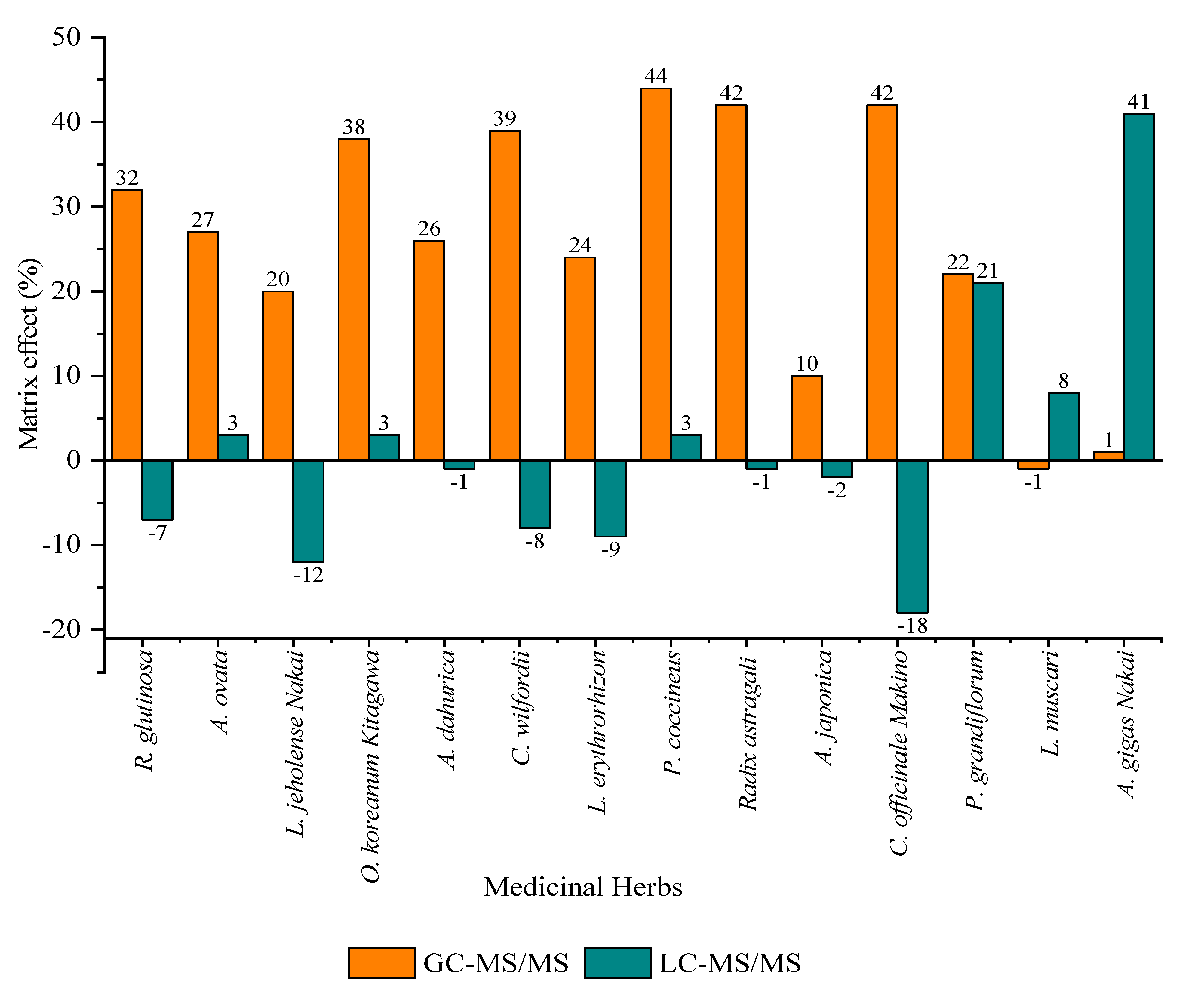
3.3.4. Matrix Effect
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nguyen, T.D.; Lee, K.J.; Lee, M.H.; Lee, G.H. A multiresidue method for the determination 234 pesticides in Korean herbs using gas chromatography mass spectrometry. Microchem. J. 2010, 95, 43–49. [Google Scholar] [CrossRef]
- Luo, Z.; Zhang, L.; Mou, Y.; Cui, S.; Gu, Z.; Yu, J.; Ma, X. Multi-residue analysis of plant growth regulators and pesticides in traditional Chinese medicines by high-performance liquid chromatography coupled with tandem mass spectrometry. Anal. Bioanal. Chem. 2019, 411, 2447–2460. [Google Scholar] [CrossRef] [PubMed]
- Rural Development Administration. 2019. Available online: http://www.rda.go.Kr (accessed on 26 July 2022).
