
Hydrophilic Interaction Liquid Chromatography to Characterize Nutraceuticals and Food Supplements Based on Flavanols and Related Compounds




Abstract

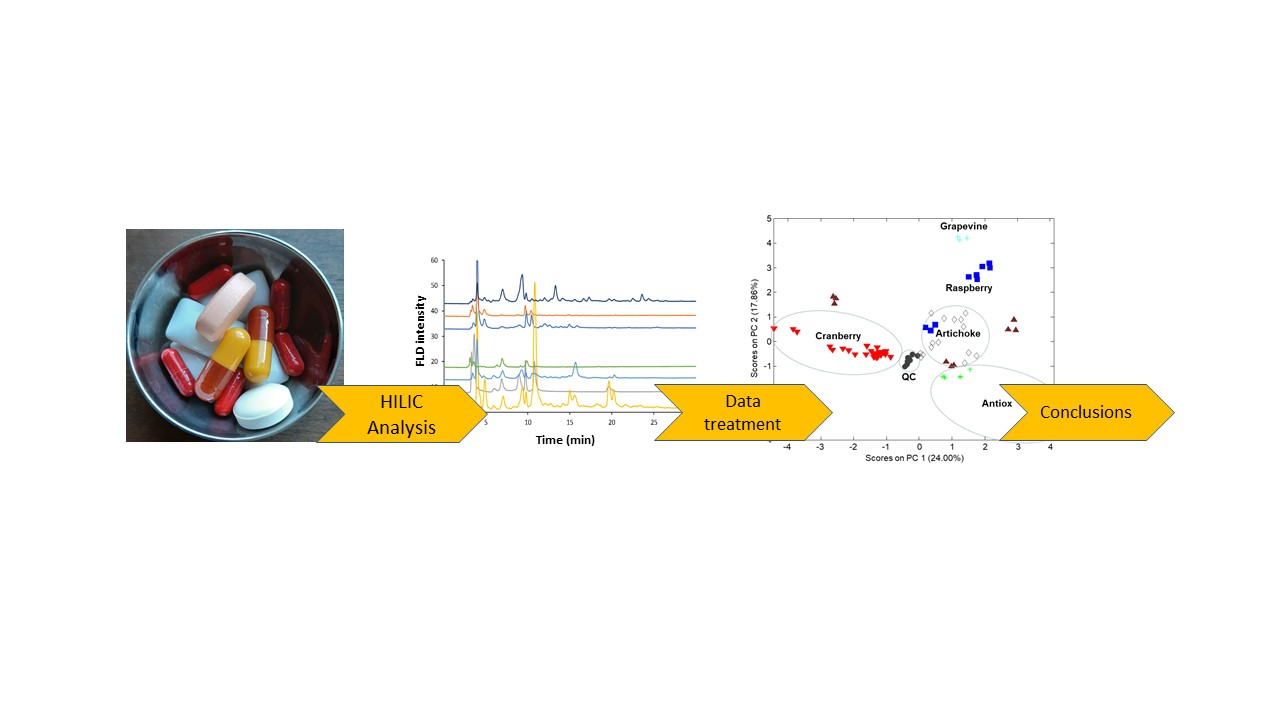{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Solutions
2.2. Samples
2.3. Instruments and Lab Equipment
2.4. Sample Treatment
2.5. Chromatographic Method
2.6. Data Analysis
3. Results and Discussion
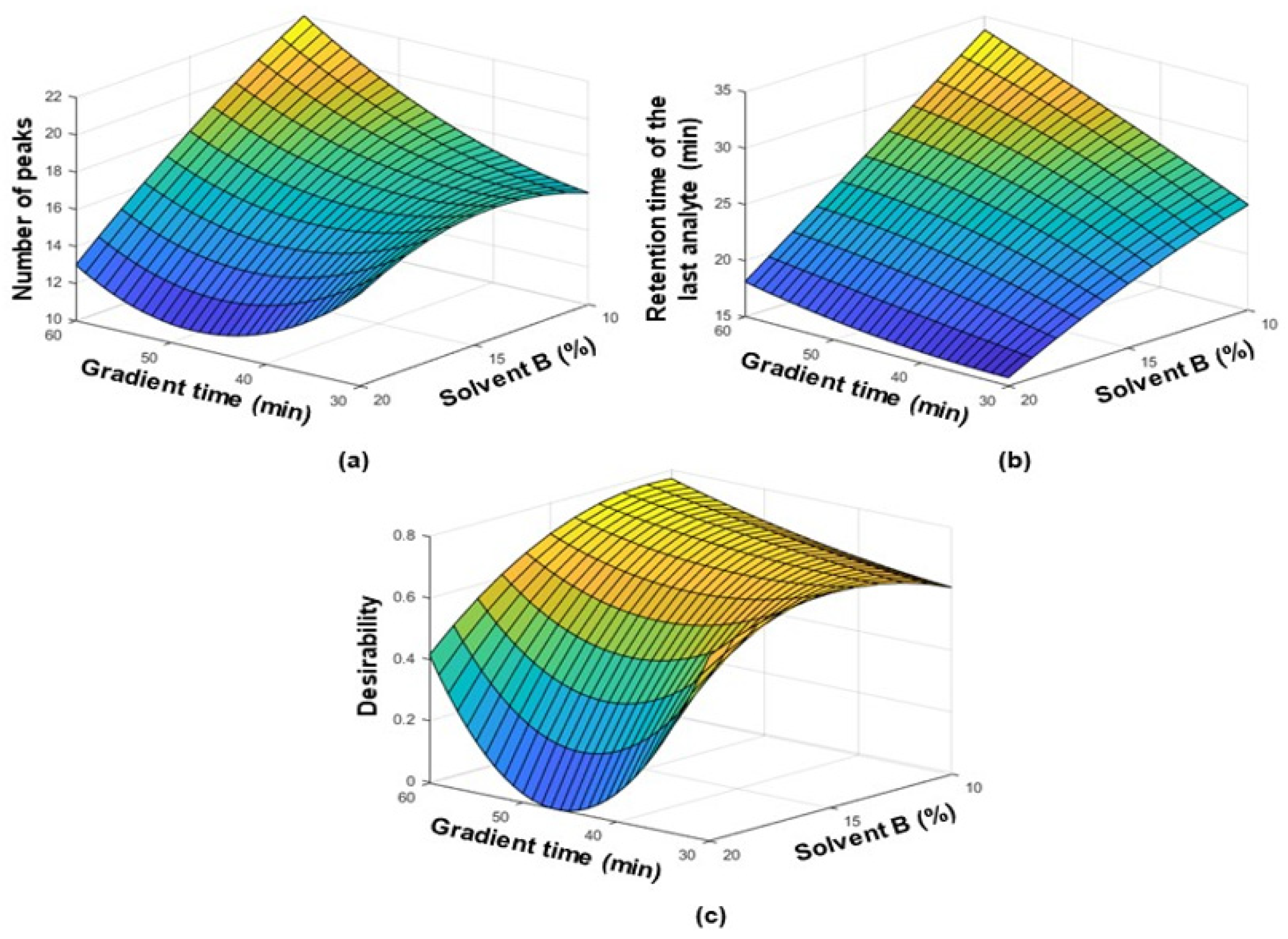
3.1. Optimization of the Separation Conditions
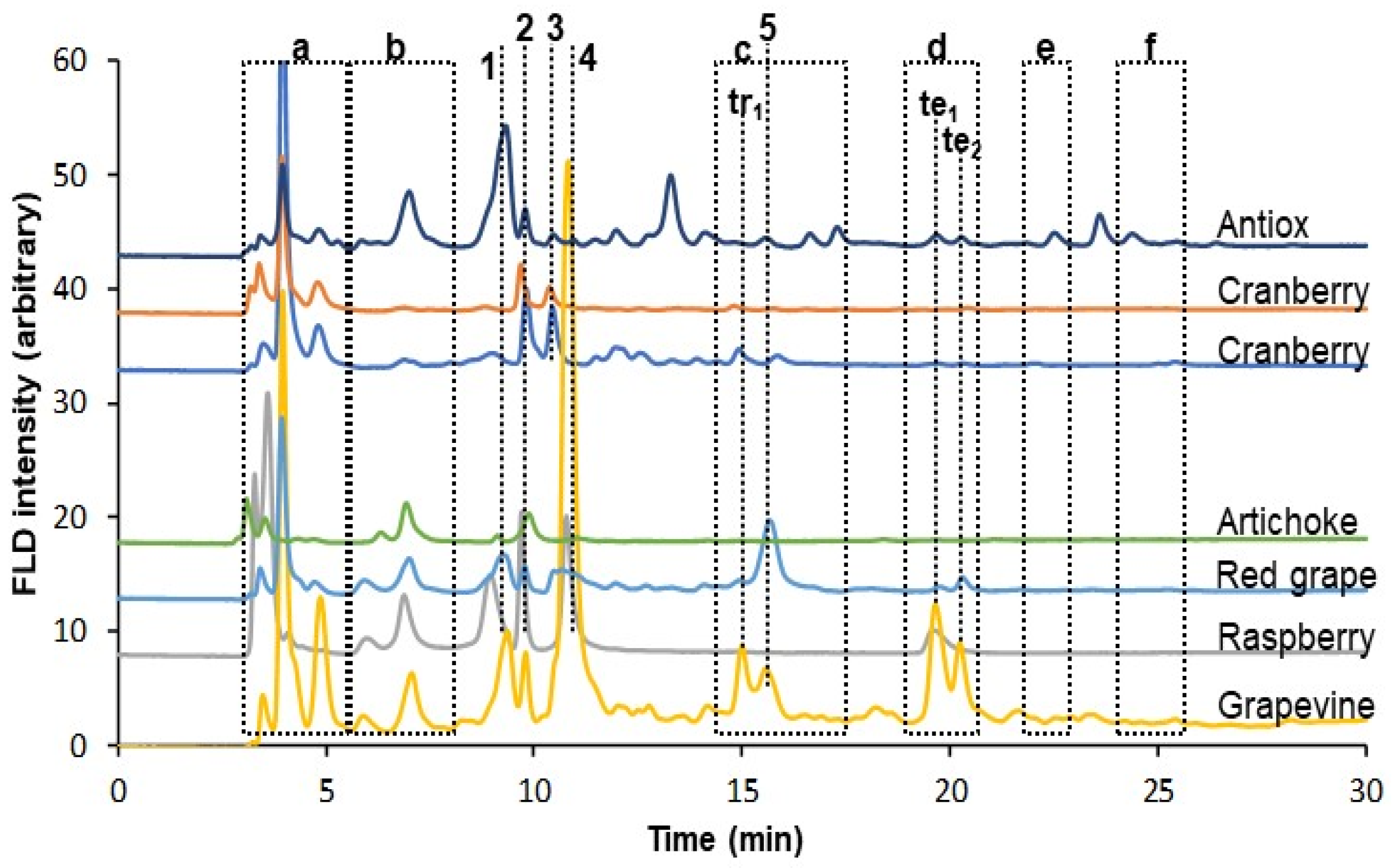
3.2. Analysis of the Sample Extracts
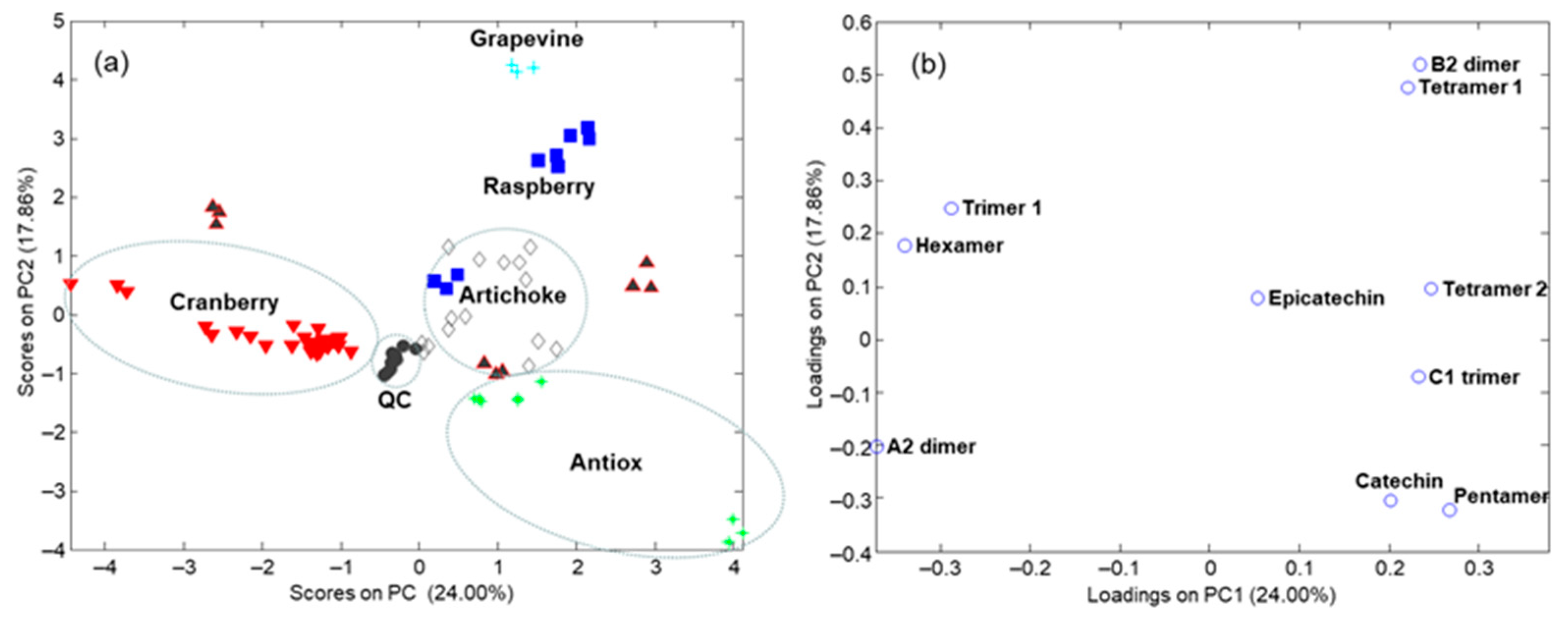
3.3. Sample Characterization by Principal Component Analysis and Related Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, A.Y.; Chen, Y.C. A Review of the Dietary Flavonoid, Kaempferol on Human Health and Cancer Chemoprevention. Food Chem. 2013, 138, 2099–2107. [Google Scholar] [CrossRef]
- Shahidi, F.; Ambigaipalan, P. Phenolics and Polyphenolics in Foods, Beverages and Spices: Antioxidant Activity and Health Effects—A Review. J. Funct. Foods 2015, 18, 820–897. [Google Scholar] [CrossRef]
- Prior, R.L.; Gu, L. Occurrence and Biological Significance of Proanthocyanidins in the American Diet. Phytochemistry 2005, 6, 2264–2280. [Google Scholar] [CrossRef]
- Caillet, S.; Côté, J.; Sylvain, J.F.; Lacroix, M. Antimicrobial Effects of Fractions from Cranberry Products on the Growth of Seven Pathogenic Bacteria. Food Control 2012, 23, 419–428. [Google Scholar] [CrossRef]
