
Homemade Pipette Tip Solid-Phase Extraction for the Simultaneous Determination of 40 Drugs of Abuse in Urine by Liquid Chromatography–Tandem Mass Spectrometry





Abstract
:1. Introduction
2. Materials and Methods
2.1. Standards and Materials
2.2. LC-MS/MS Conditions
2.3. Preparation of Homemade PT-SPE Tips
2.4. Urine Collection and PT-SPE Pre-Treatment
2.5. Validation
3. Results
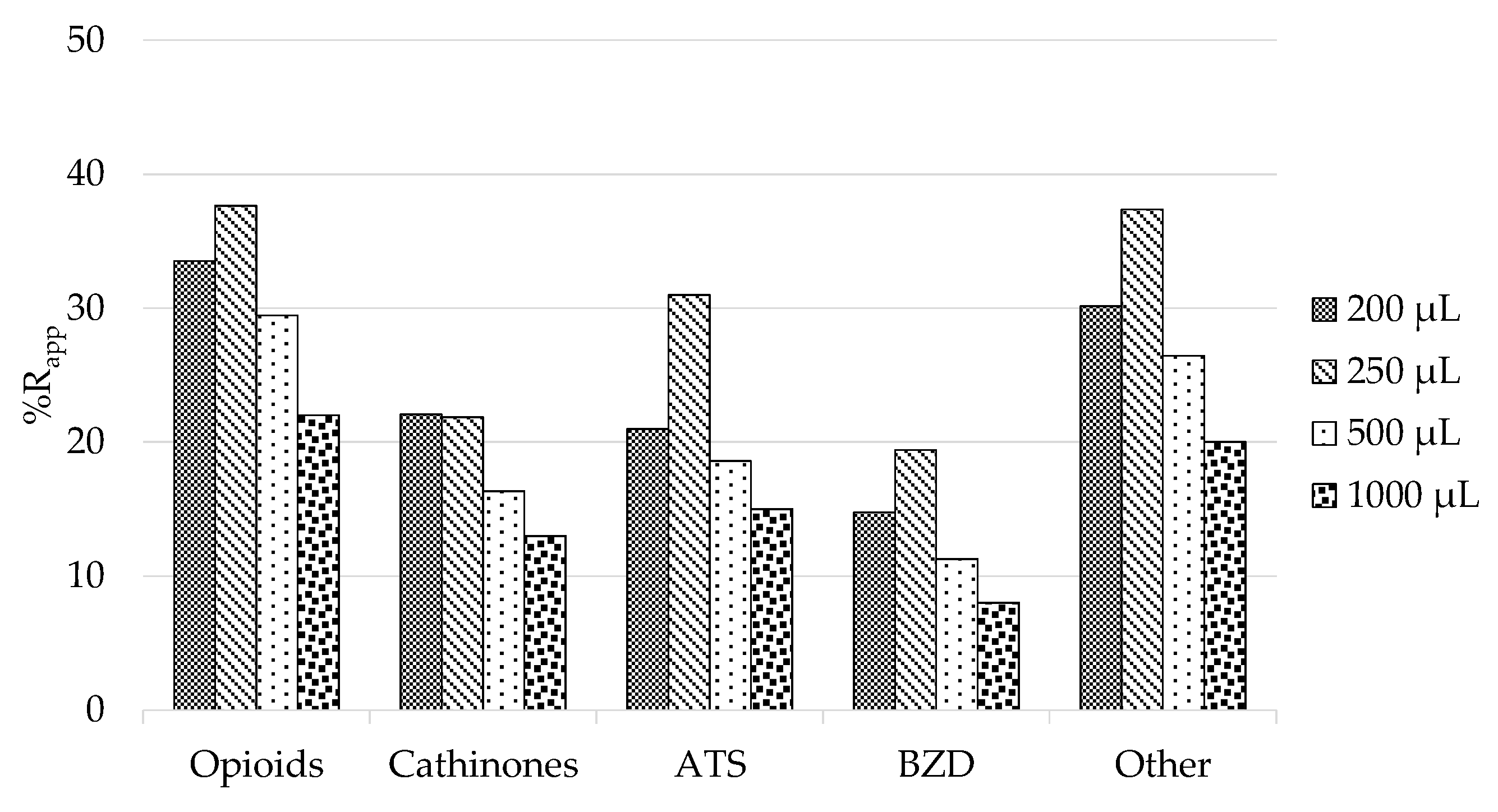
3.1. PT-SPE Optimization
3.2. Method Validation
3.3. PT-SPE LC-MS/MS Application to Real Cases
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- United Nations Office on Drugs and Crimes World Drug Report 2021. Global Overview: Drug Demand; Drug Supply. ISBN 9789211483611. 2021. Available online: https://www.unodc.org/res/wdr2021/field/WDR21_Booklet_2.pdf (accessed on 7 April 2022).
