1. Introduction
In the last decades, the market of biopharmaceuticals, including protein-, peptide- and oligonucleotide-based therapeutics, has experienced an impressive increase due to the biological properties of this new class of molecules being potentially promising for medical applications [
1,
2,
3,
4]. Recent advancements in the production strategies, such as in genomics, proteomics recombinant strategies and peptide synthesis, have pushed even further the development of these new drugs [
5,
6,
7,
8]. Compared to traditional small molecules, biopharmaceuticals exhibit higher specificity and potency, which derive from their complex three-dimensional structures [
9,
10]; in addition, their action results are effective even at very-low concentrations [
11]. Due to their structural complexity, their synthesis processes, which make use of technologies such as solid-phase peptide synthesis (SPPS) or chemo-enzymatic peptide synthesis (CEPS) [
12,
13,
14,
15], often deliver the principal product together with several unwanted impurities, lowering the overall upstream yield. Accordingly, purification steps are required to achieve the purity constraints set by regulatory agencies, such as FDA, for active pharmaceutical ingredients (APIs) [
16].
The increasing interest towards peptides as a new class of pharmaceuticals has contributed to intensify the demand for innovative and better-performing purification methods. The main technique employed for peptide purification is preparative chromatography, especially carried out in revesed-phase conditions (RPC) due to the hydrophobicity of peptides [
17]. Generally, however, a single purification step is not sufficient to separate the target molecule from all other species [
18]; therefore, different chromatographic modes are often used consecutively to get rid of several types of impurities. This approach, which is of utmost importance at an industrial level, is referred to as
orthogonality: two techniques are orthogonal if they separate compounds depending on two different types of interactions. The combination of orthogonal methods helps to address separation issues that could not be solved using a single technique [
19]. Three chromatographic modes with orthogonal selectivities, for instance, are reversed-phase chromatography, size-exclusion chromatography and ion-exchange chromatography, which separate analytes mainly based on their hydrophobicity, dimensions and charge, respectively.
In addition to applying two different techniques consecutively, orthogonal separations can be achieved within the same column when the stationary-phase particles are functionalised with two different ligands, to combine two separation mechanisms in a single chromatographic step [
20]. The technique where the stationary phase bears ligands with different reactivities is referred to as
mixed-mode chromatography [
21]. Traditional mixed-mode materials contain two functional groups with different chemistries on the same ligand, while more recent mixed-mode stationary phases exhibit two different ligands, one with a higher superficial concentration and a second one having a much lower surface density. For example, some reversed-phase stationary phases can be doped with small amounts of ion-exchange ligands, forming a so-called doped reversed phase (DRP). Since, in these cases, the ligands are two distinct functional groups, the surface concentration of each on their solid-phase particles can be accurately chosen. It has been demonstrated by Khalaf et al. that the surface density of the doping groups is linearly dependent on their concentration in the mixture used to derivatize the stationary phase [
18,
22,
23].
The doping ion-exchange ligands can work in “attractive” or “repulsive” ways, depending on the experimental conditions used [
20,
22,
24]: they exhibit repulsion towards the analytes having their same charge and attraction towards the ones with an opposite charge sign. On the other hand, the reversed-phase ligands act in an attractive way towards hydrophobic molecules, and, therefore, the whole stationary phase can work globally in “attractive–attractive” or “attractive–repulsive” ways. In traditional reversed-phase chromatography, more hydrophobic species are more retained, whereas less-hydrophobic (including charged) components elute first. When working in attractive–attractive mode, compounds charged with the opposite charge of the dopants experience an increase in retention. This leads to a decrease in resolution because those that would be the first eluting peaks in traditional RP chromatography, in this case move towards more retained ones, whose position is not influenced by the dopants. On the contrary, in attractive–repulsive mode, the repulsion performed by the dopants on the analytes with the same charge sign push them to elute earlier. Again, the hydrophobic species are not affected by the presence of doping ligands. This leads to an increase in the resolution between hydrophilic charged species and more retained hydrophobic peaks. From this purely theoretical explanation, it seems clear that DRP materials are expected to perform better in attractive–repulsive mode. When dealing with peptides, their isoelectric point (pI) defines the sign of their charge depending on the pH of the mobile phase. Particularly, when the mobile-phase pH is below the pI, the peptide is positively charged, whereas at a pH higher than the pI, the peptide is negatively charged. Therefore, to work in attractive–repulsive mode, for mobile phases where pH < pI, anion-exchange materials (AIEX) must be chosen; otherwise, for phases with pH > pI, cation-exchange resins (CIEX) must be used [
23].
