Abstract
This paper describes smart sampling paper to be used for bottom-up protein analysis. Four different manners to immobilize trypsin on cellulose were evaluated. Untreated paper, potassium-periodate-functionalized paper (with and without post-immobilization reduction) and 2-hydroxyethyl methacrylate (HEMA)/2-vinyl-4,4-dimethylazlactone (VDM)-functionalized paper were all used to immobilize trypsin. For the evaluation, Coomassie Brilliant Blue staining of proteins on paper and the BAEE trypsin activity assay needed to be modified. These methods allowed, together with data from mass spectrometric analysis of cytochrome C digestions, us to acquire fundamental insight into protein binding, and trypsin action and activity on paper. All functionalized discs bind more protein than the untreated discs. Protein binding to functionalized discs is based on both adsorption and covalent binding. Trypsin immobilized on potassium-periodate-functionalized discs exhibits the highest trypsin activity when using cytochrome C as substrate. It is proven that it is trypsin attached to paper (and not desorbed trypsin) which is responsible for the enzyme activity. The use of discs on complex biological samples shows that all functionalized discs are able to digest diluted serum; for the best-performing disc, HEMA-VDM functionalized, up to 200 high-confidence proteins are qualified, showing its potential.
1. Introduction
Sampling blood on paper for the determination of biologically interesting compounds has a history of more than a century. The use of dried blood spots (DBS) has the advantage that there is no need for trained medical personnel for sampling as it only requires a fingerprick for blood collection. It can be performed at home, even in remote areas, making it easier to carry out clinical studies. Additionally, there is limited biohazard and transport is easy [1]. The first scientific paper on DBS was published by Bang in 1913 describing the sampling of blood on filter paper for the analysis of glucose [2]. The widespread interest and use of DBS was boosted by Susie and Guthrie who established the phenylketonuria (PKU) test for newborn screening in blood collected on filter paper cards from the infant’s heel [3]. This test to determine abnormalities in the ratio of essential amino acids phenylalanine and tyrosine is still in use. Although the analytical measurement has evolved from a bacterial inhibition test [3] to state-of-the-art tandem mass spectrometric detection [4], the ease of sampling on filter cards still is the same. At present, there are numerous publications and applications on the use of DBS for small biologically active compounds. The analysis of these smaller compounds from DBS has become a routine tool which can be automated and combined with high-end analytical measurements [5,6].
The determination of large molecules such as proteins from DBS has developed less than that of the smaller molecules. There is a fair amount of publications on proteins which are determined with immunometric assays after DBS sampling. By far the most described assay is that of the CRP determination in various populations [7,8,9,10,11,12], but also immunometric assays for hemoglobin [13,14], immunoglobulins [15,16,17] and retinol-binding protein [18,19,20] after DBS sampling are well documented. In a more exploratory stage is the mass spectrometric determination of proteins from DBS. Several groups have managed to measure proteins in a top-down (such as Hb [21,22,23,24] and insulin [25,26]) as well as a bottom-up approach (such as hCG [27,28]). One of the challenges with the bottom-up approach is the time which is needed from arrival of the DBS at the laboratory to the analysis.
For the bottom-up analysis of proteins, tryptic digestion is the most used and also most time-consuming pretreatment. Integrating this step such that it already starts during the collection of the patients sample on paper is assumed to be a time-saver: there will no longer be a need for overnight protein digestion, as this already is carried out during sampling and DBS transport (typically regular mail). In this way, the ease of sampling on paper is combined with pre-lab sample preparation. It is expected that upon arrival at the analytical laboratory, most of the proteins are digested and that only a simple extraction is needed to perform the analysis, thus improving the turn-over of MS-based protein analysis from paper.
The binding of trypsin to cellulose as well as the ability to digest proteins in biological samples have been shown earlier by our group in proof-of-principle publications. Here, cellulose was first silanized, then functionalized through the polymerization of 2-hydroxyethyl methacrylate (HEMA) and 2-vinyl-4,4-dimethylazlactone (VDM), allowing trypsin to bind covalently to paper [29]. Besides this protocol, there are other possibilities to bind proteins covalently to paper [30,31,32,33,34], among which is also the simple KIO4-mediated oxidation of cellulose to create two aldehyde groups per cellulose unit, allowing subsequent protein binding. This involves the formation of a Shiff-base between the cellulose and the protein [31,33], which can be stabilized by NaCNBrH3-mediated reduction [34]. The latter protocols are easier to carry out compared to the HEMA-VDM protocol, but have never been evaluated for binding trypsin.
The long-term goal of our research is to simplify MS-based protein analysis from dried matrix samples (such as DBS), and we aim to integrate time-consuming procedures within the sampling device for a prompt start to the sample preparation upon sample collection [29,35,36,37,38].
In this particular paper, there is focus on the fundamental understanding of the behavior of immobilized trypsin on paper rather than on proof-of-concept. A comparison of untreated paper, HEMA-VDM-functionalized paper and KIO4-functionalized paper is performed in this first report of trypsin immobilization on KIO4-functionalized paper. The aim of this study is to provide simple methods to describe fundamental aspects of trypsin coupled to paper to complement the evaluation by mass spectrometry, as well as to evaluate various immobilization strategies with respect to protein binding and enzyme performance.
Cytochrome C is used as a model protein to show the digestion ability using trypsin immobilized paper as well as to estimate the enzymatic activity of the immobilized trypsin. The applicability and the potential of trypsin discs for complex samples containing many proteins is demonstrated using diluted serum.
2. Experimental
2.1. Chemicals and Reagents
Potassium periodate (KIO4), sodium cyanoborohydride (NaCNBH3), benzamide (≥95%), 2-hydroxyethyl methacrylate (HEMA, 97%, containing 200–220 ppm monomethyl ether hydroquinone), 2,2′-azobis(2-methylpropionitrile) initiator (98%, (AIBN)), 3-(trimethoxysilyl)propyl methacrylate (γ-MAPS, 98%), 2,2-diphenyl-1-picrylhydrazyl hydrate (DPPH), anhydrous N,N-dimethylformamide (DMF), Nα-benzoyl-L-arginine ethyl ester hydrochloride (BAEE), Coomassie Brilliant Blue R, cytochrome C from equine heart (≥95%), bovine serum albumin (BSA), and trypsin from bovine pancreas TPCK treated (≥10,000 BAEE units per mg protein) were all purchased from Sigma-Aldrich (St. Louis, MO, USA). 2-Vinyl-4,4-dimethylazlactone (VDM) was purchased from Polysciences Inc. (Warrington, PA, USA).
All other chemicals used were analytical grade or LC-MS grade.