- United Nations Office on Drugs and Crimes World Drug Report 2021. Drug Market Trends: Cocaine; Amphetamine-Type Substances. ISBN 9789211483611. 2021. Available online: https://www.unodc.org/res/wdr2021/field/WDR21_Booklet_4.pdf (accessed on 7 April 2022).
- United Nations Office on Drugs and Crime World Drug Report 2021. Drug Market Trends: Cannabis. Opioids. Available online: https://www.unodc.org/res/wdr2021/field/WDR21_Booklet_3.pdf (accessed on 7 April 2022).
- Saito, K.; Saito, R.; Kikuchi, Y.; Iwasaki, Y.; Ito, R.; Nakazawa, H. Analysis of Drugs of Abuse in Biological Specimens. J. Health Sci. 2011, 57, 472–487. [Google Scholar] [CrossRef]
- Singh, Z. Forensic Toxicology: Biological Sampling and Use of Different Analytical Techniques. Forensic Res. Criminol. Int. J. 2017, 4, 117–120. [Google Scholar] [CrossRef]
- Young, G.M.; Lurie, I.S. Recent Forensic Applications of Enhanced Chromatographic Separation Methods. J. Sep. Sci. 2022, 45, 369–381. [Google Scholar] [CrossRef]
- Liu, L.; Wheeler, S.E.; Venkataramanan, R.; Rymer, J.A.; Pizon, A.F.; Lynch, M.J.; Tamama, K. Newly Emerging Drugs of Abuse and Their Detection Methods: An ACLPS Critical Review. Am. J. Clin. Pathol. 2018, 149, 105–116. [Google Scholar] [CrossRef]
- Pascual-Caro, S.; Borrull, F.; Calull, M.; Aguilar, C. Recent Chromatographic and Electrophoretic Based Methods for Determining Drugs of Abuse in Urine and Oral Fluid: A Review from 2018 to June 2021. Trends Anal. Chem. 2022, 156, 116705. [Google Scholar] [CrossRef]
- Substance Abuse and Mental Health Services Administration Clinical Drug Testing in Primary Care. Available online: https://store.samhsa.gov/sites/default/files/d7/priv/sma12-4668.pdf (accessed on 14 February 2022).
- Institóris, L.; Árok, Z.; Seprenyi, K.; Varga, T.; Sára-Klausz, G.; Keller, É.; Tóth, R.A.; Sala, L.; Kereszty, É.; Róna, K. Frequency and Structure of Stimulant Designer Drug Consumption among Suspected Drug Users in Budapest and South-East Hungary in 2012–2013. Forensic Sci. Int. 2015, 248, 181–186. [Google Scholar] [CrossRef]
- Kraemer, T.; Roditis, S.K.; Peters, F.T.; Maurer, H.H. Amphetamine Concentrations in Human Urine Following Single-Dose Administration of the Calcium Antagonist Prenylamine—Studies Using Fluorescence Polarization Immunoassay (FPIA) and GC-MS. J. Anal. Toxicol. 2003, 27, 68–73. [Google Scholar] [CrossRef]
- Rosano, T.G.; Ohouo, P.Y.; Wood, M. Screening with Quantification for 64 Drugs and Metabolites in Human Urine Using UPLC-MS-MS Analysis and a Threshold Accurate Calibration. J. Anal. Toxicol. 2017, 41, 536–546. [Google Scholar] [CrossRef]
- De Campos, E.G.; da Costa, B.R.B.; dos Santos, F.S.; Monedeiro, F.; Alves, M.N.R.; Santos Junior, W.J.R.; de Martinis, B.S. Alternative Matrices in Forensic Toxicology: A Critical Review. Forensic Toxicol. 2022, 40, 1–18. [Google Scholar] [CrossRef]