This study is intended to be a proof of concept that, since the combination of attractive and repulsive interactions causes an increase in the resolution of peaks of analytes with different chemistry, the use of these doped stationary phases can be potentially beneficial for preparative applications. The compound that has been purified in this study is liraglutide, an analog of human GLP-1 (glucagon-like-peptide 1), a potent blood-glucose-lowering hormone physiologically secreted in the duodenum in response to food intake. It is a 31 amino acids polypeptide, acylated with a group -Glu-palmitoyl on Lys(1) (sequence: H-His-Ala-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys(1)-Glu-Phe-Ile-Ala-Trp-Leu-Val-Arg-Gly-Arg-Gly-(OH)-palmitoyl-Glu(1)-OH) with a pI of 4.9 and a molecular mass of 3751 g/mol [
25]. Derivatization with a fatty acid side chain and a glutamyl spacer is needed to improve pharmacokinetic and pharmacodynamic properties, to prolong its plasma half-life time [
26,
27]. In this study, reversed-phase and doped reversed-phase resins were tested and compared for the purification of a mixture of this peptide industrially obtained by means of SPPS. Particularly, the mixed-mode stationary phase was a reversed-phase resin loaded with a defined percentage of cation-exchange groups. By operating at a pH above the pI of liraglutide, the electrostatic interactions between the negatively charged peptide and the cation-exchange groups on the resin become repulsive, while the hydrophobic C8 chain positively interacts with the hydrophobic peptide, globally resulting in the already-mentioned “attractive–repulsive” effect.
2. Materials and Methods
2.1. Peptide
The crude mixture of liraglutide was synthesized by means of solid-phase synthesis by Fresenius Kabi iPSUM (Villadose, Rovigo, Italy). The peptide constitutes 34% of the crude mixture based on weight. All crude peptide samples (from now on called feed) were prepared in order to obtain a concentration of 1 g/L; in addition, the feed was filtered before injection using a Millipore apparatus with 0.45
m membrane filters produced by Carlo Erba Reagents (Milan, Italy). Liraglutide in the feed has a chromatographic purity of 49%, measured with the analytical method reported in
Section 2.4.
For the HPLC calibration curve (area vs. concentration), standard solutions of liraglutide were prepared by dissolving pure liraglutide, provided by Fresenius Kabi iPSUM (Villadose, Rovigo, Italy), as described for the feeds, in a concentration range from 0.1 to 2 g/L.
2.2. Columns and Buffers
Two different reversed-phase resins with the same column dimensions (250 × 4.6 mm) were employed in this study: a Daisogel-SP-10 m-C8-Bio column with pore size 120 Å, by Dr. Maisch Daisogel (Ammerbuch, Germany), functionalised with C8 chains, and a doped-reversed phase ZEOsphere-DRP-10 m-C8-C10 column with pore size 120 Å, produced by Zeochem AG (Uetikon am See, Switzerland), functionalized with both C8 chains and 10% of sulfonates group (CIEX ligands). All reagents for buffers preparation were purchased from Sigma-Aldrich (St. Louis, MI, USA), including acetonitrile, triethylamine, phosphoric acid 85%, trifluoroacetic acid. Particularly, for preparative applications, the mobile phases used were MP-A: triethylamine phosphate buffer 25 mM, with pH corrected to 8.5 using orthophosphoric acid 85%, and MP-B: ACN. The mobile phases used for analytical experiments were MP-A: 0.05% TFA in water, and MP-B: methanol:water:acetonitrile:TFA 80:15:5:0.05.