- Gramza-Michałowska, A.; Sidor, A.; Kulczyński, B. Berries as a Potential Anti-Influenza Factor—A Review. J. Funct. Foods 2017, 37, 116–137. [Google Scholar] [CrossRef]
- Vidal-Casanella, O.; Nuñez, O.; Hernández-Cassou, S.; Saurina, J. Assessment of Experimental Factors Affecting the Sensitivity and Selectivity of the Spectrophotometric Estimation of Proanthocyanidins in Foods and Nutraceuticals. Food Anal. Methods 2020. [Google Scholar] [CrossRef]
- Sertić, M.; Mornar, A.; Nigović, B. A Rapid Profiling of Hypolipidemic Agents in Dietary Supplements by Direct Injection Tandem Mass Spectrometry. J. Food Compos. Anal. 2014, 34, 68–74. [Google Scholar] [CrossRef]
- Sá, R.R.; Matos, R.A.; Silva, V.C.; da Cruz Caldas, J.; da Silva Sauthier, M.C.; dos Santos, W.N.L.; Magalhães, H.I.F.; de Freitas Santos Júnior, A. Determination of Bioactive Phenolics in Herbal Medicines Containing Cynara Scolymus, Maytenus Ilicifolia Mart Ex Reiss and Ptychopetalum Uncinatum by HPLC-DAD. Microchem. J. 2017, 135, 10–15. [Google Scholar] [CrossRef]
- Lucci, P.; Saurina, J.; Núñez, O. Trends in LC-MS and LC-HRMS Analysis and Characterization of Polyphenols in Food. TrAC Trends Anal. Chem. 2017, 88, 1–24. [Google Scholar] [CrossRef]
- Andersen, O.M.; Markham, K.R. Flavonoids: Chemistry, Biochemistry and Application, 1st ed.; Taylor & Francis: Boca Raton, FL, USA, 2006; pp. 553–617. [Google Scholar]