- Ramos, L. Critical Overview of Selected Contemporary Sample Preparation Techniques. J. Chromatogr. A 2012, 1221, 84–98. [Google Scholar] [CrossRef]
- Bylda, C.; Thiele, R.; Kobold, U.; Volmer, D.A. Recent Advances in Sample Preparation Techniques to Overcome Difficulties Encountered during Quantitative Analysis of Small Molecules from Biofluids Using LC-MS/MS. Analyst 2014, 139, 2265–2276. [Google Scholar] [CrossRef]
- Bordin, D.C.M.; Alves, M.N.R.; de Campos, E.G.; de Martinis, B.S. Disposable Pipette Tips Extraction: Fundamentals, Applications and State of the Art. J. Sep. Sci. 2016, 39, 1168–1172. [Google Scholar] [CrossRef]
- Meyer, M.R.; Maurer, H.H. Current Status of Hyphenated Mass Spectrometry in Studies of the Metabolism of Drugs of Abuse, Including Doping Agents. Anal. Bioanal. Chem. 2012, 402, 195–208. [Google Scholar] [CrossRef]
- Kole, P.L.; Venkatesh, G.; Kotecha, J.; Sheshala, R. Recent Advances in Sample Preparation Techniques for Effective Bioanalytical Methods. Biomed. Chromatogr. 2011, 25, 199–217. [Google Scholar] [CrossRef]
- Li, N.; Zhang, T.; Chen, G.; Xu, J.; Ouyang, G.; Zhu, F. Recent Advances in Sample Preparation Techniques for Quantitative Detection of Pharmaceuticals in Biological Samples. Trends Anal. Chem. 2021, 142, 116318. [Google Scholar] [CrossRef]
- Turoňová, D.; Kujovská Krčmová, L.; Švec, F. Application of Microextraction in Pipette Tips in Clinical and Forensic Toxicology. Trends Anal. Chem. 2021, 143, 116404. [Google Scholar] [CrossRef]
- Kataoka, H. Recent Developments and Applications of Microextraction Techniques in Drug Analysis. Anal. Bioanal. Chem. 2010, 396, 339–364. [Google Scholar] [CrossRef]
- Seidi, S.; Tajik, M.; Baharfar, M.; Rezazadeh, M. Micro Solid-Phase Extraction (Pipette Tip and Spin Column) and Thin Film Solid-Phase Microextraction: Miniaturized Concepts for Chromatographic Analysis. Trends Anal. Chem. 2019, 118, 810–827. [Google Scholar] [CrossRef]
- Sorribes-Soriano, A.; Valencia, A.; Esteve-Turrillas, F.A.; Armenta, S.; Herrero-Martínez, J.M. Development of Pipette Tip-Based Poly(methacrylic acid-co-ethylene glycol dimethacrylate) Monolith for the Extraction of Drugs of Abuse from Oral Fluid Samples. Talanta 2019, 205, 120158. [Google Scholar] [CrossRef]
- Carasek, E.; Morés, L.; Huelsmann, R.D. Disposable Pipette Extraction: A Critical Review of Concepts, Applications, and Directions. Anal. Chim. Acta 2021, 1192, 339383. [Google Scholar] [CrossRef]
- He, X.; Sun, T.; Wang, L.; Jiang, X. Pipette-Tip Micro-Solid Phase Extraction Based on Melamine-Foam@polydopamine Followed by Ultra-High-Performance Liquid Chromatography–Quadrupole–Time-of-Flight Mass Spectrometry for Detection of Psychotropic Drugs in Human Serum. J. Chromatogr. B 2021, 1163, 122499. [Google Scholar] [CrossRef]