2.3. Preparative Chromatographic Equipment and Methods
Preparative chromatography experiments were performed at room temperature using an ÅKTA pure 25L instrument (GE Healthcare, Uppsala, Sweden), equipped with a fraction collector and operated through the Unicorn software. The detector wavelength was set at 300 nm.
The purification method used included an equilibration step at 15% MP-B 1 CV long, where CV is the geometrical column volume, corresponding to 4.2 mL for a 250 × 4.6 mm column. Then the sample was loaded into the column in order to obtain a concentration of 10 mg of peptide per mL of stationary phase, which corresponds to a loaded volume of 42 mL of feed with a concentration of 1 g/L. After the loading, the column was washed for 2 CVs with 15% MP-B. A first gradient was used to increase rapidly the MP-B percentage (from 15 to 34.5% in 1 CV). A second, much-shallower gradient was used for the main purification step, where MP-B percentage increased from 34.5 to 39.5% in 6 CVs at a very low flow rate (0.5 mL/min). The washing step lasted for 10 CVs, at a constant percentage of MP-B (75%). In all the steps but the second gradient, the flow rate was 2 mL/min.
During the elution, fractions were periodically collected (1 fraction every mL eluted), diluted with water, if necessary, and analysed offline (see
Section 2.4).
2.4. Analytical Chromatographic Equipment and Methods
Analytical chromatographic analyses were performed on an Agilent 1100 Series Capillary LC (Agilent, Santa Clara, CA, USA), equipped with a binary pump system, an autosampler and a diode array detector set at 220 nm. The column thermostat was set at 25 C and the injection volume was 8 L. The column employed for analytical experiments was a Cortecs C18+, 150 × 4.6 mm, 2.7 m (Waters, Milford, MA, USA).
The elution was performed in gradient conditions, with MP-B varing from 80 to 95% in 30 min, followed by a second gradient from 95 to 100% B in 5 min, a washing step with 100% B and then a re-equilibration step, at the initial conditions.