- Turner, J.A. The Pesticide Manual, 17th ed.; BPC Publications: Alton, UK, 2015. [Google Scholar]
- Li, Y.H.; Wang, X.Y.; Hua, W.; Zhang, H. Studies on Dissipations and Residues of Indoxacarb under Different Field and Environmental Conditions. J. Anal. Methods Chem. 2020, 2020, 8874759. [Google Scholar] [CrossRef] [PubMed]
- Rural Development Administration. 2021. Available online: http://www.rda.go.Kr (accessed on 26 July 2022).
- Naik, H.R.; Pallavi, M.S.; Chawan, R.; Bheemanna, M.; Naik, A.; Paramasivam, M. Method Development and Validation for Determination of Indoxacarb Using LC-ESI-MS/MS and Its Dissipation Kinetics in Pigeonpea. Food Anal. Methods 2020, 13, 647–657. [Google Scholar] [CrossRef]
- Hajjo, R.M.; Afifi, F.U.; Battah, A.H. Multiresidue pesticide analysis of the medicinal plant Origanum syriacum. Food Addit. Contam. 2007, 24, 274–279. [Google Scholar] [CrossRef]
- Parrilla Vázquez, P.; Ferrer, C.; Martínez Bueno, M.J.; Fernández-Alba, A.R. Pesticide residues in spices and herbs: Sample preparation methods and determination by chromatographic techniques. TrAC Trends Anal. Chem. 2019, 115, 13–22. [Google Scholar] [CrossRef]
- Saha, A.; Makwana, C.; Manivel, P. QuEChERS-based gas chromatography-electron capture/flame photometric detection method for multi-pesticide residues analysis in Andrographis paniculata: A popular Indian medicinal herb. Int. J. Environ. Anal. Chem. 2020, 100, 1669–1690. [Google Scholar] [CrossRef]
- Tripathy, V.; Saha, A.; Kumar, J. Detection of pesticides in popular medicinal herbs: A modified QuEChERS and gas chromatography–mass spectrometry based approach. J. Food Sci. Technol. 2017, 54, 458–468. [Google Scholar] [CrossRef]
- Yang, R.; Liu, W.; Wang, J. Application of the QuEChERS Sample Preparation Method for the Determination of Carbamate Pesticides in Flos Carthami Samples by UPLC-MS/MS. Int. J. Sci. 2016, 3, 162–170. [Google Scholar]
- Anastassiades, M.; Lehotay, S.J.; Štajnbaher, D.; Schenck, F.J. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J. AOAC Int. 2003, 86, 412–431. [Google Scholar] [CrossRef]
- Yudan, W.; Yanwei, F.; Yunyun, W.; Qian, L.; Haonan, R.; Jiaoyang, L.; Meihua, Y. A comprehensive review on the pretreatment and detection methods of neonicotinoid insecticides in food and environmental samples. Food Chem. X 2022, 15, 100375. [Google Scholar] [CrossRef]
- Grahovac, N.L.; Stojanović, Z.S.; Kravić, S.Ž.; Orčić, D.Z.; Suturović, Z.J.; Kondić-Špika, A.Đ.; Vasin, J.R.; Šunjka, D.B.; Jakšić, S.P.; Rajković, M.M.; et al. Determination of Residues of Sulfonylurea Herbicides in Soil by Using Microwave-Assisted Extraction and High Performance Liquid Chromatographic Method. Hem. Ind. 2017, 71, 289–298. [Google Scholar] [CrossRef]
- Đurović, A.; Stojanović, Z.; Kravić, S.; Grahovac, N.; Bursić, V.; Vuković, G.; Suturović, Z. Development and Validation of Chronopotentiometric Method for Imidacloprid Determination in Pesticide Formulations and River Water Samples. Int. J. Anal. Chem. 2016, 2016, 5138491. [Google Scholar] [CrossRef]
- Stojanović, Z.; Đurović, A.; Kravić, S.; Grahovac, N.; Suturović, Z.; Bursić, V.; Vuković, V.; Brezoa, T. A simple and rapid electrochemical sensing method for metribuzin determination in tap and river water samples. Anal. Methods 2016, 8, 2698–2705. [Google Scholar] [CrossRef]
- Ghosh, S.; AlKafaas, S.S.; Bornman, C.; Apollon, W.; Hussien, A.M.; Badawy, A.E.; Amer, M.H.; Kamel, M.B.; Mekawy, E.A.; Bedair, H. The application of rapid test paper technology for pesticide detection in horticulture crops: A comprehensive review. Beni-Suef Univ. J. Basic Appl. Sci. 2022, 11, 73. [Google Scholar] [CrossRef]
