1. Introduction
The measurement of radioactive fission products from nuclear events has implications for nuclear data production, environmental monitoring, and nuclear forensics. While many of the peak yield fission products are easily measured via gamma spectroscopy in unseparated radioactive samples, there are some key analytes of interest where the radiometric interferences prevent low uncertainty measurements and/or have sufficiently low fission yields hindering detection in the presence of multiple peak yield fission products. In these cases, either chemical separation or alternative detection methodologies are required, such is the case for the lanthanide, namely the rare earth elements (REEs), which are defined here as the lanthanides and yttrium.
Previous efforts have developed methods to isolate and measure fission products of interest; however, isotopes with poor decay structure (i.e., short half-life, low branching ratio gamma emissions) have proven to be challenging [
1,
2,
3,
4,
5,
6]. Among these challenging isotopes are multiple REEs, including
91Y,
144Ce,
153Sm,
155Eu,
156Eu, and
161Tb. REEs with low fission yields and short half-lives, including
153Sm,
155Eu,
156Eu, and
161Tb, require chemical separations and/or advanced detector technologies for analysis in fission samples. Short-lived fission product analysis benefits greatly from separations, which minimize radiometric impurities by lowering the detection limit and/or shortening the analysis time, while the elimination of radiometric impurities can permit analysis using additional radiometric detection methods such as liquid scintillation counting, which has high counting efficiency but little to no energy resolution. Some high fission yield REEs, including
91Y and
144Ce, can be quantified from fission product samples with little to no separation using gamma spectroscopy, but minimizing the presence of other radionuclides can improve the data quality and/or analysis time.
Since the REEs are typically in the +3 oxidation state, chemical separation is based typically using the decreasing ionic radii across the lanthanide series, which results in an increase in complex stability for the heavy members of the f-block versus the lighter members [
7,
8]. The minimal change in ionic radii among neighboring lanthanides makes the intra-group separation challenging; yttrium has the same oxidation state and ionic radii of some of the lanthanides adding to the separation difficulty [
8].
Extraction chromatography separates analytes of interest using organic extractants coated onto polymer beads. One of four different extractants classes can be used to separate the REEs, including quaternary ammonium salts, acidic organophosphorus, neutral organophosphorus, and diglycolamide [
9]. Acidic organophosphorus-based extractants provide optimal separation among the REEs, and this is the basis of LN resin from Eichrom Technologies
®, Inc. (Lisle, IL, USA) [
10,
11]; LN resin is comprised of polymeric beads coated with di-(2-ethylhexyl)-phosphoric acid (HDEHP). LN resin has been used for intra-group REEs separations with both HCl and HNO
3 elution profiles [
12,
13,
14,
15]. REEs sorb to LN resin in low molarity acid, and as the acid concentration is increased, the REEs are sequentially eluted lightest (La) to heaviest (Lu). The stability of the REEs-extractant complex is determined by the REEs ionic radius and charge; this results in Y co-eluting with the heavy lanthanides.
An optimized separation scheme was developed by Arrigo et al. using LN resin employing increasing HNO
3 concentration for REEs separation [
12]. With the focus on a fission product analysis, the method developed by Arrigo et al. focused on maximizing the radiometric purity and chemical yield for Sm, Eu, Tb, and Y; parameters investigated during the optimization included LN resin mass, resin mesh size, lanthanide carrier mass loading, and nitric acid elution gradient [
12].
The purpose of this work is to evaluate the separation developed by Arrigo et al. to test its efficacy in conjunction with Eichrom’s UTEVA and TRU resins to isolate and measure multiple short-lived REEs fission products. The separation was tested utilizing a fission sample produced from a highly enriched uranium (HEU) source irradiated with thermal neutrons.
2. Materials and Methods
2.1. Reagents and Materials
Bulk LN resin (50–100 µm), UTEVA resin (50–100 µm, 2 mL cartridges), and TRU resin (50–100 µm, 2 mL cartridges) were purchased from Eichrom Technologies®, Inc. Reagent or Optima™ grade HNO3 and HCl were purchased from Fisher Scientific and used without further purification. Standards for the lanthanides and yttrium were purchased from Inorganic Ventures, Ultra Scientific, or High-Purity Standards Corporation as 1000 ppm single-element solutions. A 152Eu radiometric standard was purchased from Eckert and Zeigler. Iron powder, sodium nitrite, and ascorbic acid were purchased from Acros Organics and used without further purification. Sulfamic acid was purchased from Ricca and used without further purification. All solutions and samples were prepared with Milli-Q water (Millipore, Burlington, MA, USA, resistivity > 18 MΩ cm-1). Disposable polypropylene columns (0.8 cm inner diameter, part number AC-141-AL) were purchased from Eichrom® Technologies, Inc. along with the corresponding caps, tip closures, and frits; the materials were used as purchased. Polypropylene Falcon and/or Corning 50 mL centrifuge tubes and Falcon 15 mL sample tubes were purchased from Fisher Scientific. Fractions were collected in 20 mL polyethylene liquid scintillation counting (LSC) vials from Perkin Elmer.