3. Results and Discussion
The purification method described in
Section 2.3 was used to purify the liraglutide crude mixture in order to increase the peptide purity from 49 to at least 90% after collecting and pooling the fractions eluted along the gradient. All the fractions were analysed offline in order to obtain, for each, the peptide concentration, purity and recovery. By identifying the concentration and the volume of every fraction (see
Section 2.3), the mass of the peptide could be determined.
For a single fraction, purity, which is the most-important parameter to consider when evaluating the outcome of a purification, is the area of the target peak (
) divided by the total area of all the peaks integrated (
) in that fraction, which also includes impurities:
Besides purity, recovery can also be evaluated for each fraction. It is defined as
which is the mass of the peptide contained in a fraction divided by the total peptide mass injected.
Both recovery and purity were calculated as percentage.
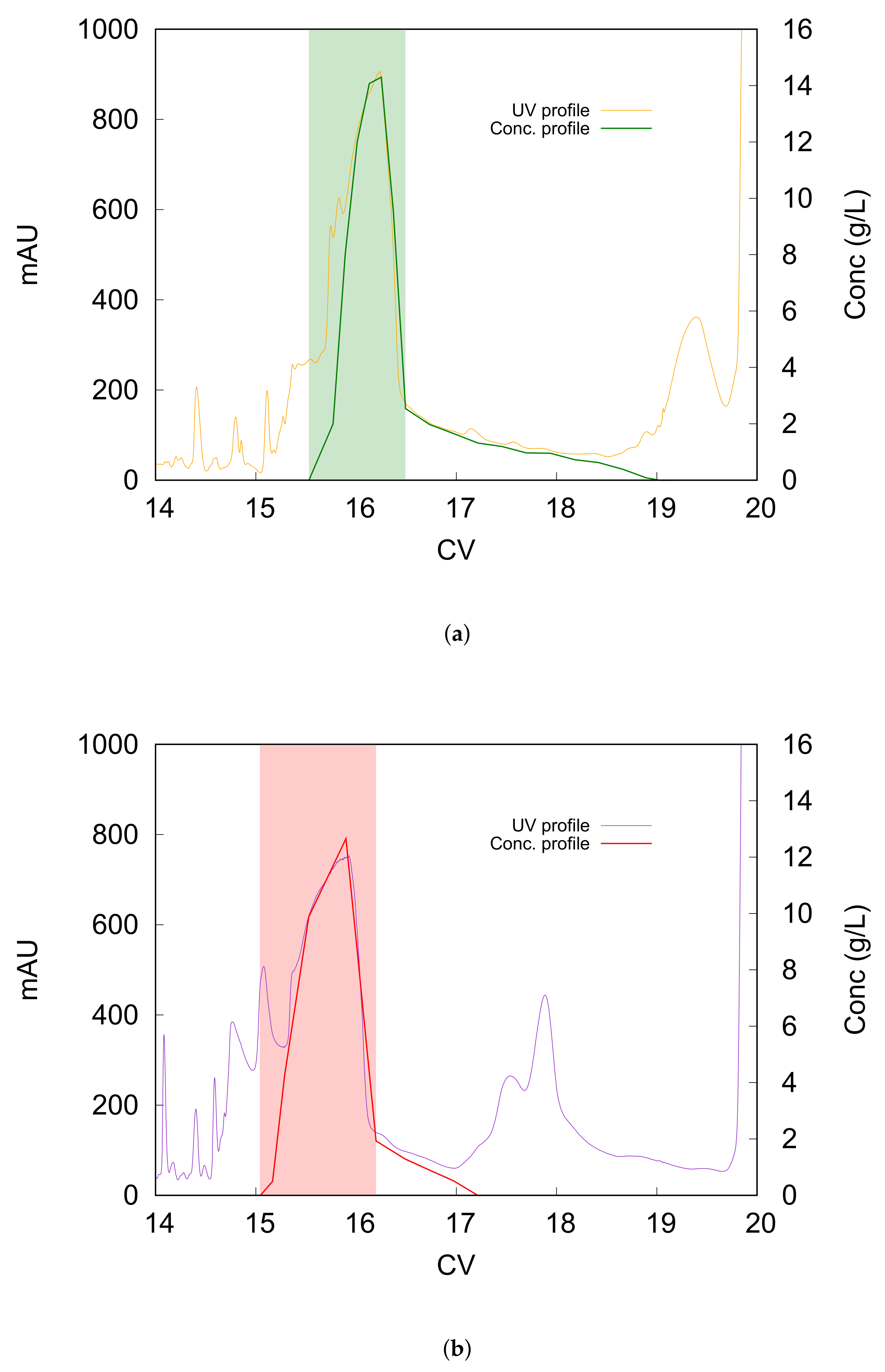
Figure 1a shows the preparative chromatogram, zoomed between 14 and 20 CV elution, obtained by using the traditional reversed-phase column (C8), in orange, and the concentration profile of liraglutide along the gradient in green. Right before CV = 20, the UV signal increases sharply because the gradient has ended and the stripping procedure is being performed. As can be seen, the peptide concentration profile is very broad and tailed. On the other side, the main peak is much less tailed when using a mixed-mode column (C8+ 10% of cation-exchange groups), as shown in
Figure 1b, where the UV profile is in violet and the concentration profile is shown in red. By comparing the two concentration profiles, it can be noted that, with the doped column, the whole peak elutes in about 2 CVs; whereas with the C8 column it elutes with in 3.5 CVs, results that are much broader.