- Vasileiou, I.; Katsargyris, A.; Theocharis, S.; Giaginis, C. Current Clinical Status on the Preventive Effects of Cranberry Consumption against Urinary Tract Infections. Nutr. Res. 2013, 33, 595–607. [Google Scholar] [CrossRef]
- Hammerstone, J.F.; Lazarus, S.A.; Mitchell, A.E.; Rucker, R.; Schmitz, H.H. Identification of Procyanidins in Cocoa (Theobroma Cacao) and Chocolate Using High-Performance Liquid Chromatography/Mass Spectrometry. J. Agric. Food Chem. 1999, 47, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Prior, R.L.; Lazarus, S.A.; Cao, G.; Muccitelli, H.; Hammerstone, J.F. Identification of Procyanidins and Anthocyanins in Blueberries and Cranberries (Vaccinium Spp.) Using High-Performance Liquid Chromatography/Mass Spectrometry. J. Agric. Food Chem. 2001, 49, 1270–1276. [Google Scholar] [CrossRef]
- Saurina, J.; Sentellas, S. Liquid Chromatography Coupled to Mass Spectrometry for Metabolite Profiling in the Field of Drug Discovery. Expert Opin. Drug Discov. 2019, 14, 469–483. [Google Scholar] [CrossRef]
- Racine, K.C.; Lee, A.H.; Stewart, A.C.; Blakeslee, K.W.; Neilson, A.P. Development of a Rapid Ultra Performance Hydrophilic Interaction Liquid Chromatography Tandem Mass Spectrometry Method for Procyanidins with Enhanced Ionization Efficiency. J. Chromatogr. A 2019, 1594, 54–64. [Google Scholar] [CrossRef]
