Abstract
This paper presents a novel miniaturized analytical method for the determination of carminic acid, a natural red food dye, in complex food and pharmaceutical matrices by microchip electrophoresis (MCE) with photometric detection. MCE has become a very attractive microscale separation technique because it offers high-speed, high-throughput, small sample injection volume and low reagents consumption. Fast determination of carminic acid in less than 5 min was achieved on a poly(methyl methacrylate) microchip in anionic separation mode at pH 6. Photometric detector based on light-emitting diode technology was set to a wavelength of 490 nm. Using a sample injection volume of 900 nL, a limit of detection of 69 nmol L−1 was achieved. A wide linear dynamic range over four orders of magnitude (from nmol L−1 to mmol L−1) was observed for peak area. Developed method provided favorable intra- and inter-day repeatability of the migration time (up to 2.5% RSD), as well as the repeatability of the peak area (less than 1.9% RSD), regardless of the sample type. The content of carminic acid was determined in various foodstuffs and pharmaceuticals, such as candies, saffron, non-alcoholic drink, and sore throat lozenges with good recoveries (92.5–104.0%).
1. Introduction
One of the most important organoleptic properties of foodstuff is its appearance, e.g., size, shape, structure or color, which significantly affects consumer choice and thus the success of the product on the market. Various natural and synthetic dyes are added to food as well as pharmaceutical products to: (i) compensate for a loss of natural color by different environmental factors (sun, air, moisture or temperature); (ii) enhance the natural color; (iii) mask unpleasant color of the product; or (iv) increase attractiveness of the product for consumers [1,2]. On the other hand, an intensive use of dyes in food and pharmaceutical industry can have an adverse effect on the human body, and therefore their monitoring and strict control in a variety of products is very important. Dyes authorized for use in foodstuffs in EU are regulated according to Regulation (EC) No. 1333/2008 [3]. Based on Directive 2009/35/EC [4], dyes approved for use in foodstuff can also be used in medicinal products. Over time, certain synthetic food dyes have been banned due to their toxicity or even carcinogenic effects after short-term or long-term ingestion [1,5]. Nowadays, synthetically prepared dyes are being replaced by their natural alternatives, which present safer choice and whose health risks are lower [5,6].
Carminic acid (CA; E120; C.I. 75470), a β-d-glucopyranosyl derivative of anthraquinone, is a natural red dye extracted from the female Dactylopius coccus Costa insect. The color of CA solutions is known to be highly pH dependent: at acidic pH it is yellow to orange and at alkaline pH it turns into violet [7]. This natural dye is widely used in food, pharmaceutical, cosmetic and textile industry, due to its natural character and stability [6]. CA can also be present in consumer products in the form of aluminum chelates, carmines. The content of CA in commercial products has to be strictly controlled because of the potential health problems caused by ingestion of CA, e.g., anaphylactic shock, rhinorrhea, angioedema, urticaria and dyspnea [8]. The European Food Safety Authority (EFSA) has established an acceptable daily intake of 2.5 mg/kg of body weight/day for CA [9]. In addition to health problems, the presence of CA in foodstuffs should be monitored because of, e.g., religious beliefs, dietary trends or environmental considerations.
Several analytical methods for the determination of CA in complex food matrices have been reported in the literature [10,11]. The official method for determination of the CA, proposed by Joint FAO/WHO Expert Committee on Food Additives, is based on spectrophotometric measurement of sample in dilute hydrochloric acid at 494 nm [9,12]. Spectrophotometry [13], stripping voltammetry [14] and differential pulse polarography [15] have been applied successfully to the determination of CA or carmines in various foodstuffs. However, these methods are not suitable for the determination of structurally similar dyes, and therefore possible interferences present in food products have to be strictly controlled. These disadvantages can be solved by including a separation step into the analytical procedure.
The most commonly used separation technique for the determination of CA is high performance liquid chromatography (HPLC) coupled with ultraviolet–visible (UV–Vis) spectrometric [16,17,18,19], fluorescence [19] and mass spectrometry detection [20]. HPLC offers many advantages such as simultaneous determination of several analytes in single run, minimal requirements for sample pretreatment, and limit of detection (LOD) for CA or carmines mostly below 5 µmol L−1 (Table S1). However, relatively high environmental impact due to large quantities of toxic waste as well as running costs make HPLC less attractive from the green chemistry standpoint.