2.2. Functionalization of Paper Discs and Protein Immobilization
In general, the paper discs were produced as described earlier in literature. In all cases, 6 mm paper discs were used. These were punched out of Grade 1 Qualitative Whatman paper from GE Healthcare (VWR, Norway) using a Philip Harris 6 mm Uni-Core puncher (Birmingham, UK).
Functionalization using KIO4: For the KIO4 functionalization, the procedure published by Chen et al. [31] was followed. In short, the discs were placed in a 0.03 M KIO4 solution at 65 °C for 2 h. The discs were four times washed by dipping them in MilliQ water. After the last wash, the discs were blotted on filter paper, left to dry (air dry) at room temperature and then placed in a desiccator.
Protein immobilization on KIO4-functionalized paper/untreated paper: 10 µL BSA or trypsin at the desired concentration (dissolved in 1 mM HCl) was dripped on the disc. The disc was placed in a 96-well plate which was sealed immediately afterwards.
Immobilization was carried out overnight at room temperature. After the immobilization, the excess solvent was removed by blotting.
The KIO4-reduced discs were produced as described above supplied with an additional step [34]: after protein immobilization, 10 µL of 1.6 mg/mL mM NaCNBrH3 was applied to the paper and the reduction was carried out at room temperature for 30 min in darkness.
Functionalization using HEMA-VDM: For the HEMA-VDM functionalization, the procedure published by Skjærvø et al. [29,36,37,38] was slightly modified and used. In short, the fibers on the paper discs were first silanized by applying 10 µL of freshly prepared silanizing solution (this is a mixture of 2.5 mg DPPH, 330 mg DMF and 172 mg γ-MAPS prepared in a glass vial). The discs were placed carefully in a 96-well plate, sealed and baked at 100 °C for two hours. The silanized discs were then rinsed in acetonitrile (ACN) and dried at room temperature. Secondly, the discs were polymerized by soaking them with 5.5 µL of freshly prepared polymerization mixture (mixture of 3.7 mg AIBN, 100 µL VDM, 300 µL HEMA and 300 µL 1-heptanol). The discs were placed back in the 96-well plate, sealed and baked at 80 to 100 °C for two to three hours. After the polymerization, the papers were washed with ACN and dried at ambient temperature.
Protein immobilization on HEMA-VDM-functionalized paper/untreated paper: BSA or trypsin at the desired concentration was dissolved in 500 µL immobilization buffer (0.1 M phosphate buffer pH 7.8 containing 5 mM ethanolamine and 4 mM benzamidine). The paper discs were placed in 2.0 mL Eppendorf (Hamburg, Germany) LoBind tubes with the protein containing immobilization buffer. The immobilization was carried out at ambient temperatures for three hours at 700 rpm.
Notice that protein immobilization on untreated paper was carried out in two different ways (o/n immobilization in 1 mM HCl and a 3 h immobilization in immobilization buffer). This was done to make the comparison with the different functionalizations the fairest it could be.
Unless otherwise described and independent of the protocol followed, the discs were washed three times using 15 µL 50 mM ammonium bicarbonate (ABC).
2.3. Staining Procedure
Staining was performed on 6 mm untreated or functionalized discs. On these discs, 0.5 µL BSA solution (0.5% in PBS) was immobilized.
After protein immobilization, the discs were blotted to remove excess BSA. The discs were subsequently washed three times with 50 mM ABC and blotted between each wash. After the third wash, the discs were placed for 10 min in a heating oven at 80 °C (Binder, Tuttlinger, Germany) to fix the protein. The discs were placed in 200 µL 0.1% Coomassie brilliant blue solution (w/v) in the washing buffer (50% methanol and 10% glacial acid acetic in water). After 10 min, the excess of Coomassie brilliant blue was removed by blotting the discs on tissue paper. The paper discs were washed two at a time in 10 mL wash solution (50/40/10: MeOH/ Milli-Q water/ glacial acetic acid (v/v/v)). The rinsing procedure was executed in agitation at 800 rpm for 30 min, before replacing the solution and shaking an additional 30 min. Afterwards, the step was completed by blotting the discs on tissue paper to physically remove the excess of stain and wash solutions, before drying the discs at room temperature.
2.4. Evaluation of Staining
The staining was evaluated visually as well as with ImageJ (version 1.53a, NIH, USA). Pictures of the discs were taken with a cellular phone. To minimize the influence of variable light settings between pictures and cellular phones, discs to be compared were placed such that they were within the same picture. A quantitative evaluation was carried out using ImageJ. The only value which was measured was the “mean gray value”. The difference in mean gray values between the background and the stained part of the disc reflects the amount of protein immobilized. Figure 1 shows the typical position of the “oval” tool on the picture for both measurements.
Figure 1.
(A) Measured area of the protein-stained part of the disc. (B) Measured area close to the disc to correct for background. The difference in mean gray value = mean gray value (B)–mean gray value (A).
2.5. Measurement of Trypsin Activity Using a BAEE-Assay
The unbound trypsin activity measurement was carried out using a slightly modified BAEE assay (modified from https://www.sigmaaldrich.com/technical-documents/protocols/biology/enzymatic-assay-of-trypsin-inhibitor.html (accessed on 1 March 2020)). The unbound trypsin was extracted from the disc by placing it for 5 min while shaking in HulaMixer (orbital: 35 rpm, 5 s; reciprocal: 60°, 5 s) in 3 mL substrate buffer (2.8 mL 67 mM NaH2PO4 buffer, pH 7.6 and 0.2 mL 1 mM HCl). After extraction, the disc was removed and 200 µL 1.27 mg/mL BAEE solution was added to the substrate buffer and mixed manually by inversion. The activity was measured at 253 nm (Beckman DU-530, Dan Meszansky, Oslo, Norway) as defined in the protocol from Sigma-Aldrich.
Production of paper discs for measuring the unbound fraction of trypsin: To standardize the BAEE test, the same amount of trypsin was immobilized on untreated paper and KIO4 as well as HEMA-VDM-functionalized paper. Immobilization was carried out with 10 µL (0.05–0.2%) trypsin. Where needed, the KIO4 discs with trypsin immobilized were reduced (see above). After immobilization, the discs were dried without a washing step before being subjected to extraction.
2.6. Measurement of Trypsin Action or Activity Using LC-MS/MS
For the measurement of the action or activity of trypsin on disc as well as the unbound trypsin (in-solution), cytochrome C was used as a model protein. After production of the trypsin immobilized discs, they were subjected to washing. Trypsin action is defined as the ability to digest proteins, while trypsin activity is defined as the amount of substrate processed/products produced within a certain timespan.