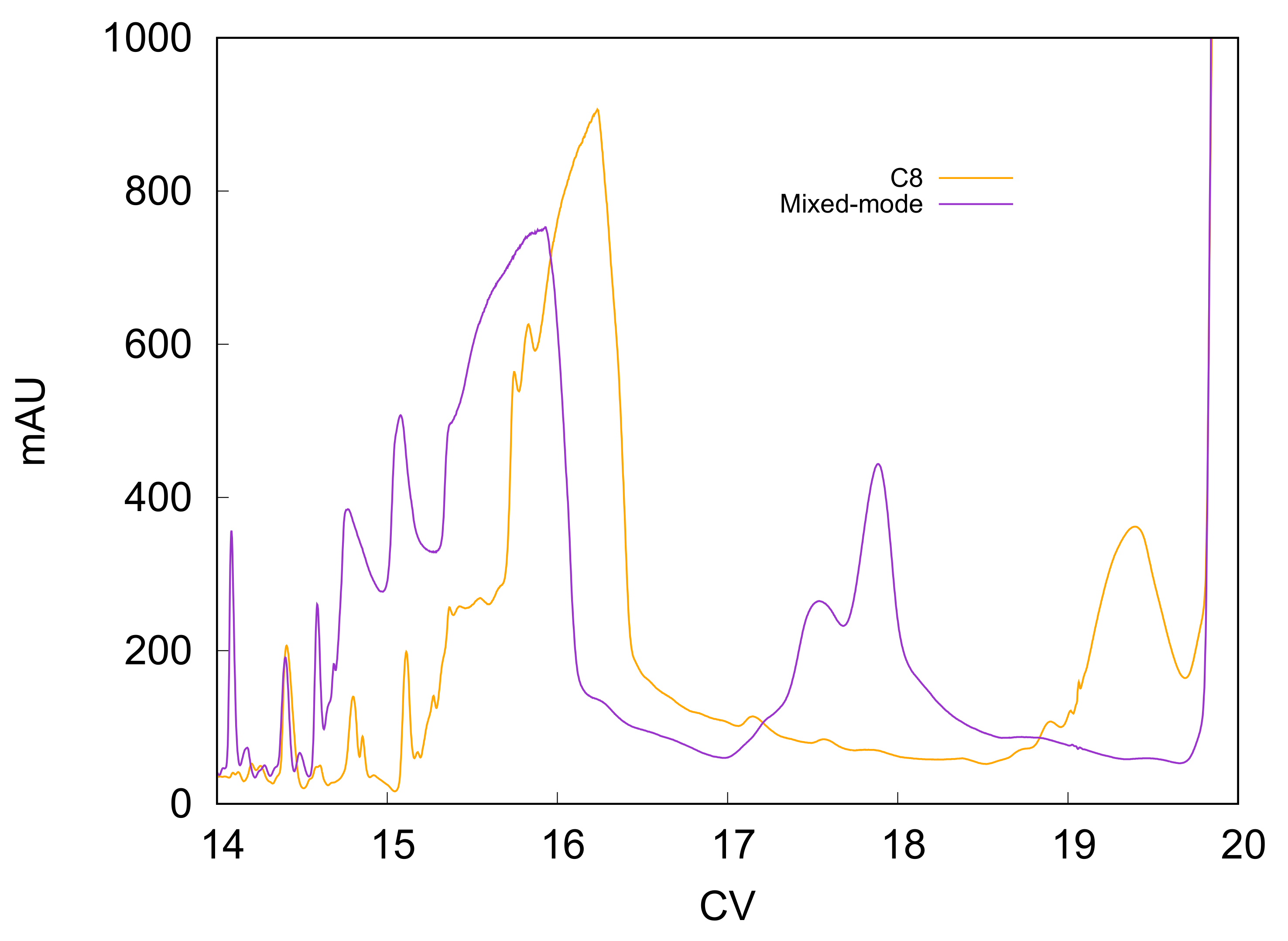
Beside being narrower, the peak also elutes earlier when using the mixed-mode column, as can be seen by superimposing the two chromatograms (see
Figure 2). This is due to the repulsive effects established between the charged ionic groups on the stationary phase and the peptides, both negatively charged. Indeed, the pH of the mobile phase, which was 8.5, was higher than the isoelectric point of liraglutide (4.9), so the peptide exhibits a negative charge in these chromatographic conditions. Therefore, in these experiments, the column was employed in attractive–repulsive mode. Beside the main peak, the decrease in retention using the doped column is also quite evident when considering a late-eluting group of impurities. These species elute just before the stripping phase, around 19 CVs, with the C8 column, but they elute earlier, between 17 and 18 CVs, with the mixed-mode column.