2.2. Resin Hydration and Column Preparation
Prior to use, each column was prepared by transferring 0.7800 ± 0.0005 g Eichrom® LN resin (50–100 µm) to a 50 mL centrifuge tube by weight, hydrated with 25–30 mL of 0.1 M HNO3 over the course of 24 h with gentle agitation. Samples were centrifuged at 3500 RPM for 5 min. The liquid phase was decanted and discarded. The resin slurry was loaded into 2 mL polypropylene columns with a 0.8 cm inner diameter purchased from Eichrom® Technologies, Inc. The columns were fitted with a top frit leaving approximately 0.5 cm liquid between the resin bed and the frit; the final bed volume was 2 mL. Each column was rinsed with 10–15 mL 0.01 M HNO3 and then stored in 0.01 M HNO3 prior to use.
2.3. Irradiated HEU Sample Production
A 93% HEU metal target was irradiated with thermal neutrons at the MITR-II reactor at the Massachusetts Institute of Technology. The sample was dissolved with 6 M nitric acid and slightly warmed; the final solution was diluted to give an acid concentration of 3 M HNO3. Four aliquots of the sample were taken, with each containing approximately 1 × 1012 fissions (0.9 g solution, 0.69 mg of U) spiked with 360 Bq 152Eu and 50 µg each of Sm, Eu, and Tb from 1000 µg/mL single element ICP standards.
2.4. Separation Chemistry Using UTEVA, TRU and LN Resins
Samples were processed through an initial separation using Eichrom’s UTEVA and TRU resin to remove actinides and select fission products from the sample prior to the REEs separation. This process has been published previously by Morley et al. with a short summary provided here [
5]. After adding the appropriate tracers and carriers to the aliquots of irradiated HEU solution, 2 mL of 0.6 M iron (II) sulfamate, 1 drop of 1 M ammonium thiocyanate as an indicator, and 1 mL of 1 M ascorbic acid were added. The samples were allowed to sit for 20 min to allow for the reduction of Np(V) to Np(IV) and Pu(IV) to Pu(III) (if present). The samples were loaded onto stacked UTEVA and TRU vacuum cartridges and eluted at 1 mL/min using a vacuum box. The samples were quantitatively transferred to the cartridge stack by rinsing the sample container three times with 2 mL of 3 M HNO
3. The cartridges were then rinsed with 10 mL of 3 M HNO
3. Following the rinse, the UTEVA and TRU cartridges were separated, and the REEs were eluted from the TRU resin with 5 mL of 3 M HNO
3—0.1 M NaNO
2 followed by 6 mL of 2 M HCl. The nitrite oxidizes Pu to the +4 oxidation state to prevent Pu(III) from co-eluting.
The REEs fractions were transposed to 0.01 M HNO
3 then underwent an intra-group separation using 2 mL LN resin (50–100 µm) columns; the flow rate was 8.6–12 bed volumes per hour (BV/hr) or 5–7 min per mL eluent (min/mL). The samples, in 1 mL of 0.01 M HNO
3, were loaded onto the LN resin columns, quantitatively transferred with three 2 mL rinses of 0.01 M HNO
3, then eluted by gravity with increasing concentrations of HNO
3 up to 8 M using the elution gradient summarized in
Table 1. The fractions containing Tb (18a, 18b, and 18c in
Table 1) were co-collected and transposed to 0.5 M HNO
3 for a second separation. The Tb containing samples, in 1 mL of 0.5 M HNO
3, were loaded onto the LN resin columns, quantitatively transferred with three 2 mL rinses of 0.5 M HNO
3 and eluted by gravity with increasing concentrations of HNO
3 up to 8 M using the elution gradient summarized in
Table 2. The fractions containing Tb (4a, 4b, and 4c in
Table 2) were co-collected and processed for analysis.
Separated Ce, Sm, and Eu fractions were diluted to a total of 10 mL such that the final acid concentration was 2% HNO3; the fractions were sent for gamma spectroscopy followed by inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis for the Sm fractions. Separated Tb fractions were split for radiometric counting and yielding, 90% and 10%, respectively. Tb samples for radiometric counting were evaporated to <0.25 mL and stippled onto a filter, covered in Kaptan tape, and counted by low energy photon spectroscopy (LEPS). The chemical recoveries were calculated by comparing the amount of each element in the purified fraction to the amount added (by mass). Chemical yields were determined for Ce by gamma spectroscopy for 141Ce and 143Ce in the HEU solution, for Sm by ICP-OES analysis in the separated fractions, for Eu by gamma spectroscopy of 152Eu radiotracer, and for Tb by inductively coupled plasma mass spectrometry (ICP-MS) analysis. All samples were prepared by weight using a four-place balance from Mettler Toledo (Columbus, OH, USA).