Due to ionogenic character of the CA, capillary electrophoresis (CE) is a good alternative to the above-mentioned techniques for the determination of CA even in complex matrices. As for HPLC, UV–Vis is the most widely used detection technique for the CE determination of CA [21,22,23,24,25] (Table S1). Even though most of the CE separations were carried out in alkaline borate buffers [21,22,23,24], acidic phosphate buffers were also used [24,25]. The LOD achieved for CA using CE methods was in the range 0.05–4.3 µmol L−1 depending on the sample pretreatment, mostly solid phase extraction, and/or preconcentration method used (Table S1).
In the field of simplification and automation of the analytical process, miniaturization is commonly denoted as the fastest growing field in terms of new analytical equipment development. For example, a lab-on-a-chip technology integrates several analytical steps, such as sample pretreatment, separation and detection, on a miniaturized device, microchip, and this enables significant reduction of the sample volume and working solutions. Microchip electrophoresis (MCE) is based on the same separation principles as CE. When compared to, e.g., liquid chromatography, it offers advantages similar to other electrodriven separation techniques, such as reduced time and cost of analysis, instrumental simplicity, high separation efficiency and low waste production, which meet the concept of green analytical chemistry [26,27]. All these benefits make MCE an attractive miniaturized analytical technique, as well as an alternative technique for the determination of CA in complex samples.
The aim of this work was to develop a novel miniaturized analytical method, requiring only minimal sample pretreatment, for the determination of CA in complex matrices. Selectivity of the method was attained by on-line combination of MCE with photometric detection (PD) at specific wavelength of 490 nm and demonstrated by the analysis of food and pharmaceutical samples. To the best of our knowledge, this is the first report of the application of MCE for the determination of CA in complex samples.
2. Materials and Methods
2.1. Chemicals and Solutions
Analytical grade chemicals used to prepare background electrolytes (BGEs) and model samples were purchased from Merck-Millipore (Darmstadt, Germany) and Sigma-Aldrich (Steinheim, Germany). Methylhydroxyethylcellulose 30,000 (MHEC; Serva, Heidelberg, Germany) was added to the BGE solutions and acted as an electroosmotic flow suppressor. The stock solution of CA (Sigma-Aldrich) was prepared at 1 mmol L−1 concentration. Water purified by a Pro-PS system (Labconco, Kansas City, KS, USA) and then deionized by Simplicity deionization unit (Millipore, Molsheim, France) was used to prepare sample and BGE solutions (resistivity of 18.2 MΩ cm at 25 °C). The sample and BGE solutions were stored at 4 °C and used for a maximum of one week. The BGE solutions were filtered prior to the use through a 0.8-µm membrane filter (Merck-Millipore) and degassed in an ultrasonic bath K5 (Ecotest, Topoľčany, Slovakia) for 5 min.
The optimal BGE used for MCE-PD separations was found to be a solution composed of 15 mmol L−1 2-(N-morpholino)ethanesulfonic acid (MES), 15 mmol L−1 l-histidine (HIS) and 0.1% (w/v) MHEC at pH 6.0 (see Section 3.1).
2.2. Food and Pharmaceutical Samples
The analyzed samples were purchased at local supermarket and pharmacy. Samples of lozenges, soft candy, hard candy and medicinal lollipops were analyzed after simple pretreatment, which included dissolution of three individual pieces in 30, 30, 45 and 120 mL of ultrapure water, respectively, followed by filtration through a 0.45-µm glass fiber filter (Merck-Millipore). One gram of saffron was ground. The powder was suspended in 100 mL of ultrapure water, sonicated in the ultrasonic bath (K5) and filtered through a 0.45-µm glass fiber filter. Ten milliliters of nonalcoholic carbonated drink (radler) were degassed in the ultrasonic bath (K5). The pH of prepared samples was adjusted to the pH of BGE. Prior to their injection on the microchip, the sample solutions were appropriately diluted and homogenized using vortex REAX 2000 (Heidolph Instruments, Schwabach, Germany) for 1 min.