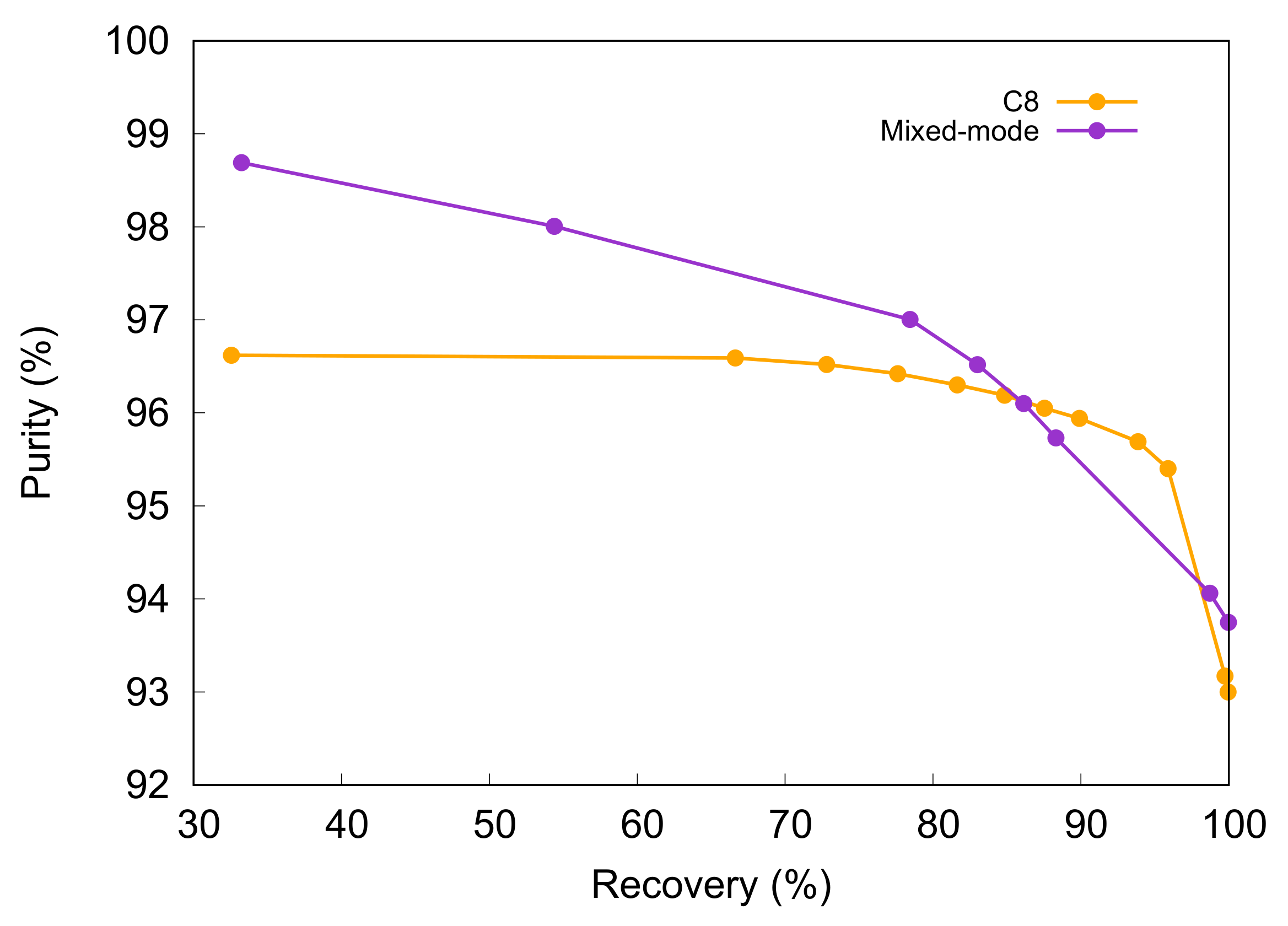
For each column, a so-called Pareto curve was also obtained, starting from the offline analysis of the fractions collected along the gradient. This curve reports the variation in purity with recovery [
2,
16,
28]. These two parameters vary inversely: the purest fraction of the peak only contains a small percentage of the peptide mass injected. On the other hand, when the collection window is broadened, the recovery increases, but some side impurities are collected together with the target compound. This results in a decrease in the purity. The Pareto curve is obtained by considering first the purest fraction, which also exhibits the lowest recovery, and then pooling it with the purest adjacent fraction. This pool is then, again, pooled with the purest adjacent fraction in order to increase the recovery, and so on and so forth until the whole target peak has been collected. This corresponds to the case where the purity is as low as possible.