2.5. Sample Analysis
Samples were analyzed by gamma spectroscopy, LEPS, ICP-OES, and/or ICP-MS; the sample analyses were monitored for impurities that might result in spectral or mass interferences.
2.5.1. HPGe Analysis
Gamma emitting radionuclides in the irradiated HEU solution and separated fractions were counted on multiple high purity germanium detectors (HPGe) from Canberra or Ortec. Samples were analyzed in a calibrated geometry consisting of 10 mL solution (in this case, 2% HNO
3) in 20 mL polyethylene LSC vials. All gamma data was collected and analyzed using Canberra Genie 2000 V3.4.1 and APEX software. The data were corrected for decay during irradiation and counting and were reported at the end of the irradiation time. Corrections are applied for cascade summing in all counting geometries. The detectors were calibrated using a NIST-traceable mixed gamma standard from Eckert and Zeigler with certified gamma peaks at 13 energies between 46.5 and 1836 keV; isotopes include
210Pb,
241Am,
109Cd,
57Co,
139Ce,
203Hg,
113Sn,
85Sr,
137Cs,
88Y, and
60Co. Control checks are performed daily for energy calibration, peak resolution, and efficiency calibration using control sources containing
241Am,
137Cs, and
60Co. Nuclear data, including half-lives and gamma branching ratios, were adopted from the NUDAT2 database at the National Nuclear Data Center at Brookhaven National Laboratory. For isotopes with more than one gamma line, activities reported were based on the weighted mean values. Separated fractions for Sm were counted for 1.42 h, and the principal gamma ray detected from
153Sm was 103.18 keV. Separated fractions for Eu were counted for 17–19 h, the principal gamma rays detected from
152Eu were 121.78, 778.9, 867.37, 1085.87, 1112.07, and 1408.01 keV, and the principal gamma rays detected from
156Eu were 646.29, 723.47, 1065.14, 1079.16, 1153.8, 1230.71, 1242.42, 1277.43, 1366.41, and 1965.95 keV. The separated fractions for Ce were counted for 17 h; the principle gamma ray(s) detected from
141Ce was 145.44 keV, from
143Ce were 57.36, 231.55, 293.27, 350.62, 490.37, 664.57, 721.93, and 880.46 keV, and from
144Ce was 133.51 keV. Uncertainties in counting statistics were based on Currie [
16].
2.5.2. Low Energy Photon Spectroscopy Analysis
Terbium-161 was quantified in the purified Tb fractions using HPGe detectors designed for LEPS from Ortec or Canberra; samples were counted for up to 10 days. The principal gamma rays and X-rays detected from
161Tb were at 25.6, 45.5, 48.9, and 74.6 keV and weaker X-rays were detected at 52.0 and 57.2 keV. The individual peaks had at least 1000 counts for a statistical uncertainty of approximately 3%. All gamma data was collected and analyzed using Genie 2000 V3.4.1 (Mirion Technologies, Meriden, CT, USA) and APEX-GAMMA software (Mirion Technologies, Meriden, CT, USA). The data were corrected for decay during irradiation and counting and were reported at the end of the irradiation time. The detectors were calibrated using the NIST-traceable mixed gamma standard from Eckert and Zeigler described for the HPGe detectors as well as single isotope standards to avoid interferences. Control checks are performed daily for energy calibration, peak resolution, and efficiency calibration using control sources containing
241Am,
137Cs, and
60Co. Uncertainties in counting statistics were based on Currie [
16].
2.5.3. ICP-OES Analysis
The analysis of Sm was performed using a Perkin-Elmer Optima 5300 DV ICP-OES. Calibration solutions ranged from 2–1000 µg/mL in 2% HNO3 for radial mode and 0.5–10 µg/mL in 2% HNO3 for axial mode. Samples prepared using a separate NIST traceable standard were included during the analysis run every 10 samples to verify instrument stability and accuracy of the calibration. The solution was aspirated using a peristaltic pump, and the sample introduction included a quartz Scott-type spray chamber, GemCone high dissolved solids nebulizer, and a 2.0 mm inner diameter alumina injector. The acquisition parameters included data collection based on peak area with 1–5 s auto-read and three replicates, RF power 1350 W, peristaltic pump 1.5 mL/min, with carrier gases 20 L/min plasma, 0.3 L/min auxiliary, and 0.65 L/min to the nebulizer.