- Carneiro Cruz, J.; Cabral Miranda, L.F.; Costa Queiroz, M.E. Pipette Tip Micro-Solid Phase Extraction (Octyl-Functionalized Hybrid Silica Monolith) and Ultra-High-Performance Liquid Chromatography-Tandem Mass Spectrometry to Determine Cannabidiol and Tetrahydrocannabinol in Plasma Samples. J. Sep. Sci. 2021, 44, 1621–1632. [Google Scholar] [CrossRef]
- Ellison, S.T.; Brewer, W.E.; Morgan, S.L. Comprehensive Analysis of Drugs of Abuse in Urine Using Disposable Pipette Extraction. J. Anal. Toxicol. 2009, 33, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.L.; Marinetti, L.J.; Smith, R.K.; Brewer, W.E.; Clelland, B.L.; Morgan, S.L. The Analysis of Δ9-Tetrahydrocannabinol and Metabolite in Whole Blood and 11-nor-Δ9-Tetrahydrocannabinol-9-Carboxylic Acid in Urine Using Disposable Pipette Extraction with Confirmation and Quantification by Gas Chromatography-Mass Spectrometry. J. Anal. Toxicol. 2008, 32, 659–666. [Google Scholar] [CrossRef]
- Vlčková, H.; Pilařová, V.; Novák, O.; Solich, P.; Nováková, L. Micro-SPE in Pipette Tips as a Tool for Analysis of Small-Molecule Drugs in Serum. Bioanalysis 2017, 9, 887–901. [Google Scholar] [CrossRef]
- Shi, J.W.; Zhou, J.F.; He, X.; Zhang, Y. Rapid Analysis of Four Amphetamines in Urine by Self-Made Pipette-Tip Solid-Phase Extraction Followed by GC-MS/MS. J. Chromatogr. Sci. 2020, 58, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Mozaner Bordin, D.C.; Alves, M.N.R.; Cabrices, O.G.; de Campos, E.G.; de Martinis, B.S. A Rapid Assay for the Simultaneous Determination of Nicotine, Cocaine and Metabolites in Meconium Using Disposable Pipette Extraction and Gas Chromatography-Mass Spectrometry (GC-MS). J. Anal. Toxicol. 2014, 38, 31–38. [Google Scholar] [CrossRef]
- Montesano, C.; Sergi, M.; Odoardi, S.; Simeoni, M.C.; Compagnone, D.; Curini, R. A μ-SPE Procedure for the Determination of Cannabinoids and Their Metabolites in Urine by LC-MS/MS. J. Pharm. Biomed. 2014, 91, 169–175. [Google Scholar] [CrossRef]
- Kumazawa, T.; Hasegawa, C.; Lee, X.P.; Hara, K.; Seno, H.; Suzuki, O.; Sato, K. Simultaneous Determination of Methamphetamine and Amphetamine in Human Urine Using Pipette Tip Solid-Phase Extraction and Gas Chromatography-Mass Spectrometry. J. Pharm. Biomed. 2007, 44, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Napoletano, S.; Montesano, C.; Compagnone, D.; Curini, R.; D’Ascenzo, G.; Roccia, C.; Sergi, M. Determination of Illicit Drugs in Urine and Plasma by Micro-SPE Followed by HPLC-MS/MS. Chromatographia 2012, 75, 55–63. [Google Scholar] [CrossRef]
- Chen, X.; Wu, X.; Luan, T.; Jiang, R.; Ouyang, G. Sample Preparation and Instrumental Methods for Illicit Drugs in Environmental and Biological Samples: A Review. J. Chromatogr. A 2021, 1640, 461961. [Google Scholar] [CrossRef] [PubMed]