- Yang, W.; Ma, X.; Laaksonen, O.; He, W.; Kallio, H.; Yang, B. Effects of Latitude and Weather Conditions on Proanthocyanidins in Blackcurrant (Ribes Nigrum) of Finnish Commercial Cultivars. J. Agric. Food Chem. 2019, 67, 14038–14047. [Google Scholar] [CrossRef]
- Yang, W.; Laaksonen, O.; Kallio, H.; Yang, B. Effects of Latitude and Weather Conditions on Proanthocyanidins in Berries of Finnish Wild and Cultivated Sea Buckthorn (Hippophaë rhamnoides L. Ssp. Rhamnoides). Food Chem. 2017, 216, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Hollands, W.J.; Voorspoels, S.; Jacobs, G.; Aaby, K.; Meisland, A.; Garcia-Villalba, R.; Tomas-Barberan, F.; Piskula, M.K.; Mawson, D.; Vovk, I.; et al. Development, Validation and Evaluation of an Analytical Method for the Determination of Monomeric and Oligomeric Procyanidins in Apple Extracts. J. Chromatogr. A 2017, 1495, 46–56. [Google Scholar] [CrossRef]
- Sommella, E.; Pepe, G.; Pagano, F.; Ostacolo, C.; Tenore, G.C.; Russo, M.T.; Novellino, E.; Manfra, M.; Campiglia, P. Detailed Polyphenolic Profiling of Annurca Apple (M. pumila Miller Cv Annurca) by a Combination of RP-UHPLC and HILIC, Both Hyphenated to IT-TOF Mass Spectrometry. Food Res. Int. 2015, 76, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Kosińska-Cagnazzo, A.; Kerr, W.L.; Amarowicz, R.; Swanson, R.B.; Pegg, R.B. Separation and Characterization of Phenolic Compounds from Dry-Blanched Peanut Skins by Liquid Chromatography-Electrospray Ionization Mass Spectrometry. J. Chromatogr. A 2014, 1356, 64–81. [Google Scholar] [CrossRef] [PubMed]