- Đurović, A.; Stojanović, Z.; Kravić, S.; Rakić, D.; Grahovac, N. Novel electrochemical procedure for the determination of metamitron. Int. J. Environ. Anal. Chem. 2018, 98, 369–385. [Google Scholar] [CrossRef]
- Nunes, G.S.; Toscano, I.A.; Barceló, D. Analysis of pesticides in food and environmental samples by enzyme-linked immunosorbent assays. TrAC Trends Anal. Chem. 1998, 17, 79–87. [Google Scholar] [CrossRef]
- Tong, H.; Tong, Y.; Xue, J.; Liu, D.; Wu, X. Multi-residual Pesticide Monitoring in Commercial Chinese Herbal Medicines by Gas Chromatography-Triple Quadrupole Tandem Mass Spectrometry. Food Anal. Methods 2014, 7, 135–145. [Google Scholar] [CrossRef]
- He, P.; Aga, D.S. Comparison of GC-MS/MS and LC-MS/MS for the analysis of hormones and pesticides in surface waters: Advantages and pitfalls. Anal. Methods 2019, 11, 1436–1448. [Google Scholar] [CrossRef]
- Choi, J.Y.; Lee, Y.J.; Ham, H.J.; Ishag, A.A.E.S.; Hur, J.H. A Study on Improvement of the Analytical Method of Chlorantraniliprole Residue in Herbal Medicine (Rehmannia glutinosa Libosch) using HPLC-UVD. Korean J. Pestic. Sci. 2021, 25, 196–211. [Google Scholar] [CrossRef]
- Sardar, S.W.; Byeon, G.D.; Choi, J.Y.; Ham, H.J.; Ishag, A.E.S.A.; Hur, J.H. Residual characteristics and safety assessment of the insecticides spiromesifen and chromafenozide in lettuce and perilla. Sci. Rep. 2022, 12, 4675. [Google Scholar] [CrossRef] [PubMed]
- Ham, H.-J.; Leeb, B.-g.; Choi, J.-Y.; Ishag, S.A.A.; Hur, J.-H. Establishment of Analyzing Method of Fenpropathrin Residue in Angelica dahurica Radix and Osterici radix. Curr. Tradit. Med. 2022, 8, e010722206556. [Google Scholar] [CrossRef]
- Łozowicka, B.; Rutkowska, E.; Jankowska, M. Influence of QuEChERS modifications on recovery and matrix effect during the multi-residue pesticide analysis in soil by GC/MS/MS and GC/ECD/NPD. Environ. Sci. Pollut. Res. 2017, 24, 7124–7138. [Google Scholar] [CrossRef]
- Choung, M.G. Development of analytical method for valinamide carbamate fungicide iprovalicarb residue. J. Korean Soc. Int. Agric. 2019, 31, 25–33. [Google Scholar]
- Tengfei, L.; Minghui, D.; Fengjie, Z.; Daifeng, Y.; Xueming, Z. Development and validation of an analytical method for detecting chlorantraniliprole residues in fresh tea leaves. Food Sci. Hum. Wellness 2019, 8, 362–367. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, H.; Liu, Y.; Wang, J.; Zhang, Y.C.; Dong, A.J.; Zhao, H.T.; Sun, C.H.; Cui, J. Multiresidue method for determination of 88 pesticides in berry fruits using solid-phase extraction and gas chromatography-mass spectrometry: Determination of 88 pesticides in berries using SPE and GC-MS. Food Chem. 2011, 127, 855–865. [Google Scholar] [CrossRef]
- Acikara, B.Ö. Ion-Exchange Chromatography and Its Applications. In Column Chromatography; Martin, D.F., Martin, B.B., Eds.; IntechOpen: London, UK, 2013. [Google Scholar] [CrossRef]
- Stachniuk, A.; Fornal, E. Liquid Chromatography-Mass Spectrometry in the Analysis of Pesticide Residues in Food. Food Anal. Methods 2016, 9, 1654–1665. [Google Scholar] [CrossRef]
- SANTE. Guidance SANTE 11312/2021—Analytical Quality Control and Method Validation Procedures for Pesticide Residues Analysis in Food and Feed. Directorate General Health and Consumer Protection, European Commission. Available online: https://www.accredia.it/en/documento/guidance-sante-11312-2021-analytical-quality-control-and-method-validation-procedures-for-pesticide-residues-analysis-in-food-and-feed/ (accessed on 1 March 2022).