2.3. MCE-PD Instrumentation
MCE-PD equipment consisted of microchip, electrolyte and electronic control units as well as photometric detector (Figure 1). A poly(methyl methacrylate) microchip (IonChip™ 3.0, Merck) with coupled channels was used for the MCE analyses. Photometric detector K-2001 (Knauer, Berlin, Germany) was modified and equipped with light-emitting diode L490-01 (Muevo-Vertrieb, Wipperfürth, Germany) working at a 490 nm wavelength. Photometric detector was coupled to the microchip by two optical fibers M22L01 (400 µm core diameter; ThorLabs, Newton, NJ, USA) facing each other, in the wider part of the first separation channel (C1) of the microchip in order to increase detection sensitivity (Figure 1). Channels C1–C3 were filled with BGE while channel CS was filled with sample, as shown in Figure 1. The inlets of the microchip channels were connected to the electrolyte unit via polyether ether ketone capillaries of 500 µm inner diameter (i.d.) (IDEX Health & Science, Wertheim, Germany).
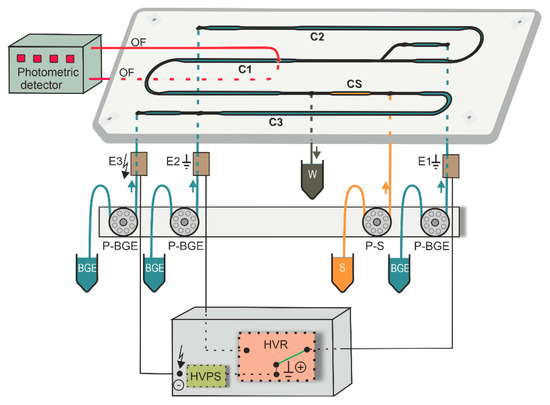
Figure 1.
A schematic of MCE-PD instrument. OF, optical fibers (red lines); C1, the first separation channel (4.5 µL, 59 × 0.2–0.5 × 0.14–0.2 mm (length × width × depth)), C2, the second separation channel (4.3 µL, 56 × 0.2–0.5 × 0.14–0.2 mm); C3, auxiliary channel (9.8 µL, 60 × 0.2–0.5 × 0.2–0.38 mm); CS, sample (S) channel (0.9 µL, 12 × 0.2–0.5 × 0.2 mm); W, permanently opened outlet to waste container; E1 and E2, membrane driving electrodes for C1 and C2, respectively; E3, membrane driving electrode connected to high voltage power supply (HVPS); P-BGE and P-S, peristaltic micropumps for filling corresponding channels with BGE or S solutions, respectively; HVR, high voltage relay.
The electrolyte control unit consisted of four peristaltic micropumps (P-BGE and P-S) and membrane driving electrodes (E1–E3, Figure 1), which were interconnected via fluorinated ethylene propylene capillaries of 500 µm i.d. (IDEX Health & Science). The primary function of the peristaltic micropumps was to transport BGE and sample solutions to the corresponding microchip channels. The other function of the micropumps was to suppress hydrodynamic flow in the microchip channels by closing its inlets. Membrane driving electrodes were used to eliminate interference caused by bubbles generated during the separation process [28].
The electronic control unit consisted of high voltage power supply (HVPS) and high voltage relay (HVR). HVPS which delivers driving current to the microchip, was used in voltage-stabilized mode working in the range 0–7 kV; a maximum applied current was 50 µA. The function of HVR was to switch direction of the driving current in the channel during the MCE analysis. To achieve fast analyses, the separation was performed only in the channel C1, when driving current was flowing between driving electrodes E3 and E1.
MicroITP software (Merck) was used for automatic filling of individual microchip channels and time-programmed control of MCE runs. The software was also used to process data acquired during MCE analysis. OriginPro 9.0 (OriginLab, Northampton, MA, USA) software was used to evaluate the electropherograms and perform necessary calculations.
2.4. Microchip Maintenance
Before the first run of the day, microchip channels were rinsed with ultrapure water for 5 min. Between the runs which utilized the same BGE and the same sample solutions, microchip channels were rinsed only with BGE. Prior to a replacement of BGE and/or sample solutions, channels were rinsed with ultrapure water for 5 min to remove residues, and then microchip channels were flushed with next BGE and/or sample solutions for 2 min.