- Salazar Mogollón, N.G.; Quiroz-moreno, C.D.; Prata, P.S.; de Almeida, J.R.; Sof, A.; Augusto, F. New Advances in Toxicological Forensic Analysis Using Mass Spectrometry Techniques. J. Anal. Methods Chem. 2018, 2018, 4142527. [Google Scholar] [CrossRef]
- Pascual-Caro, S.; Borrull, F.; Aguilar, C.; Calull, M. Development of a Liquid Chromatography—Tandem Mass Spectrometry Method for the Simultaneous Determination of 40 Drugs of Abuse in Human Urine: Application to Real Cases. J. Anal. Toxicol. 2022, bkac020. [Google Scholar] [CrossRef] [PubMed]
- Taskinen, S.; Beck, O.; Bosch, T.; Brcak, M.; Carmichael, D.; Fucci, N.; George, C.; Piper, M.; Salomone, A.; Schielen, W.; et al. European Guidelines for Workplace Drug Testing in Urine. Drug Test. Anal. 2017, 9, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Official Journal of the European Communities EC/96/23 Commission Decision of 12 August 2002 Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results. ISBN 9788578110796. 2002. Available online: http://extwprlegs1.fao.org/docs/pdf/eur49615.pdf (accessed on 7 April 2022).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Optimum Condition |
|---|---|
| Type of sorbent | ExtraBond SCX |
| Amount of sorbent | 10 mg |
| Conditioning | 1 cycle of 500 µL of MeOH and 1 cycle of 500 µL of H2O at pH 4 |
| Loading sample volume and cycles | 1 cycle of 250 µL of the mixture urine:H2O at pH 4 (50:50, v/v) |
| Washing volume and cycles | 1 cycle of 500 µL of MeOH |
| Elution volume and cycles | 1 cycle of 1000 µL of 5% NH4OH in MeOH |
| Compound | MDL a | MQL a | Linear Range a | %Rapp b | %ME b | %Rapp c | %MEc | %Rapp d | %ME d | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | MOR | 0.050 | 0.250 | 0.25–5 5–100 | 55 | −50 | 55 | −42 | 54 | −47 |
| 2 | COD | 0.050 | 0.250 | 0.25–5 5–75 | 42 | −41 | 58 | −36 | 53 | −27 |
| 3 | Methylephedrine | 0.500 | 1.500 | 1.5–75 | 40 | −34 | 42 | −35 | 47 | −34 |
| 4 | DMC | 0.250 | 1.000 | 1–75 | 15 | −22 | 12 | −24 | 16 | −21 |
| 5 | Methylone | 0.050 | 0.250 | 0.25–5 5–75 | 45 | −31 | 45 | −27 | 52 | −24 |
| 6 | Flephedrone | 0.250 | 1.000 | 1–75 | 19 | −46 | 21 | −48 | 19 | −45 |
| 7 | AMP | 0.250 | 1.000 | 1–75 | 25 | −33 | 31 | −27 | 29 | −26 |
| 8 | Ethcathinone | 0.250 | 1.000 | 1–75 | 29 | −24 | 33 | −19 | 36 | −22 |
| 9 | Hyoscine | 0.150 | 1.000 | 1–75 | 46 | −18 | 48 | −14 | 51 | −15 |
| 10 | 6-AM | 0.250 | 0.750 | 0.75–75 | 43 | −38 | 53 | −34 | 56 | −24 |
| 11 | MDA | 0.250 | 1.000 | 1–75 | 34 | −24 | 36 | −25 | 42 | −22 |
| 12 | MAMP | 0.250 | 0.750 | 0.75–5 5–100 | 32 | −33 | 31 | −26 | 36 | −16 |
| 13 | Methedrone | 0.100 | 0.500 | 0.5–75 | 45 | −28 | 47 | −26 | 52 | −18 |
| 14 | Ethylone | 0.250 | 0.750 | 0.75–100 | 48 | −32 | 45 | −29 | 49 | −21 |