- Bakhytkyzy, I.; Nuñez, O.; Saurina, J. Determination of Flavanols by Liquid Chromatography with Fluorescence Detection. Application to the Characterization of Cranberry-Based Pharmaceuticals through Profiling and Fingerprinting Approaches. J. Pharm. Biomed. Anal. 2018, 156, 206–213. [Google Scholar] [CrossRef]
- Bakhytkyzy, I.; Nuñez, O.; Saurina, J. Size Exclusion Coupled to Reversed Phase Liquid Chromatography for the Characterization of Cranberry Products. Food Anal. Methods 2019, 12, 604–611. [Google Scholar] [CrossRef]
- Gu, L.; Kelm, M.; Hammerstone, J.F.; Beecher, G.; Cunningham, D.; Vannozzi, S.; Prior, R.L. Fractionation of Polymeric Procyanidins from Lowbush Blueberry and Quantification of Procyanidins in Selected Foods with an Optimized Normal-Phase HPLC-MS Fluorescent Detection Method. J. Agric. Food Chem. 2002, 50, 4852–4860. [Google Scholar] [CrossRef] [PubMed]
- Navarro, M.; Nunez, O.; Saurina, J.; Hernandez-Cassou, S.; Puignou, L. Characterization of Fruit Products by Capillary Zone Electrophoresis and Liquid Chromatography Using the Compositional Profiles of Polyphenols: Application to Authentication of Natural Extracts. J. Agric. Food Chem. 2014, 62, 1038–1046. [Google Scholar] [CrossRef]