The washing procedure for the measurement of trypsin action: the discs were placed in 2 mL washing solution (10 mM HCl) and shaken for 5 min with a HulaMixer (Thermofisher Scientific, Oslo, Norway) setted on orbital (35 rpm, 5 s) and reciprocal (60°, 5 s). After 5 min the discs were transferred to a fresh washing solution and the wash was repeated. After 5 min, this was repeated once more. After 5 min, the discs were transferred to 2 mL milliQ and shaken for 5 min with a HulaMixer. The last step in the washing procedure was carried out by transferring the discs to 100 µL 50 mM ABC and shaking for 5 min at 600 rpm. After the removal of the disc, this aliquot was used to measure the released (or desorbed/extracted/unbound) trypsin activity. This was done by simply adding 10 µL of cytochrome C (in fresh 50 mM ABC, varying concentrations cytochrome C were tested). The solution was placed at room temperature. The digestion reaction was stopped by adding 10 µL 1% formic acid.
The washing procedure for the measurement of trypsin activity consisted of 9 cycles. For the first 8 cycles, the discs were transferred to a 2 mL washing solution (0.05% Tween in PBS) and shaken for 5 min with a HulaMixer setted on orbital (35 rpm, 5 s) and reciprocal (60°, 5 s). After this was done 8 times, the discs were transferred to 2 mL water for a last wash. After this, the discs were transferred to 100 µL 50 mM ABC and shaken for 5 min at 600 rpm. In this way, the released (or desorbed/extracted/unbound) trypsin activity could be measured as in in-solution digestion by simply adding 10 µL of cytochrome C (in fresh 50 mM ABC, varying concentrations of cytochrome C were tested). The solution was placed at room temperature. The digestion reaction was stopped by adding 10 µL 1% formic acid.
Digestion of cytochrome C on disc was performed by dripping 10 µL of cytochrome C (in fresh 50 mM ABC, varying concentrations cytochrome C were tested) on the disc. The discs were either dried, placed in a 96-well plate followed by sealing or placed in a 96-well plate followed by sealing and drying. Both cytochrome C concentration, time of sealing and time of drying varied with different experiments. The digestion was stopped either when the discs became dry or when the tryptic peptides were extracted. Extraction was carried out by placing the discs (either wet or dry) in a 110 µL 0.1% formic acid solution followed by shaking (600 rpm) for 5 min.
2.7. Serum Digestion on Functionalized Paper with Immobilized Trypsin
For the digestion of serum on paper, 3 paper discs of each functionalization and immobilization procedure are used (three KIO4 discs, three KIO4-reduced and three HEMA-VDM discs). In all cases, 0.1% trypsin was used during the immobilization. After immobilization, the discs were washed using the following procedure: 10 wash cycles were used, for the first 8 cycles the discs were transferred to a 2 mL washing solution (0.1% formic acid) and shaken for 5 min with a HulaMixer setted on orbital (35 rpm, 5 s) and reciprocal (60°, 5 s). Formic acid (0.1%) was the preferred washing solution here (compared to 0.05% Tween in PBS in the previous wash) to avoid too high background on the full scan mass spectrometric analysis. After this, the discs were transferred to 2 mL water for a 9th wash. After this, the discs were transferred to 100 µL 50 mM ABC and shaken for 5 min at 600 rpm. The discs were taken out of the vials and air-dried before use.
Twenty microliters of ten times diluted human serum (diluted with fresh 50 mM ABC) was dripped on the discs and sealed for an hour. After this, the discs were left to air-dry at room temperature (took up to one hour). Each of the dried discs were placed in its own eppendorf vial and 100 µL freshly prepared 50 mM ABC containing 5 mM DDT was added to each of the vials. The vials were placed at 50 °C and shaken at 800 rpm for 15 min. After this, the vials were allowed to cool down followed by the addition of 10 µL 250 mM IAA. The discs were placed in the dark at room temperature for 15 min.
Prior to injection, clean-up was carried out on 100 µL of the sample using in-house-made solid phase extraction as described by [29]. After drying, the residue was reconstituted using 100 µL 0.1% formic acid and transferred to the injection vial.
2.8. LC-MS Conditions for Determination of Cytochrome C Digestion Products
Five tryptic peptides (see Table 1) were determined using high-performance liquid chromatography-tandem tandem mass spectrometry (LC-MS/MS). The LC-MS/MS system consisted of a Vanquish UPLC coupled to an Altis triple quadrupole mass spectrometer (ThermoFisher, Nerliens, Oslo, Norway). XCaliburTM version 4.2.28.14 was used to manage the system and acquire and process data. Positive electrospray ionization and selected reaction monitoring were performed to determine peptides. The spray voltage was set to 3.5 kV. Sheath gas, auxiliary gas and sweep gas were set to 25, 5 and 0 arbitrary units, respectively. The temperatures for the ion transfer tube and vaporizer were set to 325 and 75 °C, respectively. The collision energy was 20 V and the CID gas pressure was 1.5 Torr. The dwell time was 54 ms per transition. The mass-to-charge (m/z) ratio of the parent and the products as well the nature of the fragments are presented in Table 1.

Table 1.
Tryptic peptides and SRM transitions.
Separation was carried out on an Aquasil C18 (3 µm), 50 × 1 mm reversed phase column (ThermoFisher). Mobile phase A consisted of 5% acetonitrile and 95% 20 mM formic acid, whereas mobile phase B consisted of 95% acetonitrile and 5% 20 mM formic acid. The flow rate was set to 0.05 mL/min. The gradient started at 0% mobile phase B for 3 min. After 3 min, the share of mobile phase B was increased linearly to 50% until t = 23 min. From 23 to 23.1 min, the system switched to 100% mobile phase B and was kept there until t = 28 min. At t = 28.1 min, the system switched back to starting conditions to re-equilibrate the column. The total run time was 35 min.
2.9. LC-MS Conditions for Determination of Serum Digestion Products
Peptides were analyzed using an Ultimate 3000 nano-UHPLC system (Dionex, Sunnyvale, CA, USA) connected to a Q Exactive mass spectrometer (ThermoElectron, Bremen, Germany) equipped with a nano electrospray ion source. For liquid chromatography separation, an Acclaim PepMap 100 column (C18, 3 µm beads, 100 Å, 75 μm inner diameter) (Dionex, Sunnyvale, CA, USA) capillary of 50 cm bed length was used. A flow rate of 300 nL/min was employed with a solvent gradient of 3–35% B in 50 min, to 50% B in 3 min and then to 80% B in 1 min. Solvent A was 0.1% formic acid and solvent B was 0.1% formic acid/90% acetonitrile.