| 15 | Buphedrone | 0.250 | 0.750 | 0.75–75 | 22 | −33 | 26 | −30 | 29 | −21 |
| 16 | MDMA | 0.100 | 0.500 | 0.5–5 5–100 | 41 | −29 | 39 | −24 | 48 | −19 |
| 17 | Oxo-LSD | 0.500 | 1.500 | 1.5–75 | 17 | −26 | 35 | −22 | 43 | −18 |
| 18 | 2-MMC | 0.250 | 1.000 | 1–75 | 18 | −31 | 22 | −26 | 26 | −23 |
| 19 | Butylone | 0.250 | 0.750 | 0.75–100 | 38 | −25 | 37 | −22 | 48 | −20 |
| 20 | Mephedrone | 0.100 | 0.500 | 0.5–5 5–100 | 33 | −35 | 38 | −32 | 45 | −25 |
| 21 | Norketamine | 0.250 | 1.000 | 1–100 | 20 | −31 | 29 | −27 | 35 | −25 |
| 22 | KET | 0.250 | 1.000 | 1–100 | 35 | −32 | 38 | −24 | 43 | −20 |
| 23 | BZE | 0.075 | 0.250 | 0.25–5 5–100 | 37 | −33 | 48 | −21 | 51 | −21 |
| 24 | 4-MEC | 0.075 | 0.250 | 0.25–5 5–100 | 27 | −21 | 32 | −20 | 41 | −18 |
| 25 | Pentedrone | 0.100 | 0.500 | 0.5–5 5–100 | 20 | −33 | 25 | −27 | 35 | −21 |
| 26 | 3,4-DMMC | 0.100 | 0.500 | 0.5–5 5–100 | 36 | −36 | 37 | −35 | 42 | −23 |
| 27 | HER | 0.100 | 0.500 | 0.5–5 5–75 | 12 | −33 | 14 | −28 | 17 | −26 |
| 28 | Alpha-PVP | 0.025 | 0.100 | 0.1–5 5–75 | 29 | −34 | 33 | −30 | 41 | −22 |
| 29 | COC | 0.030 | 0.100 | 0.1–5 5–100 | 52 | −19 | 58 | −12 | 62 | −11 |
| 30 | MDPV | 0.030 | 0.100 | 0.1–5 5–100 | 47 | −25 | 50 | −22 | 54 | −16 |
| 31 | LSD | 0.030 | 0.100 | 0.1–5 5–100 | 39 | −29 | 45 | −27 | 54 | −19 |
| 32 | Pyrovalerone | 0.030 | 0.100 | 0.1–5 5–75 | 31 | −19 | 33 | −17 | 36 | −11 |
| 33 | Fentanyl | 0.100 | 0.500 | 0.5–5 5–100 | 49 | −22 | 51 | −22 | 53 | −17 |
| 34 | Bromazepam | 0.500 | 1.500 | 1.5–75 | 54 | −32 | 58 | −26 | 61 | −22 |
| 35 | Buprenorphine | 0.500 | 1.500 | 1.5–75 | 34 | −29 | 36 | −27 | 38 | −25 |
| 36 | EDDP | 0.075 | 0.250 | 0.25–5 5–100 | 40 | −29 | 44 | −25 | 49 | −21 |
| 37 | Methadone | 0.150 | 0.500 | 0.5–5 5–100 | 30 | −28 | 34 | −26 | 41 | −17 |
| 38 | Lorazepam | 0.500 | 1.500 | 1.5–100 | 11 | −32 | 19 | −28 | 21 | −18 |
| 39 | Alprazolam | 0.500 | 1.500 | 1.5–100 | 47 | −26 | 53 | −24 | 57 | −21 |
| 40 | Diazepam | 0.025 | 0.100 | 0.1–5 5–100 | 46 | −19 | 47 | −18 | 53 | −16 |
| Specimen | Analyte Detected | Concentration Found with the PT-SPE Method (ng mL−1) | Concentration Found with the SPE Method (ng mL−1) [37] |
|---|---|---|---|
| 1 | Lorazepam | <MQL | 0.531 ± 0.020 |
| Diazepam | 137 ± 4 | 144 ± 3 | |
| 2 | COC | 2.45 ± 0.05 | 2.11 ± 0.06 |
| BZE | 16.2 ± 0.5 | 15.6 ± 0.4 | |
| HER | 3.0 ± 0.1 | 2.64 ± 0.06 | |
| 3 | COC | 0.45 ± 0.05 | 0.48 ± 0.06 |
| BZE | 17.8 ± 0.5 | 18.2 ± 0.4 | |
| 4 | COC | <MQL | <MQL |
| BZE | 7.5 ± 0.5 | 7.64 ± 0.11 | |
| 5 | BZE | 4.4 ± 0.1 | 4.56 ± 0.07 |
| Lorazepam | 4.91 ± 0.08 | 4.73 ± 0.03 | |
| 6 | COC | 5.2 ± 0.3 | 4.90 ± 0.09 |
| BZE | 1970 ± 22 | 1993 ± 17 | |
| 7 | COC | 2.92 ± 0.05 | 2.97 ± 0.07 |
| BZE | 58 ± 1 | 54.1 ± 0.4 | |
| 8 | COC | <MQL | <MQL |