- Sardar, S.W.; Choi, Y.; Park, N.; Jeon, J. Occurrence and Concentration of Chemical Additives in Consumer Products in Korea. Int. J. Environ. Res. Public Health 2019, 16, 5075. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Z.; Gao, F.; Song, D.; Lu, X. Selection of representative matrices for the multiresidue analysis of pesticides in tea by GC-MS/MS. Anal. Methods 2018, 10, 855–866. [Google Scholar] [CrossRef]
- De Sousa, F.A.; Guido Costa, A.I.; De Queiroz, M.E.L.R.; Teófilo, R.F.; Neves, A.A.; De Pinho, G.P. Evaluation of matrix effect on the GC response of eleven pesticides by PCA. Food Chem. 2012, 135, 179–185. [Google Scholar] [CrossRef]
- Rajski, Ł.; Lozano, A.; Uclés, A.; Ferrer, C.; Fernández-Alba, A.R. Determination of pesticide residues in high oil vegetal commodities by using various multi-residue methods and clean-ups followed by liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2013, 1304, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Uclés, S.; Lozano, A.; Sosa, A.; Parrilla Vázquez, P.; Valverde, A.; Fernández-Alba, A.R. Matrix interference evaluation employing GC and LC coupled to triple quadrupole tandem mass spectrometry. Talanta 2017, 174, 72–81. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Medicinal Herbs | Average Recovery (±SD *) |
|---|---|
| P. coccineus | 89.4 ± 3.51 |
| Radix astragali | 97.8 ± 12.32 |
| A. japonica | 85.6 ± 3.88 |
| P. grandiflorum | 107.4 ± 9.70 |
| L. muscari | 114.6 ± 3.54 |
| C. wilfordii | 79.7 ± 8.60 |
| R. glutinosa | 117.6 ± 3.08 |
| Medicinal Herbs | Average Recovery (±SD *) |
|---|---|
| R. glutinosa | 85.2 ± 5.6 |
| A. ovata | 83.0 ± 4.0 |
| L. jeholense Nakai | 105.9 ± 6.3 |
| O. koreanum Kitagawa | 81.5 ± 2.7 |
| A. dahurica | 86.5 ± 0.6 |
| C. wilfordii | 86.5 ± 1.0 |
| L. erythrorhizon | 85.7 ± 2.4 |
| P. coccineus | 82.6 ± 2.9 |
| Radix astragali | 100.4 ± 4.0 |
| A. japonica | 74.1 ± 2.1 |
| C. officinale Makino | 86.4 ± 5.9 |
| P. grandiflorum | 79.2 ± 1.8 |
| L. muscari | 81.2 ± 3.3 |
| A. gigas Nakai | 85.8 ± 1.8 |
| Medicinal Herbs | Average Recovery (±SD *) |
|---|---|
| R. glutinosa | 99.0 ± 3.7 |
| A. ovata | 73.0 ± 2.5 |
| L. jeholense Nakai | 91.3 ± 2.5 |
| O. koreanum Kitagawa | 75.8 ± 2.2 |
| A. dahurica | 75.2 ± 1.6 |
| C. wilfordii | 90.0 ± 13.9 |
| L. erythrorhizon | 94.9 ± 2.9 |
| P. coccineus | 84.2 ± 4.3 |
| Radix astragali | 93.7 ± 3.2 |
| A. japonica | 84.5 ± 6.4 |
| C. officinale Makino | 96.0 ± 4.4 |
| P. grandiflorum | 75.5 ± 4.8 |
| L. muscari | 88.1 ± 2.0 |
| A. gigas Nakai | 80.5 ± 5.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ham, H.-J.; Sardar, S.W.; Ishag, A.E.S.A.; Choi, J.-Y.; Hur, J.-H. Optimization of an Analytical Method for Indoxacarb Residues in Fourteen Medicinal Herbs Using GC–μECD, GC–MS/MS and LC–MS/MS. Separations 2022, 9, 232. https://doi.org/10.3390/separations9090232
Ham H-J, Sardar SW, Ishag AESA, Choi J-Y, Hur J-H. Optimization of an Analytical Method for Indoxacarb Residues in Fourteen Medicinal Herbs Using GC–μECD, GC–MS/MS and LC–MS/MS. Separations. 2022; 9(9):232. https://doi.org/10.3390/separations9090232
Chicago/Turabian StyleHam, Hun-Ju, Syed Wasim Sardar, Abd Elaziz Sulieman Ahmed Ishag, Jeong-Yoon Choi, and Jang-Hyun Hur. 2022. "Optimization of an Analytical Method for Indoxacarb Residues in Fourteen Medicinal Herbs Using GC–μECD, GC–MS/MS and LC–MS/MS" Separations 9, no. 9: 232. https://doi.org/10.3390/separations9090232
APA StyleHam, H.-J., Sardar, S. W., Ishag, A. E. S. A., Choi, J.-Y., & Hur, J.-H. (2022). Optimization of an Analytical Method for Indoxacarb Residues in Fourteen Medicinal Herbs Using GC–μECD, GC–MS/MS and LC–MS/MS. Separations, 9(9), 232. https://doi.org/10.3390/separations9090232