The mass spectrometer was operated in the data-dependent mode to automatically switch between MS and MS/MS acquisition. Survey full scan MS spectra (from m/z 400 to 2000) were acquired with the resolution R = 70,000 at m/z 200, after accumulation to a target of 1e6. The maximum allowed ion accumulation times were 100 ms. The method used allowed for the sequential isolation of up to the twelve most intense ions, depending on signal intensity (intensity threshold 1.7e4), for fragmentation using higher collision induced dissociation (HCD) at a target value of 10,000 charges, a resolution R = 17,500 and a normal collision energy (NCE) of 28. Target ions already selected for MS/MS were dynamically excluded for 15 sec. The isolation window was m/z = 2 without offset. The maximum allowed ion accumulation for the MS/MS spectrum was 60 ms. For accurate mass measurements, the lock mass option was enabled in MS mode and the polydimethylcyclosiloxane ions generated in the electrospray process from ambient air were used for internal recalibration during the analysis.
2.10. Data Analysis
XCaliburTM version 4.2.28.14 was used to analyze the cytochrome C data from the Altis Triple quadrupole (ThermoFirsher).
XCaliburTM version 4.2.47 was used to analyze the data from the diluted serum samples.
Proteome Discoverer version 2.3 (ThermoFisher) was used to analyze the MS and MS/MS spectra of the diluted serum samples. A fasta-file from proteins in human serum obtained from UniProt (4 September 2019) was used as a database. The maximum amount of missed cleavages was set to 5, and the minimum peptide length and maximum peptide length were set to 6 and 144 amino acids, respectively. Tolerance was 10 ppm, the fragment tolerance 0.6 Da. As dynamic modification methionine oxidation (m/z +15.995) was chosen. The carboxymethyl modification (m/z +58.005) was static.
3. Results and Discussion
3.1. Description of the Functionalized Papers Used
In this study, a set of methods is used to assess both protein binding to paper and enzyme activity on paper. Four different paper treatments, all of which are shown in Figure 2, were evaluated with this set of methods:

Figure 2.
Binding of trypsin to cellulose (A) without treatment, (B) after KIO4 oxidation (C) after KIO4 oxidation and NaCNBrH3 reduction (D) after HEMA-VDM silanization and polymerization.
- -
- Untreated paper: On this paper, protein was immobilized by simply dripping the protein solution on paper followed by incubation for 3 h or overnight. Protein attachment to paper is believed to be caused mainly by adsorption (Figure 2A).
- -
- KIO4-mediated functionalized paper: After KIO4 treatment, aldehydes are formed, which allow for the covalent coupling of proteins through a Shiff base (Figure 2B).
- -
- KIO4-mediated functionalized paper, reduced: Production of this paper follows the same protocol as the KIO4-mediated functionalized paper. After protein immobilization, sodium cyanoborohydride reduction is carried out to stabilize the protein–paper bond (Figure 2C).
- -
- HEMA-VDM-functionalized paper: Using this approach, a polymer layer is constructed around the paper fibers and proteins are covalently bound to this polymer layer (Figure 2D).
3.2. Staining of Proteins on Paper to Assess Protein Binding
An easy way to determine if there is protein bound to the paper is by CBB staining. To do this for our purpose, a previously described staining procedure for paper was used [37,39]. It was expected that the protein immobilized on untreated paper followed by a wash would yield minimally stained discs, while protein immobilization on functionalized paper followed by a wash would yield strongly stained discs. In this way, evidence would be provided for the covalent binding of protein to the functionalized discs. Figure S1 (upper discs–lane A) in the Supplementary Materials shows the results after using the referred protocol. It is visually challenging to differentiate between the color of the stained untreated discs and the stained functionalized discs. To be able to determine differences in the protein binding of various paper treatments, some modifications of the protocol were necessary.
3.3. Protocol for Staining—Method Modification
The following modifications of the protocol were made. It should be stressed that these modifications are only to improve the method’s ability to assess protein binding.
- (A)
- Spot size on the disc: Since it is difficult to detect differences between the various treated discs, it was decided that the application volume of protein solution should be such that its boundaries were within the edges of the disc (dripping 0.5 µL BSA solution on 6 mm discs). The rationale was that the diameter of the protein spot would be less (4–5 mm) than the diameter of the disc (6 mm). When the discs were washed, the unbound protein would be washed out/wick beyond the spot boundaries. The bound protein would be within the application spot. This makes it visually easier to determine where the protein is bound.
- (B)
- Protein concentration: The protein solution applied was set to 0.5% BSA. Too low or too high concentrations do not give a staining intensity which allows for easy differentiation.
- (C)
- Number of washes: A three-times wash of the discs with 15 µL 50 mM ABC solution is carried out to remove the unbound protein, which would interfere with the staining.
The discs in the lower part of Figure S1 (lane B) in the Supplementary Materials shows staining results of 0.5 µL 0.5% BSA solution applied on untreated and functionalized discs. From this figure, it is concluded that the visual evaluation of the stained discs is possible with the modified procedure.
3.4. Assessment of Protein Binding after Various Treatments and Washes
To assess the protein binding to paper as well as the effect of various washing solutions, the following experiment was carried out: 0.5 µL of a 0.5% BSA solution was applied to paper which underwent the four different treatments for immobilization (untreated, KIO4 functionalized, KIO4 functionalized and reduced, HEMA-VDM functionalized). After immobilization, the paper discs were washed three times with either water, 20 mM ABC buffer, 10 mM HCl or 10 mM NaOH. Figure 3 shows the result of the paper staining after these treatments and a subsequent HCl wash.

Figure 3.
(Left) Representative stained discs after protein (BSA) immobilization on paper that was KIO4 functionalized, KIO4 functionalized and reduced, untreated or HEMA-VDM functionalized. Wash: three times with 10 mM HCl. (Right) Mean gray value of the discs in the pictures on the left.
Figure S2 in the Supplementary Materials shows the effect of all washes on the various discs.
Visual assessment of the stained discs in Figure 3 shows that all papers contain protein after a 10 mM HCl wash. The untreated paper has expectedly the least color intensity; however, the initial spot still is slightly visible, indicating unspecific binding (adsorption) of BSA to the paper. KIO4 and KIO4-reduced paper showed a higher color intensity with a distinct circle of the spot indicating more protein bound. The HEMA-VDM paper shows the highest color intensity of the four paper treatments. This could indicate that this treatment gives the most protein binding. It should, however, be noted that the spot circle on the HEMA-VDM discs is more irregular in shape and that the color intensity is not evenly distributed throughout the spot. Additionally, there is a larger variation in color intensity for the HEMA-VDM discs than for the other functionalized discs. In general, it can be said that, for the functionalized paper, the protein binding probably is due to both adsorption and covalent binding.
An ImageJ analysis of the color intensity of these discs (Figure 3 right) supports the visual assessment of the amount protein bound: The difference in mean gray value (measured in calibrated units) between the spot and background of the HEMA-VDM is the highest, followed by the KIO4-functionalized discs. The lowest difference in mean gray value between the spot and background was detected for the untreated discs. The larger the difference in mean gray value, thus, the more protein is attached to the disc.