- Barbosa, S.; Pardo-Mates, N.; Hidalgo-Serrano, M.; Saurina, J.; Puignou, L.; Nunez, O. Detection and Quantitation of Frauds in the Authentication of Cranberry-Based Extracts by UHPLC-HRMS (Orbitrap) Polyphenolic Profiling and Multivariate Calibration Methods. J. Agric. Food Chem. 2018, 66, 9353–9365. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vidal-Casanella, O.; Arias-Alpizar, K.; Nuñez, O.; Saurina, J. Hydrophilic Interaction Liquid Chromatography to Characterize Nutraceuticals and Food Supplements Based on Flavanols and Related Compounds. Separations 2021, 8, 17. https://doi.org/10.3390/separations8020017
Vidal-Casanella O, Arias-Alpizar K, Nuñez O, Saurina J. Hydrophilic Interaction Liquid Chromatography to Characterize Nutraceuticals and Food Supplements Based on Flavanols and Related Compounds. Separations. 2021; 8(2):17. https://doi.org/10.3390/separations8020017
Chicago/Turabian StyleVidal-Casanella, Oscar, Kevin Arias-Alpizar, Oscar Nuñez, and Javier Saurina. 2021. "Hydrophilic Interaction Liquid Chromatography to Characterize Nutraceuticals and Food Supplements Based on Flavanols and Related Compounds" Separations 8, no. 2: 17. https://doi.org/10.3390/separations8020017
APA StyleVidal-Casanella, O., Arias-Alpizar, K., Nuñez, O., & Saurina, J. (2021). Hydrophilic Interaction Liquid Chromatography to Characterize Nutraceuticals and Food Supplements Based on Flavanols and Related Compounds. Separations, 8(2), 17. https://doi.org/10.3390/separations8020017