2.5.4. ICP-MS Analysis
The analysis of 159Tb was performed using an Agilent 7700X quadrupole ICP-MS (Santa Clara, CA, USA). Calibration solutions ranging from 0.1 to 5 ng/g in 2% HNO3 were prepared by gravimetric dilution of a 1000 µg/mL single element standard from Ultra Scientific (North Kingston, RI, USA); all dilutions were made with 2% HNO3 prepared from Fisher Optima™ acid. Samples prepared using a separate NIST traceable standard were included during the analysis run every 10 samples to verify instrument stability and accuracy of the calibration. The solution was aspirated using a peristaltic pump, and the sample introduction included a standard quartz micromist nebulizer and quartz spray chamber (Glass Expansion, Pocasset, MA, USA). The background equivalent concentration (BEC) was 0.003 ng/g for Tb. The acquisition parameters include peak pattern 1, replicates 3, integration time 0.50 s, RF power 1550 W, nebulizer pump 0.1 rps, and carrier gas 1.00 L/min.
3. Results and Discussion
An HEU metal sample was irradiated with thermal neutrons and dissolved nitric acid with gradual heat. The solution was evaporated to soft dryness and reconstituted to give a solution containing 1.15 × 10
12 ± 5% fissions per gram HEU solution in 3 M HNO
3. The number of fissions was calculated based on the average atoms/g of HEU solution as determined by gamma spectroscopy for the peak yield fission products
95Zr,
97Zr,
99Mo,
103Ru,
140Ba/La,
132Te/I,
137Cs,
141Ce,
143Ce,
144Ce, and