Both visual assessment as well as the mean gray value measurement show that the HEMA-VDM discs contain the most protein, followed by KIO4 and KIO4-reduced. The untreated paper has the least protein attached.
3.5. Trypsin Immobilization and Its Activity
Coupling trypsin to paper is carried out the same way as the coupling of BSA to paper. However, as the above experiments show the ability to bind protein to the various forms of functionalized paper, instead of measuring the protein presence or content, the enzyme activity was measured to get an impression on the efficiency of immobilized trypsin.
The trypsin activity was determined in two ways:
- -
- The indirect way, by measuring the activity of unbound trypsin (portion of trypsin not attached after immobilization). In this way, the amount of trypsin coupled to paper can be estimated. This measurement is carried out using a BAEE assay.
- -
- The direct way, by measuring the formation of tryptic peptides after applying a protein sample to the discs. This measurement is carried out by LC-MS/MS.
3.6. BAEE Assay of Unbound Trypsin—Indirect Measure for Amount Trypsin Bound
The binding of trypsin to paper can happen in two ways, by adsorption as well as by covalent binding. It is expected that for functionalized paper there is a portion of trypsin which will bind covalently as well as a portion which will bind via adsorption. For untreated paper, adsorption is the only way trypsin will be bound. Either way, both untreated paper and functionalized paper will contain trypsin after immobilization. The immobilization yield is less than 100%, leaving a share of the trypsin unbound. This unbound share can be measured. For this purpose, a generic protocol for the BAEE assay was slightly modified for activity measurement of the unbound trypsin. The unbound trypsin was extracted from the discs in the substrate buffer without substrate being present. After the removal of the discs, only the portion of unbound and released trypsin remains in solution, hence the activity measured can solely originate from this. Thus, the activity is a measure for the unbound trypsin.
Figure 4 shows the activity of unbound trypsin after overnight immobilization on the KIO4-functionalized paper, KIO4-functionalized paper followed by reduction and untreated paper (Figure 4A) as well as 3 h immobilization on HEMA-VDM-functionalized paper and untreated paper (Figure 4B). In all cases, there is a limited and known amount of trypsin present for immobilization: 10 µL containing either 0.2%, 0.1% or 0.05% trypsin. These concentrations were chosen on the basis of upper and lower detection levels when the BAEE method is used. If more than 0.2% trypsin is used, the activity of the unbound trypsin is so high that it exhausts the substrate within less than a minute, which makes it impossible to determine the enzyme activity. Concentrations lower than 0.05% did not show any activity of unbound trypsin at all.
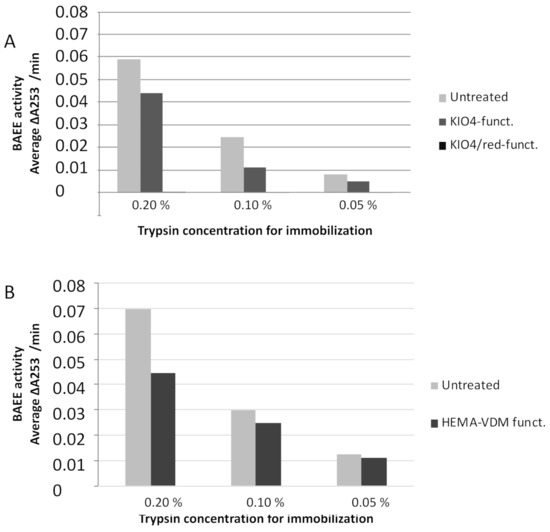
Figure 4.
Indirect measurement of the trypsin activity. Activity in wash fraction as a function of immobilized trypsin concentration for (A) untreated paper, KIO4-functionalized paper and KIO4/reduced functionalized paper. Immobilization was carried out overnight using 10 µL (0.05–0.2%) trypsin (B) untreated paper and HEMA-VDM-functionalized paper. Immobilization was carried out for 3 h using 10 µL (0.05–0.2%) trypsin.
It can be seen that in all cases and for all three trypsin concentrations the amount of unbound trypsin is higher for the untreated paper (this results in lower activity in the BAEE assay). This is in accordance with the staining experiment in Figure 3: the functionalized paper binds more protein than the untreated paper. It, therefore, seems logical to conclude that the less unbound trypsin seen for all functionalized papers is due to a portion of the trypsin binding covalently to the paper in addition to the non-specific adsorption. From this experiment, it is not possible to know how much of the trypsin is immobilized on functionalized paper through adsorption and how much through covalent binding. The only conclusion which indirectly can be drawn is that more trypsin is immobilized on the functionalized discs.
Another interesting observation from Figure 4A was that the extract of the KIO4/reduced discs does not exhibit any trypsin activity, which suggests that all the trypsin is bound to the paper. This is probably not the case, as explained by the following:
- -
- The experiments shown earlier (Figure 3) do not suggest that all protein is bound to the paper by including the reduction step in the KIO4 functionalization, even though slightly more protein is bound compared to the non-reduced KIO4-functionalized paper.
- -
- There is a slight effect, if any, of the reducing agent NaCNBrH3 on the activity measurement BAEE method (measured in solution—data not shown).
- -
- Dripping the 10 µL NaCNBrH3 on the disc probably extracts some of the trypsin and will thus be lost for a measurement.
A combination of these factors will probably cause the activity to be below the detection limit of the BAEE method.
To get a yield estimate on how much of the trypsin was attached to the discs, the activity results from the 0.1% trypsin concentration immobilization (as described above) were compared to the activity of 10 µL freshly prepared 0.1% trypsin as well as 0.1% trypsin solutions which were left on the bench at room temperature for 3 h and overnight. The activity (Table S1 in the Supplementary Materials) varies between these three trypsin solutions but is in the same order of magnitude. The activity of the unbound trypsin of the discs (by means of the Δ A253/min) is between 0.011 and 0.031. The yield of the immobilization is 72.8% and 77.2% for the untreated and HEMA-VDM-functionalized paper, respectively, and 73.7% and 88.4% for the untreated and KIO4-functionalized paper, respectively. This shows that much of the trypsin is bound to the paper, either by adsorption or by a combination of adsorption and covalent binding. These data support the findings shown in Figure 3. It should, however, be noted that, in Figure 3, protein amounts are measured, while in Table B (Supplementary Materials) enzyme activities are measured.
3.7. MS Analysis of Protein Digestion on Paper
The disc’s ability to digest protein was monitored in several ways by LC-MS. Initially, the trypsin action was evaluated by checking for cytochrome C digestion products. The trypsin activity was assessed by monitoring tryptic peptide production only a short time (10 min exactly) after the digestion has started. This reflects a situation with substrate excess.