| BZE | <MQL | 0.39 ± 0.05 | |
| Lorazepam | 14.1 ± 0.3 | 13.71 ± 0.08 | |
| Diazepam | 5.3 ± 0.2 | 5.3 ± 0.3 | |
| 9 | - | - | - |
| 10 | COC | 7.6 ± 0.3 | 7.72 ± 0.14 |
| BZE | 9.2 ± 0.4 | 8.8 ± 0.1 | |
| 11 | COC | <MQL | <MQL |
| BZE | 43.2 ± 0.8 | 42.4 ± 0.4 | |
| 12 | COC | 122 ± 4 | 119 ± 2 |
| BZE | 262 ± 5 | 257.8 ± 0.4 | |
| Diazepam | 2.45 ± 0.15 | 2.21 ± 0.09 | |
| 13 | HER | 57 ± 1 | 58.3 ± 0.8 |
| MOR | 2.1 ± 0.3 | 1.78 ± 0.11 | |
| 6-AM | 9.9 ± 0.7 | 9.4 ± 0.3 | |
| COC | 6.0 ± 0.3 | 6.34 ± 0.10 | |
| BZE | 232 ± 4 | 235.5 ± 0.4 | |
| Diazepam | 9.8 ± 0.6 | 10.3 ± 0.3 | |
| 14 | BZE | 1.8 ± 0.1 | 1.69 ± 0.04 |
| HER | <MQL | <MQL | |
| Bromazepam | 3.4 ± 0.3 | 3.74 ± 0.14 | |
| 15 | COC | 1.87 ± 0.18 | 1.94 ± 0.11 |
| BZE | 111.8 ± 0.9 | 112.4 ± 0.4 | |
| HER | 6.1 ± 0.6 | 6.5 ± 0.3 | |
| 16 | COC | 1.4 ± 0.1 | 1.31 ± 0.09 |
| BZE | 49 ± 1 | 47.8 ± 0.4 | |
| HER | 33 ± 2 | 30.9 ± 0.8 | |
| Lorazepam | 3.3 ± 0.4 | 3.03 ± 0.03 | |
| 17 | COC | 10.7 ± 0.3 | 10.9 ± 0.3 |
| BZE | 260 ± 4 | 257.9 ± 0.4 | |
| Diazepam | 2.6 ± 0.4 | 2.8 ± 0.2 | |
| 18 | COC | 540 ± 5 | 535 ± 2 |
| BZE | 9585 ± 24 | 9572 ± 1 | |
| HER | 5.9 ± 0.6 | 6.5 ± 0.3 | |
| MDMA | 1.5 ± 0.1 | 1.52 ± 0.03 | |
| MDA | <MQL | <MQL | |
| 19 | BZE | 23.1 ± 0.8 | 22.2 ± 0.4 |
| HER | 6.0 ± 0.7 | 6.5 ± 0.3 | |
| Diazepam | 12.0 ± 0.6 | 12.1 ± 0.4 | |
| 20 | COC | <MQL | <MQL |
| BZE | 24.5 ± 0.4 | 24.4 ± 0.4 | |
| Diazepam | 14.7 ± 0.7 | 15.1 ± 0.5 | |
| 21 | COC | 14.7 ± 0.7 | 14.5 ± 0.6 |
| BZE | 61 ± 1 | 60.4 ± 0.4 | |
| Fentanyl | 40 ± 1 | 42.2 ± 0.7 | |
| 22 | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pascual-Caro, S.; Borrull, F.; Calull, M.; Aguilar, C. Homemade Pipette Tip Solid-Phase Extraction for the Simultaneous Determination of 40 Drugs of Abuse in Urine by Liquid Chromatography–Tandem Mass Spectrometry. Separations 2022, 9, 233. https://doi.org/10.3390/separations9090233
Pascual-Caro S, Borrull F, Calull M, Aguilar C. Homemade Pipette Tip Solid-Phase Extraction for the Simultaneous Determination of 40 Drugs of Abuse in Urine by Liquid Chromatography–Tandem Mass Spectrometry. Separations. 2022; 9(9):233. https://doi.org/10.3390/separations9090233
Chicago/Turabian StylePascual-Caro, Sergi, Francesc Borrull, Marta Calull, and Carme Aguilar. 2022. "Homemade Pipette Tip Solid-Phase Extraction for the Simultaneous Determination of 40 Drugs of Abuse in Urine by Liquid Chromatography–Tandem Mass Spectrometry" Separations 9, no. 9: 233. https://doi.org/10.3390/separations9090233
APA StylePascual-Caro, S., Borrull, F., Calull, M., & Aguilar, C. (2022). Homemade Pipette Tip Solid-Phase Extraction for the Simultaneous Determination of 40 Drugs of Abuse in Urine by Liquid Chromatography–Tandem Mass Spectrometry. Separations, 9(9), 233. https://doi.org/10.3390/separations9090233