MCE analysis was initiated by filling the microchip channels with BGE and sample solutions in the order: C2, C1, C3 and CS (Figure 1). Only one channel was filled at the time to prevent mixing of individual solutions. Excesses of solutions were removed from the channels via permanently opened outlet to a waste container. The whole filling procedure lasted 3 min.
Clean-up process of the microchip was carried out after the last run of the day, and consisted of 5 min rinsing with 2% (v/v) aqueous solution of neutral detergent (Extran MA02, Merck-Millipore) and then with ultrapure water for 5 min. The microchip channels were filled with ultrapure water when not in use.
3. Results and Discussion
3.1. Separation Conditions
Optimization of MCE separation conditions started with selection of the BGE in terms of pH, ionic strength and buffer capacity. Since CA is a weak acid with three experimentally confirmed pKa values (pKa1 = 2.81; pKa2 = 5.43; pKa3 = 8.10 [29]; Figure S1), its mobility highly depends on the pH of the BGE. The second criterion applied for the selection of the pH of BGE was to maximize the signal corresponding to the individual ionic forms of CA at selected detection wavelength of 490 nm. As shown in the literature [7], the absorption maxima of CA shifts from 490 nm at acidic pH to 567 nm at alkaline pH. Based on these observations, a pH range of the BGEs from 5 to 9 was inspected.
Electropherograms in Figure 2 show different peak profiles and intensities depending on the composition of the BGE and the pH. At pH 5.0, the peak corresponding to CA is relatively broad and asymmetric (Figure 2A). With increasing pH of the BGE, the migration time is shortened, and the peak of CA becomes narrower. The most symmetrical peak was observed at pH 6.0 (Figure 2B). At this pH, the carboxylic acid group is deprotonated and the second most acidic hydrogen in CA molecule, coming from the hydroxyl group, is dissociated to the extent of 79% (Figure S1). The fastest migration of CA was achieved in the BGE of pH 9 (Figure 2F), however the peak intensity of CA was the lowest. The decrease of peak height is probably due to the change in the absorption maximum of CA, i.e., it shifted to higher value (528 nm [7]). In general, the disadvantage of using alkaline BGE is absorption of carbon dioxide from air moisture in form of carbonates. This phenomenon makes it more difficult to reproducibly prepare BGE and it adversely affects separation process leading to worse reproducibility of migration time. Therefore, the BGE at pH 6.0 was chosen as optimal because of narrow and symmetric peak of CA migrating under 5 min with adequate absorbance signal (Figure 2B).
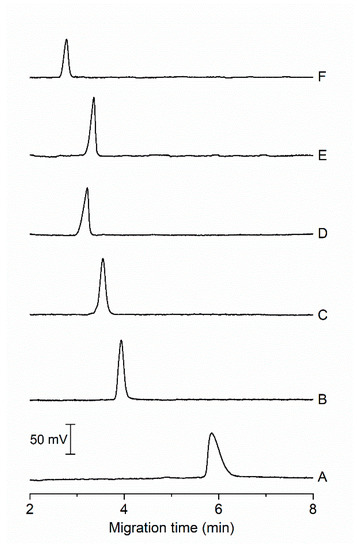
Figure 2.
Electropherograms from MCE-PD analysis of model samples under different pH of BGE. Composition of BGE: (A) 15 mmol L−1 propionic acid, 20 mmol L−1 creatinine, 0.1% (w/v) MHEC, pH 5.0; (B) 15 mmol L−1 MES, 15 mmol L−1 HIS, 0.1% (w/v) MHEC, pH 6.0; (C) 25 mmol L−1 MES, 35 mmol L−1 2,2-bis(hydroxymethyl)-2,2′,2″-nitrilotriethanol, 0.1% (w/v) MHEC, pH 6.5; (D) 25 mmol L−1 N-[tris-(hydroxymethyl)methyl]-2-aminoethanesulfonic acid, 35 mmol L−1 imidazole, 0.1% (w/v) MHEC, pH 7.5; (E) 25 mmol L−1 N-[tris(hydroxymethyl)methyl]-3-aminopropanesulfonic acid, 15 mmol L−1 2-amino-2-(hydroxymethyl)-1,3-propanediol, 0.1% (w/v) MHEC, pH 8.0; and (F) 25 mmol L−1 glycine, 30 mmol L−1 1,3-bis[tris(hydroxymethyl)methylamino]propane, 0.1% (w/v) MHEC, pH 9.0. Injected sample: 20 µmol L−1 CA. Separation voltage: 4 kV. Detection wavelength: 490 nm.