147Nd compared to the cumulative fission yields (CFY) for thermal irradiation of
235U reported by England and Rider [
17]. To ensure near quantitative yield, the HEU sample was analyzed by gamma spectroscopy before and after dissolution, which showed minimal loss of activity to the quartz irradiation ampoule.
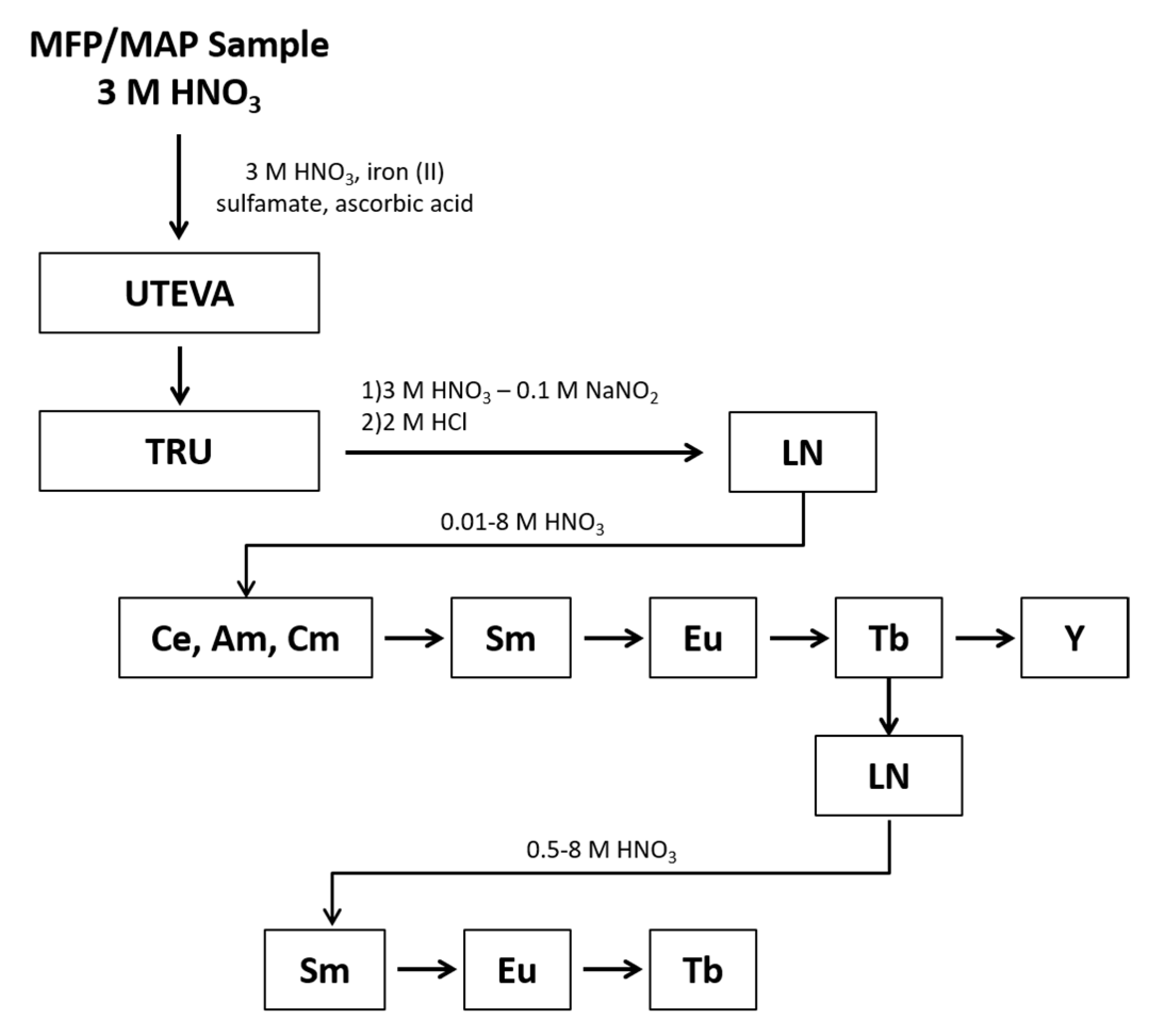
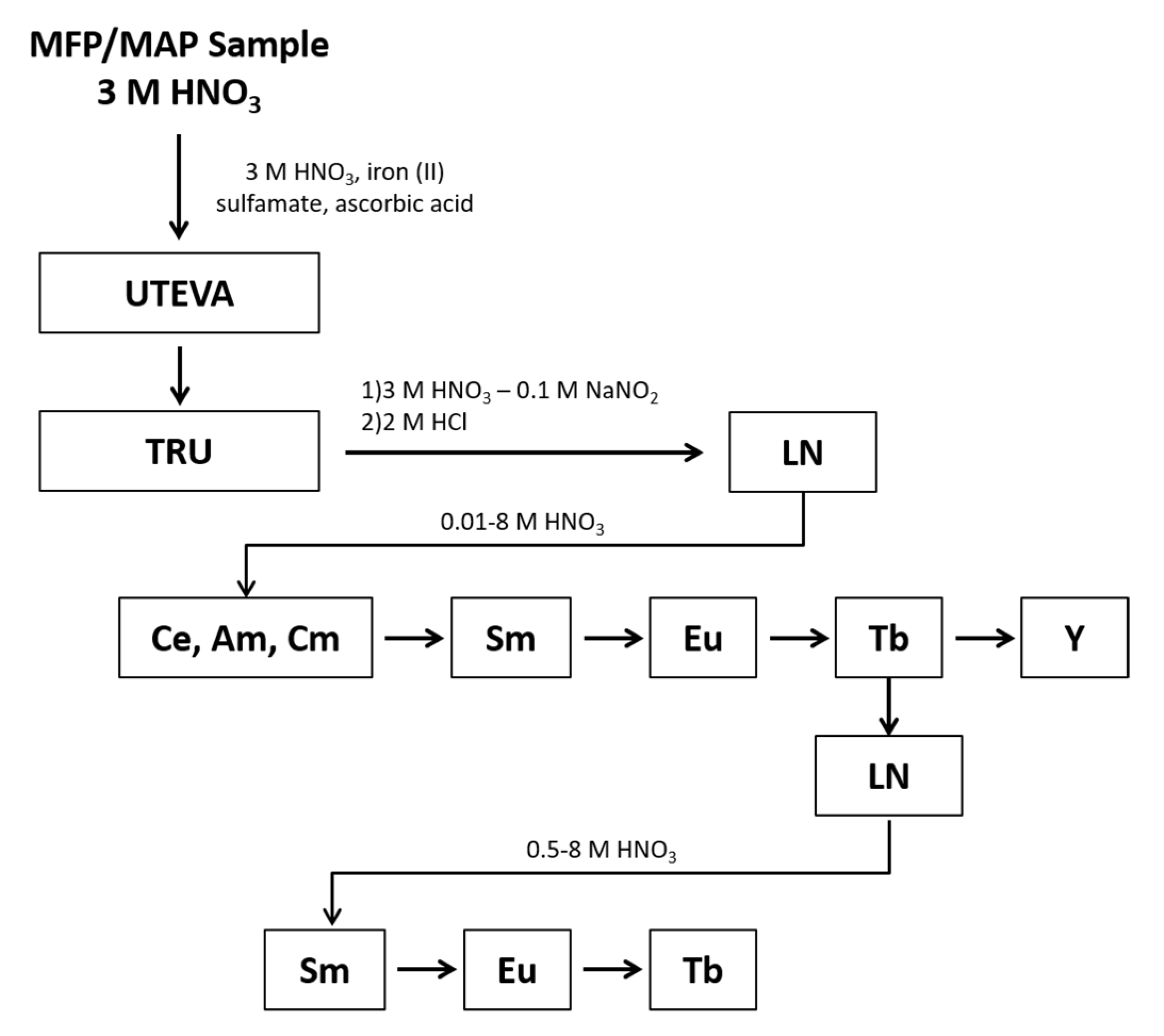
Samples were prepared in quadruplicate and processed using the chemical separation scheme shown in
Figure 1. The REEs were separated as a group from the rest of the sample using Eichrom’s UTEVA and TRU resins. When the samples were loaded onto the UTEVA and TRU resins, an Fe(II) reductant was used to reduce Pu to Pu(III), which sorbs to the TRU resin along with other tri-valent actinides and REEs. During the REEs elution, nitrite was added to oxidize Pu to Pu(IV) to prevent co-elution with the REEs. Under the conditions of this experiment, the REEs are present in the +3 valence as determined during optimization studies where the yield of each REE was tracked following elution from the TRU and LN resins to determine the REEs were quantitatively eluted as expected for the +3 valence. Following elution from the TRU resin, the REEs fraction underwent an intergroup separation using Eichrom’s LN resin to isolate separate fractions containing the light lanthanides (La through Nd), Sm, Eu, Tb, and Y. The Tb fraction underwent an additional separation using LN resin to remove low-level radiometric contaminants that would hinder LEPS analysis.