3.7.1. Trypsin Action: Presence of Digestion Products
The discs’ ability to digest a protein was evaluated by simply applying 10 µL of a 25 µg/mL CytC solution to discs which were pre-washed with 10 mM HCl (3 × 2 mL), water (1 × 2 mL) and 50 mM ABC (100 µL). After incubation and drying, the tryptic peptides were extracted into 0.1% FA and analyzed with LC-MS/MS. Figure 5A shows the extracted ion chromatograms of five target peptides, all which are zero missed cleaved peptides. From the figure, it is clear that trypsin on the disc was able to digest CytC. However, the production of tryptic CytC peptides was similar in the last wash fraction (see Figure 5B). The question thus raised if the action of trypsin was caused by the trypsin bound on the paper, by unbound trypsin which in a dried condition is situated between the fibers of the paper and is released when a sample is applied or by a combination of bound and unbound trypsin. With the experimental conditions chosen here, it was not possible to do that differentiation: the amount of CytC compared to the amount of trypsin is very low. This will easily lead to exhaustion of the substrate (complete digest of CytC) even if only 1% of the original amount of trypsin applied to the discs is present.


Figure 5.
Ion chromatograms of five “zero missed-cleaved” peptides of Cytochrome C. m/z values of the traces measured are shown in Table 1. (A) A total of 10 µL 25 µg/mL CytC applied on a dry KIO4-functionalized trypsin disc which was washed three times with 2 mL 10 mM HCl followed by 2 mL water and concluded with a 100 µL 50 mM ABC extraction. The disc was kept in a sealed container for 1 h. After the seal was taken off, the discs were left to air dry (1 h,) followed by extraction in 110 µL 0.1% formic acid. (B) A total of 10 µL 25 µg/mL CytC added to the 100 µL 50 mM ABC extract of a washed KIO4-functionalized trypsin disc. Incubation 2 h, reaction stopped with 10 µL 1% FA. Both (A) and (B) were carried out at room temperature.
3.7.2. Trypsin Activity Testing by LC-MS
From the experiments above, the following question was raised: is it the trypsin which is attached to the paper or is it trypsin in solution which is desorbed from the paper at the moment the sample is applied to the paper which causes digestion of CytC. In the latter case, the digestion would be an in-solution digestion rather than an on-disc digestion. An on-disc digestion by bound trypsin is desired since it is anticipated to lead to less auto-digestion products. The following experiment to determine the trypsin activity was therefore designed: after the immobilization of trypsin on the discs, a thorough washing procedure with PBS/Tween, water and 50 mM ABC was carried out (10 wash steps in total). This thorough wash was chosen to ensure that minimal trypsin would remain in the unbound state on paper. The activity of the trypsin on both the disc and in the final wash solution (50 mM ABC) was tested by adding a high amount cytochrome C (100 µg) and incubating it for only 10 min. In this way, it was assured that there was no substrate exhaustion and that the digestion rate was close to the initial reaction rate of the enzyme, the production of the tryptic peptides thus being a measure for the activity of the trypsin. Figure 6 shows that there is some (albeit very little) trypsin activity in the extracts (visible in the 100× enlargement of Figure 6). From this, it is concluded that there is only a very small portion of trypsin desorbing from the discs and that the production of peptides with a high degree of probability is caused by trypsin bound to the paper. This conclusion is not only based on the production of MIFAGIK (Figure 6) but also on the production of the other tryptic peptides monitored (see Figure S3 in the Supplementary Materials).

Figure 6.
Signal intensity of MIFAGIK. UH: untreated paper, trypsin immobilized for 3 h. H: HEMA-VDM-functionalized paper, trypsin immobilized for 3 h. UK: untreated paper, trypsin immobilization o/n. K: KIO4-functionalized paper, trypsin immobilization o/n. R: KIO4-functionalized paper, trypsin immobilization o/n followed by reduction. -s: in-solution digestion, -d: on-disc digestion.
The KIO4-functionalized paper exhibits overall the highest trypsin activity: it was at least 10 times higher than on untreated paper and approximately 4 times higher than on the KIO4-reduced paper. Considering all the peptides, there was little to no difference between activity on the HEMA-VDM-functionalized paper and on the untreated paper. Figure S4 in the Supplementary Materials shows a typical ion chromatogram of a CytC digestion on KIO4-functionalized paper.
3.7.3. Disc Performance on Complex Biological Samples
The discs’ ability to digest a complex sample like diluted serum was tested by dripping sample on the disc and allow for air drying according to the method described in the experimental section. The analytical workflow is shown in Figure 7.

Figure 7.
(A): smart sampler; (B): analytical workflow of serum digestion using immobilized trypsin on paper.
Figure 8 shows the base peak chromatogram of the tryptic peptides generated by one of the HEMA-VDM discs. It is clear that the paper-immobilized trypsin was able to digest proteins to peptides. Table 2 shows the number of high-confidence peptides and proteins found in these complex samples.

Figure 8.
Base peak chromatogram of a 10× diluted serum sample digested on HEMA-VDM paper immobilized with 0.1% trypsin. Reduction and alkylation was performed post-digestion.
Table 2.
Number of peptides and proteins measured in the HEMA-VDM-, KIO4- and KIO4-reduced discs.
From Table 2, it can be seen that up to 200 protein groups have been identified with high confidence for the HEMA-VDM discs, whereas lower numbers are seen for KIO4-treated discs. This is also seen in the number of high-confidence peptides qualified. As in earlier studies [35], it is seen that carboxymethylated tryptic peptides are found in expected amounts [40]. The latter is of interest since the reduction and alkylation take place after the digestion.
Although differences in performance on complex biological samples between the discs are seen, this experiment was merely designed to show the potential of the discs. A thorough investigation of experimental conditions would be needed to evaluate the discs’ performance for complex biological samples.
3.8. Future Perspectives
Although the experiments and results described are purely fundamental in nature, we believe smart samplers have potential in routine clinical analysis. The smart samplers integrate time-consuming sample preparation in the sampling and transport and by such they will reduce the overall sample processing time and improve the sample throughput. In the specific case of trypsination, very complex samples are generated from blood and serum on the modified paper and high-end analytical instrumentation for peptide separation and determination will still be needed. One potential use in clinical practice might be in personal proteomic profiling where larger populations sample and send their blood samples for general proteomic analysis. It also can be used in routine clinical practice for a targeted approach using LC-SRM-MS/MS when arrays of high- and medium abundant proteins are analyzed simultaneously. For the lower abundant proteins, it can be combined with peptide immunoaffinity clean-up and enrichment followed by LC-SRM-MS/MS determination. From an exploratory point of view, these smart samplers can function as small reactors, which can be integrated in a proteomic workflow.
In a broader perspective, the coupling of reagent proteins (other enzymes, antibodies) might even allow not only for sample preparation during sampling, but also for the easy on-spot determination of certain target compounds.