Based on the results obtained from the optimization of MCE separation, the optimal MCE-PD conditions for determination of CA were as follows: anionic MCE separation under voltage stabilized at 4 kV; using BGE at pH 6.0 consisting of 15 mmol L−1 MES, 15 mmol L−1 HIS and 0.1% (w/v) MHEC; and monitoring with PD at 490 nm. These optimal conditions were applied to the analysis of model samples as well as various food and pharmaceutical samples.
3.2. Method Performance Parameters
The performance of the MCE-PD method for the determination of the CA was verified. Analytical parameters of the MCE-PD method were evaluated under the optimal working and separation conditions as described above. The verification procedure included evaluation of linearity, sensitivity, precision and accuracy [30].
3.2.1. Linearity
Linearity of the method was investigated by analysis of four replicates of standard solutions of CA prepared at 15 concentration levels which covered the range from 200.0 nmol L−1 to 1.0 mmol L−1. The linear dynamic range (LDR) was evaluated for the peak height of CA as well as for its peak area. The peak areas were not normalized by the migration times. The LDR obtained for the peak height was from 200.0 nmol L−1 to 100.0 µmol L−1, expressed by correlation coefficient (R) of 0.999 (Figure S2a). The LDR for the peak area with R of 0.999 was from 200.0 nmol L−1 to 1.0 mmol L−1 (Figure S2b), i.e., over four orders of magnitude. From inspection of residuals (Figure S3), we concluded that distribution is random, and no trend is observed which indicates good linearity. In addition, the y-axis intercept is in the range 1.05–8.59% of the signal corresponding to the maximum permitted level (MPL; see Section 3.3), which indicates good linearity over the entire test range.
3.2.2. Limit of Detection and Limit of Quantitation
To assess the LOD, the calibration curve was created by plotting peak height of CA versus concentration of CA (Table 1). The LOD was calculated as [30]:
where σ is the standard deviation (SD) of the blank sample (noise) and S is the slope of the calibration curve constructed from the peak height of CA.

Table 1.
Parameters for calculation of detection and quantitation limits.
The LOD of 69 nmol L−1 for CA was obtained by the MCE-PD method (Table 1). In comparison to the conventional CE methods, coupled to UV–Vis detection and employing extraction sample pretreatment (Table S1), this method provides much better LOD [22,23,24,25]. The MCE-PD method has approximately the same sensitivity as CE-UV–Vis method with large volume sample stacking used as preconcentration technique [21]. Such favorable LOD was achieved mainly by: (1) injecting a relatively large volume of the sample onto the microchip (0.9 µL); (2) using a wider part of the channel with a 500 µm i.d. for detection; and (3) selecting a specific wavelength for detection of CA (490 nm).
The limit of quantitation (LOQ; Table 1) was calculated based on the same parameters as LOD:
3.2.3. Precision
Precision of the method was expressed as relative standard deviation (RSD) of the migration time and the peak area of CA [30]. The intra-day precision also termed as repeatability was assessed from four repeated measurements at three different concentration levels of CA corresponding to LOQ, 5 times LOQ and 25 times LOQ on the same microchip in one day. The RSD of the migration time of CA was less than 2.5%, while RSD of the peak area ranged from 1.1 to 1.9%. Inter-day precision was evaluated from 20 runs at three different concentration levels of CA performed in five days using freshly prepared BGE. The inter-day precision did not differ significantly from the intra-day precision (Table 2).
Table 2.
Intra-day and inter-day precision in model samples.
3.2.4. Accuracy
The accuracy was reported as percent recovery by determination of known added concentration of CA to the sample [30]. The recovery was calculated as:
where cspiked is the concentration of CA in the spiked sample, csample is the concentration of CA in the unspiked sample, and cadded corresponds to the concentration of added amount of CA.