The chemical yields for Ce, Sm, Eu, and Tb are presented in
Table 3. Cerium elutes primarily in fraction 3, with the remaining Ce eluting in fraction 4. Samarium and Eu each elute predominately over three 2 mL fractions with Sm eluting in fractions 6–9 and Eu eluting in fractions 10–13. Because the analytes elute over multiple fractions, small changes in the elution conditions can result in large changes in yield when evaluating each fraction. To maximize separation and account for small shifts in the elution profile, each 2 mL fraction was collected separately and screened by gamma spectroscopy for 20 min to determine the elution profile. The yields for Ce, Sm, and Eu can each be maximized to > 90% if the applicable fractions are combined either during elution or following the screening by gamma spectroscopy [
12].
The chemical yield for Tb is high because this represents the complete Tb elution peak where fractions 18–20 were co-collected and processed through a second LN resin separation where fractions 4–6 were collected. The eluate collected for
161Tb radiometric analysis is slightly larger than the Tb elution peak as determined by cold carrier studies to allow for small shifts in the elution profile to maximize the Tb yield without co-eluting Eu or Y. Based on cold carrier studies, the Tb yield from the LN separation is >95% [
12]. However, there can be Tb loss during the TRU separation reducing the overall yield; cold carrier studies have shown this loss to be variable but were typically between 2% and 15%.
The quantities of radio-lanthanides present in the Sm, Eu, and Tb fractions are given in
Table 4,
Table 5 and
Table 6. In addition to the primary radio-lanthanide of interest, the quantities and the minimum detectable concentration (MDC) are given for other lanthanides which may be present in those samples. The Sm fraction contains small quantities of light lanthanides; the highest decontamination factor (DF) is achieved for the lightest lanthanides, which are expected as they elute in order of atomic number. The Eu fraction also contains small quantities of Ce and Nd, although Sm was below MDC. The quantities of light lanthanides remaining in the Sm and Eu fractions do not adversely impact quantification using gamma spectroscopy.
Optimization studies showed that the Tb fraction following the first separation using LN resin was sometimes contaminated with other radio-lanthanides, including 141,143,144Ce, 147Nd, and 153Sm. While the quantities of the impurities were low, their presence hindered 161Tb analysis via LEPS and prevented the use of beta counting. Performing a second separation using LN resin on the Tb fraction removes the radio-lanthanide impurities.
The light lanthanides including
141Ce,
143Ce,
144Ce, and
147Nd are all peak yield fission products which are present in the irradiated HEU sample at much higher levels than Sm, Eu, or Tb;
141,143,144Ce and
147Nd are present at roughly 15–40 times that of Sm, 150–400 times that of Eu, and 26,000–68,000 times that of Tb; the CFY are given in
Table 7. These radionuclides also have sufficiently long half-lives to be present during analysis following the chemical separations described here. The effectiveness of the separation to remove the light lanthanides can be quantified using a DF that is based on the number of atoms of an analyte in the samples before separation compared to the number of atoms of that analyte post separation as given in Equation (1).
The Tb samples following the second separation were found to have 8.79 × 10
5 ± 20.3% atoms
161Tb/g HEU solution with all other radionuclides below the MDC; the MDC for radio-lanthanides of interest is shown in
Table 6.