The Pareto curves obtained using the two columns with the same preparative method as described in
Section 2.3 are shown in
Figure 3. It is worth noting that, at very high values of purity, the Pareto curve obtained with the mixed-mode column lies above the curve obtained with the C8 column. This means that, in this particular region of the Pareto curve, the doped column allows for higher purities than the C8, with the recovery being similar. A similar behavior is also present at very-high recoveries, even if to a lesser extent. In the central region, however, the purity results are higher for the C8 column. The reason why, when using these particular conditions, the doped column Pareto curve does not lie above the other curve for the whole range of recovery could be that the charged peaks result is much narrower but they also elute closer to each other because of the attractive–repulsive effect. For instance, the late-eluting peaks on the right of the target peak possibly overlap with the tail of liraglutide in the case of the mixed-mode column.
One of the major advantages of using the mixed-mode column is the peak shape obtained, which is much-narrower and less-tailed. From the industrial point of view, after deciding the purity limitation that the pool collected must fulfill, the side regions of the main peak, which overlap with close-eluting impurities, must be reprocessed, in order to not waste large amounts of the target peptide. It is clear that this further purification step is not beneficial in terms of time, cost and waste. Therefore, an interesting option that shows the benefits provided by the doped column would be to collect the main peak excluding the tail, as highlighted in the chromatograms in
Figure 1a,b. The performance obtained for the two columns when pooling this portion of the chromatogram are reported in
Table 1. It results that, with the mixed-mode column, the purity can be slightly improved (+2.5%) while the recovery increases by +15%. A direct consequence is that a smaller percentage of the target product must be reprocessed in a following purification step when using the mixed-mode column (about half, with respect to the C8 column), which corresponds to the peptide recovery of the the liraglutide peak tail discarded.
Alternernatively, if the whole peak satisfies the purity requirements, it can be entirely collected. In this case, it elutes completely in 2 CVs with the mixed-mode column and 3.5 CVs with the C8 column. This results in a pool with a higher concentration, in the first case. In addition, the duration of the method could be reduced and the gradient could be stopped after the main peak elution, with a considerable saving of time and solvents.
4. Conclusions
Mixed-mode resins are alternative, promising stationary phases that exploit two retention mechanisms generally based on hydrophobicity and charge interactions. Their use can be particularly beneficial when dealing with complex mixtures such as peptide crude samples, for which more traditional stationary phases do not allow to obtain enough chemoselectivity. At pHs above the peptide isoelectric point, a repulsive effect is established between the target compound and the charged cation-exchange ligands doping the stationary phase, which are both negatively charged. As a result, the peaks become narrower and their retention decreases; in addition, the main peak is much less-tailed.
In this study, an industrial crude liraglutide mixture was purified with both a C8 column and a C8 stationary phase doped with 10% cation-exchange groups. It was shown that, if collecting the whole peak or if collecting the purest fractions, the mixed-mode column shows slightly better performance with respect to the C8. The most relevant aspect is that, when the target peak is collected by discarding the tailed part, the mixed-mode column allows to increase the recovery by +15%, at a slightly higher purity. From an industrial viewpoint, it is convenient for the target peak to be as little tailed as possible, in order to minimize the side portions of the peak, which must be recycled in a subsequent purification step. This leads to an increase in productivity and to an improvement in the overall process performance, also from the point of view of the greenness of the process, since smaller solvent amounts would be required. This research is to be intended as a proof-of-concept study for the particular purification of liraglutide, but similar outcomes are expected also for different crude-peptide mixtures when using these resins.


 ,
, 

{kind=link}
{kind=link}
{kind=link}