4. Conclusions
With CBB coloring and BAEE activity testing modified for evaluating protein binding as well as mass spectrometric analysis, we have described some fundamental aspects in the coupling of protein to paper. Paper that is functionalized to allow for covalent protein binding binds more protein than untreated paper. This goes for both the HEMA-VDM discs and the KIO4-treated discs.
From the evaluation of the activity of unbound trypsin, it indirectly concluded that a large share of the activity is captured by paper. Although the KIO4-treated discs (88%) and HEMA-VDM-treated discs (77%) capture slightly more, the untreated paper (73%) also adsorbs a considerable amount of trypsin. Trypsin bound, either covalently, by adsorption or by a combination of these to the paper is able to digest cytochrome C. Additionally, it is proven that the bound trypsin is responsible for the digestion of cytochrome C rather than trypsin which is desorbed during the sample application. The KIO4-treated discs show, for four of the five peptides measured, the best performance with at least double peptide production compared to the other discs within the short time of the assay. From the data, it does not seem to be favorable to reduce the cellulose-protein binding.
Based on our knowledge, this is the first time that KIO4-functionalized paper has been used to immobilize trypsin successfully.
For the more complex samples, it seems that all the discs are able to digest proteins. Under the conditions chosen, the HEMA-VDM-treated discs have the best performance with almost 200 protein groups identified.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/separations8050066/s1, Figure S1: Evidence for covalent binding of protein to the functionalized discs. Figure S2: Effect of wash solutions on the discs. Figure S3: Signal intensities of 5 tryptic peptides of cytochrome C. Figure S4: Extracted ion-chromatograms of 5 tryptic peptides of cytochrome C. Table S1: BAEE measurements.
Author Contributions
Conceptualization, L.R.; formal analysis, E.P., C.J.K. and L.R.; writing—original draft preparation, E.P. and L.R.; writing—review and editing, L.R., E.P., C.J.K. and T.G.H.; visualzation, E.P. and L.R.; supervision, L.R. and T.G.H.; project administration, L.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Crimmins, E.M.; Zhang, Y.S.; Kim, J.K.; Frochen, S.; Kang, H.; Shim, H.; Ailshire, J.; Potter, A.; Cofferen, J.; Faul, J. Dried blood spots: Effects of less than optimal collection, shipping time, heat, and humidity. Am. J. Hum. Biol. 2020, 32, e23390. [Google Scholar] [CrossRef] [PubMed]
- Bang, I. Blutzucker. Z. Für Anal. Chem. 1913, 52, 521–523. [Google Scholar] [CrossRef]
- Guthrie, R.; Susi, A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 1963, 32, 338–343. [Google Scholar] [PubMed]
- Chace, D.H.; Millington, D.S.; Terada, N.; Kahler, S.G.; Roe, C.R.; Hofman, L.F. Rapid diagnosis of phenylketonuria by quantitative analysis for phenylalanine and tyrosine in neonatal blood spots by tandem mass spectrometry. Clin. Chem. 1993, 39, 66–71. [Google Scholar] [CrossRef]
- Alexovič, M.; Dotsikas, Y.; Bober, P.; Sabo, J. Achievements in robotic automation of solvent extraction and related approaches for bioanalysis of pharmaceuticals. J. Chromatogr. B 2018, 1092, 402–421. [Google Scholar] [CrossRef]
- Resano, M.; Belarra, M.A.; García-Ruiz, E.; Aramendía, M.; Rello, L. Dried matrix spots and clinical elemental analysis. Current status, difficulties, and opportunities. TrAC Trends Anal. Chem. 2018, 99, 75–87. [Google Scholar] [CrossRef]
- McDade, T.W.; Hawkley, L.C.; Cacioppo, J.T. Psychosocial and behavioral predictors of inflammation in middle-aged and older adults: The Chicago health, aging, and social relations study. Psychosom. Med. 2006, 68, 376–381. [Google Scholar] [CrossRef]
- Holmes, L.M.; Marcelli, E.A. Neighborhoods and systemic inflammation: High CRP among legal and unauthorized Brazilian migrants. Health Place 2012, 18, 683–693. [Google Scholar] [CrossRef]
- Hadley, C.; Decaro, J.A. Testing Hypothesized Predictors of Immune Activation in Tanzanian Infants and Children: Community, Household, Caretaker, and Child Effects. Am. J. Hum. Biol. 2014, 26, 523–529. [Google Scholar] [CrossRef]
- Kuzawa, C.W.; Fried, R.L.; Borja, J.B.; McDade, T.W. Maternal pregnancy C-reactive protein predicts offspring birth size and body composition in metropolitan Cebu, Philippines. J. Dev. Orig. Health Dis. 2017, 8, 674–681. [Google Scholar] [CrossRef]
- Swartz, J.R.; Prather, A.A.; Hariri, A.R. Threat-related amygdala activity is associated with peripheral CRP concentrations in men but not women. Psychoneuroendocrinology 2017, 78, 93–96. [Google Scholar] [CrossRef]
- Goetz, S.M.; Lucas, T. C-reactive protein in saliva and dried blood spot as markers of stress reactivity in healthy African–Americans. Biomark. Med. 2020, 14, 371–380. [Google Scholar] [CrossRef]
- Fokkema, M.R.; Bakker, A.J.; de Boer, F.; Kooistra, J.; de Vries, S.; Wolthuis, A. HbA1c measurements from dried blood spots: Validation and patient satisfaction. Clin. Chem. Lab. Med. 2009, 47, 1259–1264. [Google Scholar] [CrossRef]
- Lacher, D.A.; Berman, L.E.; Chen, T.C.; Porter, K.S. Comparison of dried blood spot to venous methods for hemoglobin A1c, glucose, total cholesterol, high-density lipoprotein cholesterol, and C-reactive protein. Clin. Chim. Acta 2013, 422, 54–58. [Google Scholar] [CrossRef]
- Condorelli, F.; Scalia, G.; Stivala, A.; Gallo, R.; Marino, A.; Battaglini, C.M.; Castro, A. Detection of immunoglobulin-g to measles-virus, rubella-virus, and mumps-virus in serum samples and in microquantities of whole-blood dried on filter-paper. J. Virol. Methods 1994, 49, 25–36. [Google Scholar] [CrossRef]