The absolute CFY for each analyte was calculated based on the atoms of each radionuclide and the fissions, which were produced in the sample as shown in Equation (2). The CFY for the radionuclides quantified at PNNL with relative uncertainties, as well as the CFY reported by England and Rider [
17] and ENDF/B-II.1 [
18], are shown in
Table 7. The CFY for
140La,
141Ce,
143Ce, and
147Nd is based on the gamma radiometric analysis of the unseparated HEU solution, while
144Ce,
153Sm,
156Eu, and
161Tb are based on radiometric analysis of the separated fractions following the LN resin separation. The calculated results based on the thermally irradiated HEU are consistent with the values reported in England and Rider [
17] and ENDF/B-VII.1 [
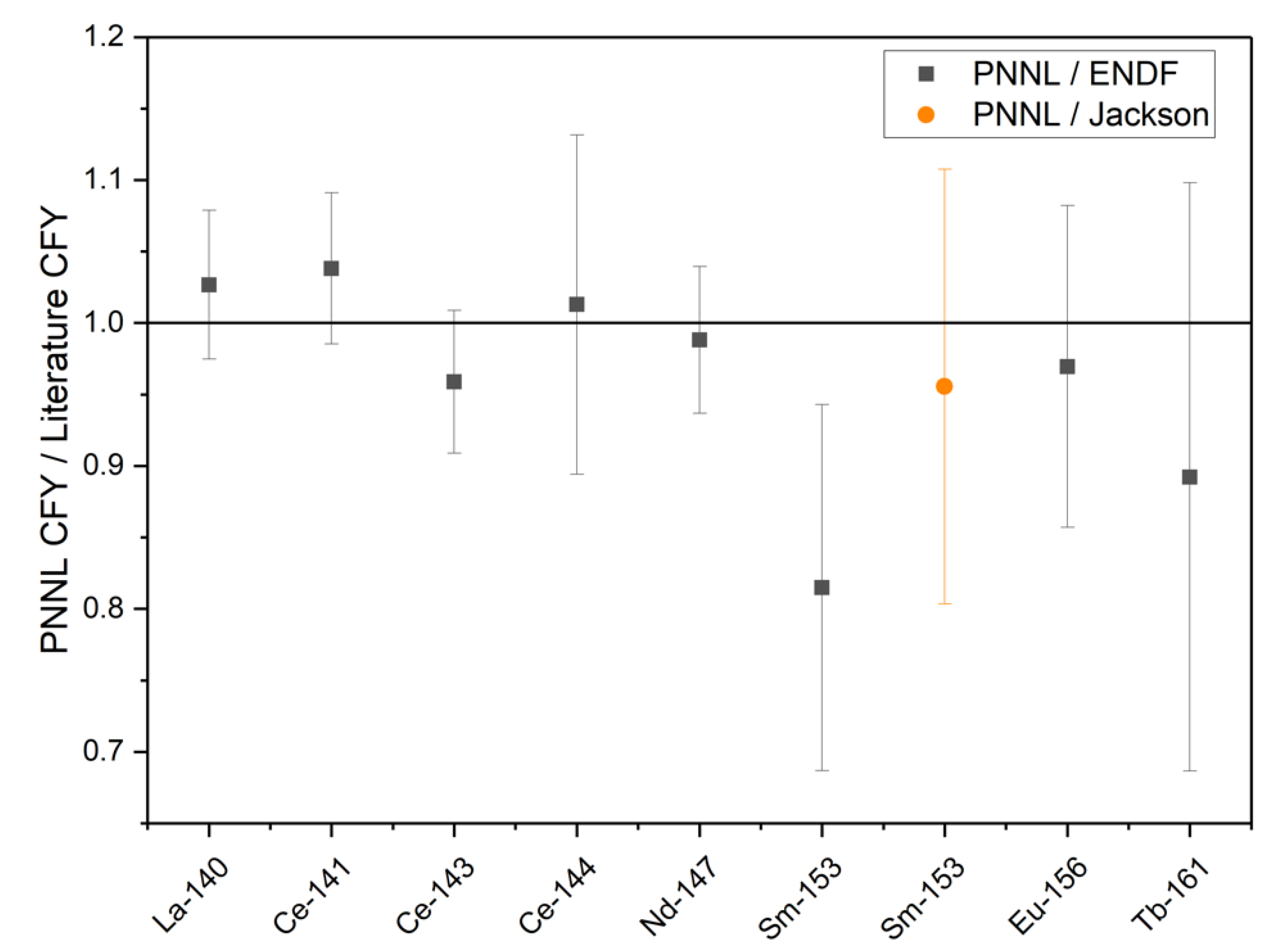
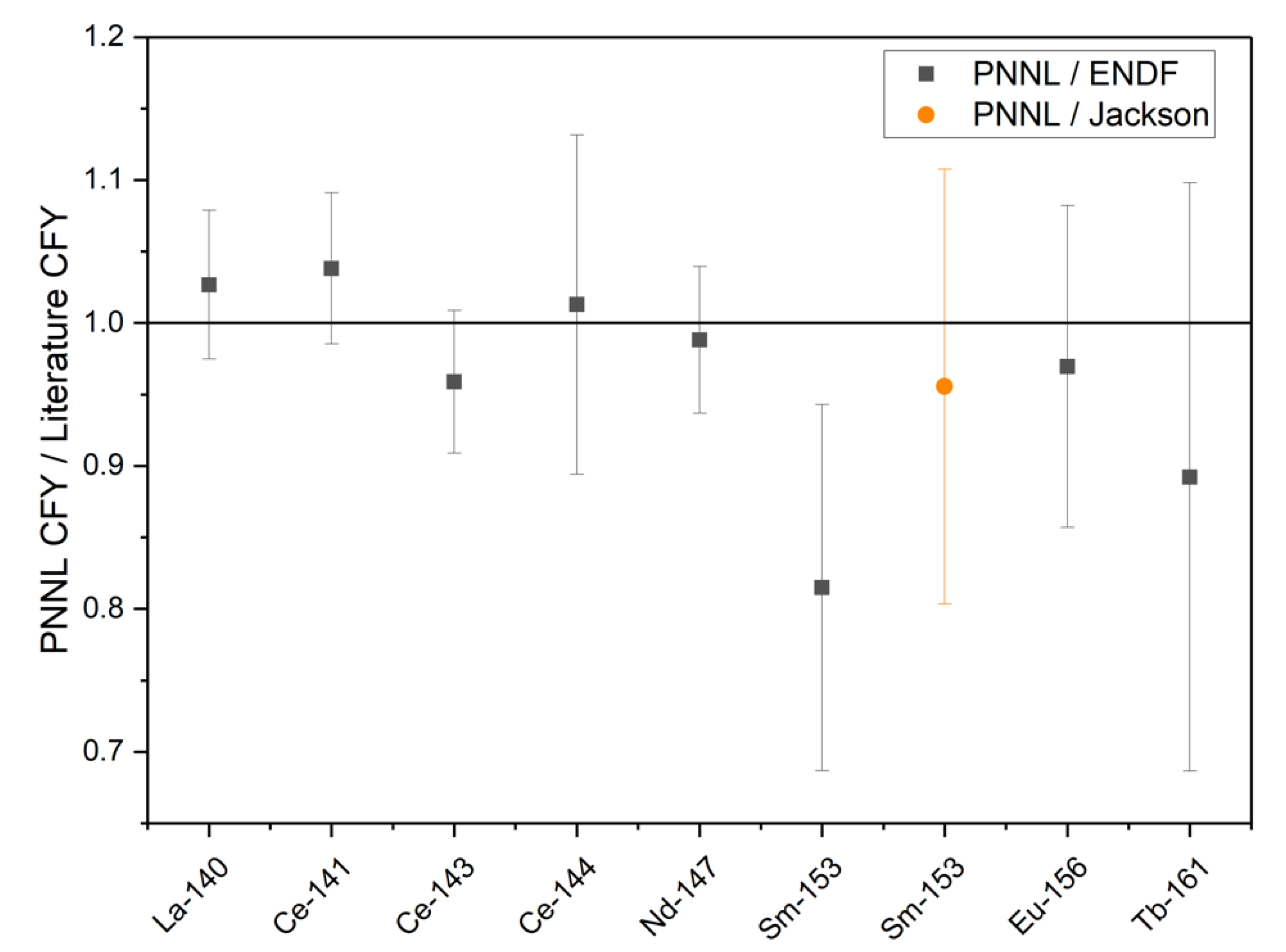
18] within a 1σ uncertainty for the radionuclides other than
153Sm. During an interlaboratory calibration and comparison study for
153Sm, Jackson et al. found that the
153Sm thermal
235U CFY reported by England and Rider (1.58 × 10
−3) is most likely 15% high and should be closer to 1.35 × 10
−3 [
17]. While the
153Sm CFY calculated based on the experimental results reported here is low compared to England and Rider, it agrees well with the value reported by Jackson et al. [
19]. The ratios of the PNNL CFY and the CFY reported in ENDF/B-II.1 or by Jackson et al. are shown in
Figure 2.
4. Conclusions
With the methods developed for REEs separation reported here, it is possible to obtain separated fractions containing Ce, Sm, Eu, and Tb from a single aliquot of a fission product sample. The radiometric purity of the Sm and Eu fractions is greatly increased, thereby decreasing the detection limit and/or radiometric analysis time. The improved radiometric purity generates Tb fractions suitable for LEPS or beta analysis. An additional benefit of the new separation method is a light lanthanide fraction suitable for quantification of Ce. Cerium-144 has a relatively long half-life and can be quantified in the irradiated HEU solution after the shorter-lived fission products have decayed; however, with this new method, a low uncertainty measurement by gamma spectroscopy is possible after the chemical separation. This method is based on gravity column chromatography, so it is possible to run many separations in parallel. The method reported here uses HNO3 as the eluent, the extractant that enables the separation remains on the column removing the need for additional processing prior to analysis, and the eluent does not require pH adjustment. In contrast, anion exchange separations, including high-pressure ion chromatography methods, add organic ions to the sample to greatly improve the intra-group separation, and the separation is highly dependent on the eluent pH. The separation of REEs using LN resin with an HNO3 eluent offers an additional option to the available intra-group REEs separation methods.



{kind=link}
{kind=link}