For model samples, CA spiked into sample diluent, the accuracy was assessed from four repeated measurements of the sample at three different concentrations of CA corresponding to LOQ, 5 times LOQ and 25 times LOQ. The recovery data of CA in real samples were assessed from four repeated measurements of diluted samples spiked with two standard additions. The concentrations of standard additions were chosen so that the peak area of CA in the spiked sample is approximately within 50–150% of its peak area in the diluted sample. Since no signal was observed in the position of CA in the blank samples, correction of the peak area was not necessary. Mean recoveries ranged from 98.4% to 100.7% for model samples and from 92.5% to 104.0% for real samples, as presented in Table 3. The obtained recoveries in the real samples indicate minimal matrix effects as well as accurate determination of CA in food and pharmaceutical samples under employed working conditions.
Table 3.
Accuracy in model and real samples.
3.3. Analysis of Food and Pharmaceutical Samples
Prior to analysis of real samples, we verified the performance of the system by carrying out intra-day precision of migration time and peak area in real and spiked food and pharmaceutical samples. Each sample was injected four times and the results are summarized in Table 4. Furthermore, we verified the linear response of the system by constructing the calibration line at seven concentration levels of CA corresponding to 1–25 times LOQ (0.2–5.0 µmol L−1); each calibration standard was injected four times. The calibration range was chosen based on the preliminary experiments with diluted food and pharmaceutical samples, so that the signal of CA in the injected sample lies in the middle of the calibration curve to minimize the error in the concentration calculation. Calibration standards were injected in random order; three calibration standards were injected before the real samples and four calibration standards were injected after the real samples. Equation of regression line, created by plotting peak area of CA versus concentration of CA, was as follows: y = 0.697x − 0.001. Good linearity was obtained (R = 0.999) indicating that the system performance remained unchanged during the analysis of real samples. Concentrations of CA in the analyzed samples calculated using this equation are summarized in Table 5.
Table 4.
Intra-day precision in real and spiked samples.
Table 5.
A content of CA in food and pharmaceutical samples.
The RSD values of migration times for CA in real samples were less than 1.2%, i.e., similar to the model samples (compare data in Table 2 and Table 4). On the other hand, the migration times of CA varied for different sample types (Figure 3). A likely explanation of this is that the variability depends on the actual content of ionogenic components in the sample or ionic strength of the injected sample. As evident in Figure 3c, the migration time of CA was highest in the radler sample, which was only two times diluted and contained large amount of ionogenic components. Identification of CA in the samples was carried out by the method of standard addition (Figure 3). Intra-day precision of the peak area did not significantly differ to that achieved for the model samples (compare data in Table 2 and Table 4). This indicates that matrix had no effect on the precision.
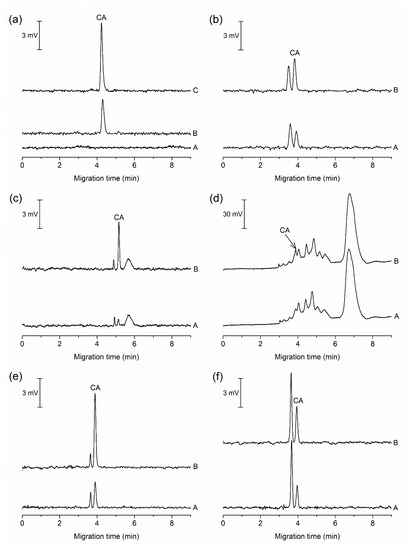
Figure 3.
Electropherograms from the MCE-PD analysis of food and pharmaceutical samples. (a) A, a blank sample (10% (v/v) BGE); B, five times diluted soft candy; C, the same as B with addition of 0.5 µmol L−1 CA; (b) A, 40 times diluted hard candy; B, the same as A with addition of 0.3 µmol L−1 CA; (c) A, two times diluted radler; B, the same as A with addition of 0.45 µmol L−1 CA; (d) A, two times saffron; B, the same as A with addition of 0.5 µmol L−1 CA; (e) A, 40 times diluted medicinal lollipops; B, the same as A with addition of 0.75 µmol L−1 CA; and (f) A, 2.5 times diluted lozenges; B, the same as A with addition of 0.3 µmol L−1 CA. For separation conditions, see Section 3.1.
The applicability of the MCE-PD method for the determination of CA was verified by the analysis of six food and pharmaceutical samples (Figure 3). In comparison to the previously published separation methods [17,18,19,20,21,23,24,25], the MCE-PD method required only minimal sample pretreatment, which included filtration or degassing. The procedure for sample preparation is described in Section 2.2. After simple pretreatment, the samples were analyzed for the content of CA.