- Parker, S.P.; Cubitt, W.D.; Ades, A.E. A method for the detection and confirmation of antibodies to hepatitis C virus in dried blood spots. J. Virol. Methods 1997, 68, 199–205. [Google Scholar] [CrossRef]
- Fachiroh, J.; Prasetyanti, P.R.; Paramita, D.K.; Prasetyawati, A.T.; Anggrahini, D.W.; Haryana, S.M.; Middeldorp, J.M. Dried-blood sampling for Epstein-Barr virus immunoglobulin G (IgG) and IgA serology in nasopharyngeal carcinoma screening. J. Clin. Microbiol. 2008, 46, 1374–1380. [Google Scholar] [CrossRef]
- Reddy, S.; May, W.; Mueller, P.; Hewett, J. Development of a dried blood spot method for retinol-binding protein (RBP): A potential indicator for vitamin A status. FASEB J. 1996, 10, 4698. [Google Scholar]
- Fujita, M.; Brindle, E.; Shofer, J.; Ndemwa, P.; Kombe, Y.; Shell-Duncan, B.; O’Connor, K.A. Retinol-binding protein stability in dried blood spots. Clin. Chem. 2007, 53, 1972–1975. [Google Scholar] [CrossRef]
- Engle-Stone, R.; Haskell, M.J.; Ndjebayi, A.O.; Nankap, M.; Erkardt, J.G.; Gimou, M.M.; Brown, K.H. Plasma Retinol-Binding Protein Predicts Plasma Retinol Concentration in Both Infected and Uninfected Cameroonian Women and Children. J. Nutr. 2011, 141, 2233–2241. [Google Scholar] [CrossRef]
- Edwards, R.L.; Griffiths, P.; Bunch, J.; Cooper, H.J. Top-Down Proteomics and Direct Surface Sampling of Neonatal Dried Blood Spots: Diagnosis of Unknown Hemoglobin Variants. J. Am. Soc. Mass Spectrom. 2012, 23, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Graca, D.C.; Hartmer, R.; Jabs, W.; Beris, P.; Clerici, L.; Stoermer, C.; Samii, K.; Hochstrasser, D.; Tsybin, Y.O.; Scherl, A.; et al. Identification of hemoglobin variants by top-down mass spectrometry using selected diagnostic product ions. Anal. Bioanal. Chem. 2015, 407, 2837–2845. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-J.C.; Ip, S.W. Age-Associated Methylation in Human Hemoglobin and Its Stability on Dried Blood Spots As Analyzed by Nanoflow Liquid Chromatography Tandem Mass Spectrometry. Chem. Res. Toxicol. 2018, 31, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Stolz, A.; Hedeland, Y.; Salzer, L.; Römer, J.; Heiene, R.; Leclercq, L.; Cottet, H.; Bergquist, J.; Neusüß, C. Capillary Zone Electrophoresis-Top-Down Tandem Mass Spectrometry for In-Depth Characterization of Hemoglobin Proteoforms in Clinical and Veterinary Samples. Anal. Chem. 2020, 92, 10531–10539. [Google Scholar] [CrossRef]
- Rosting, C.; Sae, C.O.; Gjelstad, A.; Halvorsen, T.G. Evaluation of water-soluble DBS for small proteins: A conceptual study using insulin as a model analyte. Bioanalysis 2016, 8, 1051–1065. [Google Scholar] [CrossRef]
- Thomas, A.; Thevis, M. Analysis of insulin and insulin analogs from dried blood spots by means of liquid chromatography–high resolution mass spectrometry. Drug Test. Anal. 2018, 10, 1761–1768. [Google Scholar] [CrossRef]
- Rosting, C.; Gjelstad, A.; Halvorsen, T.G. Water-Soluble Dried Blood Spot in Protein Analysis: A Proof-of-Concept Study. Anal. Chem. 2015, 87, 7918–7924. [Google Scholar] [CrossRef]
- Rosting, C.; Tran, E.V.; Gjelstad, A.; Halvorsen, T.G. Determination of the low-abundant protein biomarker hCG from dried matrix spots using immunocapture and nano liquid chromatography mass spectrometry. J. Chromatogr. B 2018, 1077, 44–51. [Google Scholar] [CrossRef]
- Skjærvø, Ø.; Halvorsen, T.G.; Reubsaet, L. Smart blood spots for whole blood protein analysis. Analyst 2018, 143, 3184–3190. [Google Scholar] [CrossRef]
- Kotel’nikova, N.E.; Mikhailova, S.A.; Vlasova, E.N. Immobilization of proteolytic enzymes trypsin and α-chymotrypsin to cellulose matrix. Russ. J. Appl. Chem. 2007, 80, 322–329. [Google Scholar] [CrossRef]
- Chen, S.; Wan, Q.; Badu-Tawiah, A.K. Mass Spectrometry for Paper-Based Immunoassays: Toward On-Demand Diagnosis. J. Am. Chem. Soc. 2016, 138, 6356–6359. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, J.Y. Enzyme immobilization on cellulose matrixes. J. Bioact. Compat. Polym. 2016, 31, 553–567. [Google Scholar] [CrossRef]
- Kim, U.J.; Lee, Y.R.; Kang, T.H.; Choi, J.W.; Kimura, S.; Wada, M. Protein adsorption of dialdehyde cellulose-crosslinked chitosan with high amino group contents. Carbohydr. Polym. 2017, 163, 34–42. [Google Scholar] [CrossRef]
- Hong, W.; Jeong, S.-G.; Shim, G.; Kim, D.Y.; Pack, S.P.; Lee, C.-S. Improvement in the Reproducibility of a Paper-based Analytical Device (PAD) Using Stable Covalent Binding between Proteins and Cellulose Paper. Biotechnol. Bioprocess Eng. 2018, 23, 686–692. [Google Scholar] [CrossRef]
- Skjærvø, Ø.; Rosting, C.; Halvorsen, T.G.; Reubsaet, L. Instant on-paper protein digestion during blood spot sampling. Analyst 2017, 142, 3837–3847. [Google Scholar] [CrossRef]
- Skjærvø, Ø.; Halvorsen, T.G.; Reubsaet, L. All-in-one paper-based sampling chip for targeted protein analysis. Anal. Chim. Acta 2019, 1089, 56–65. [Google Scholar] [CrossRef]
- Skjærvø, Ø.; Solbakk, E.J.; Halvorsen, T.G.; Reubsaet, L. Paper-based immunocapture for targeted protein analysis. Talanta 2019, 195, 764–770. [Google Scholar] [CrossRef]
- Skjærvø, Ø.; Halvorsen, T.G.; Reubsaet, L. Pre-lab proteolysis for dried serum spots—A paper-based sampling concept targeting low abundant biomarkers. Anal. Methods 2020, 12, 97–103. [Google Scholar] [CrossRef]
- McCann, L.; Benavidez, T.E.; Holtsclaw, S.; Garcia, C.D. Addressing the distribution of proteins spotted on μPADs. Analyst 2017, 142, 3899–3905. [Google Scholar] [CrossRef]
- Ren, D.; Julka, S.; Inerowicz, H.D.; Regnier, F.E. Enrichment of Cysteine-Containing Peptides from Tryptic Digests Using a Quaternary Amine Tag. Anal. Chem. 2004, 76, 4522–4530. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).