Figure 3a illustrates a typical blank run acquired before the analysis of all samples. As evident from this electropherogram, no interfering constituent is observed in the migration position of CA in the blank sample. Electropherograms from the MCE-PD analyses of food and pharmaceutical samples in Figure 3 show different matrix components migrating close to CA. The complexity of individual electropherograms depends on the nature of the sample, i.e., for a natural product we observe a more complex electropherogram with larger number of comigrating ionogenic species. It is obvious that the most complex sample in terms of the number of components absorbing at 490 nm is Saffron (Figure 3d) due to the presence of crocins, natural carotenoids of saffron, which have maximum absorbance at about 440 nm [31]. Therefore, time-consuming sample pretreatment would be required for accurate determination of CA by UV–Vis spectrophotometry without the use of separation step. This shows the limitations of direct UV–Vis analysis of complex samples for determination of artificial or natural food dyes.
The content of CA in the analyzed samples determined using MCE-PD method is summarized in Table 5. The MPL of CA corresponding to individual group of products, as proposed by EFSA [9], are also listed in Table 5. There are no MPL values for pharmaceuticals; however, as these were in the form of lozenges and lollipops, we have taken values for hard candy into consideration. Even though the producers do not provide the contents of CA in any of the analyzed samples, it is evident that the content of CA falls below the MPL in all analyzed samples.
4. Conclusions
This study demonstrated that online coupling MCE with PD offers a fast, sensitive, accurate and reproducible procedure for the determination of CA in various foodstuffs and pharmaceuticals. The LOD value achieved for CA (69 nmol L−1) using MCE-PD method under the optimized conditions (large sample volume, specific detection wavelength and large optical path) compares favorably with those reported for HPLC-UV–Vis [16,17,18,19] and conventional CE-UV–Vis [21,22,23,24,25] combined with extraction and/or other preconcentration sample pretreatment steps. We demonstrated the applicability of the MCE-PD method by the analysis of complex samples requiring only minimal sample pretreatment, filtration or degassing. The results of the work highlight the strong analytical potential of the MCE as a technique which meets the requirements of green analytical chemistry. The proposed MCE-PD method is applicable to laboratory practice and is a good alternative to previously published analytical methods dealing with the determination of CA in complex samples.
Supplementary Materials
The following are available online at https://www.mdpi.com/2297-8739/7/4/72/s1, Figure S1: Structure of carminic acid with pKa values, Figure S2: Linear dynamic range for: (a) peak height; and (b) peak area of carminic acid, Figure S3: Plot of residuals of peak area, Table S1: Some parameters of analytical methods used for determination of carminic acid.
Author Contributions
Conceptualization, J.Š., P.B. and M.M.; methodology, R.S., J.H. and E.V.; software, A.M. and J.H.; validation, J.H., A.M. and R.S.; formal analysis, E.V. and J.H.; investigation, E.V. and J.H.; resources, P.B. and M.M.; data curation, E.V. and J.H.; writing—original draft preparation, J.H., A.M. and M.M.; writing—review and editing, R.S. and J.H.; visualization, A.M. and J.H.; supervision, R.S. and J.Š.; project administration, J.H. and M.M.; and funding acquisition, M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Slovak Research and Development Agency (APVV-17-0318 and APVV-17-0373), the Slovak Grant Agency for Science (VEGA 1/0787/18), and Comenius University Grant (UK/19/2020).
Acknowledgments
This contribution is the result of the project implementation (ITMS 26240220061) supported by the OPRaD funded by the ERDF. The authors also thank to František Iványi for technical support.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| BGE | Background electrolyte |
| CA | Carminic acid |
| CE | Capillary electrophoresis |
| EFSA | European Food Safety Authority |
| FAO | Food and Agriculture Organization of the United Nations |
| HIS | L-histidine |
| HPLC | High performance liquid chromatography |
| HVPS | High voltage power supply |
| HVR | High voltage relay |
| i.d. | Internal diameter |
| LDR | Linear dynamic range |
| LOD | Limit of detection |
| LOQ | Limit of quantitation |
| MCE | Microchip electrophoresis |
| MES | 2-(N-morpholino)ethanesulfonic acid |
| MHEC | Methylhydroxyethylcellulose |
| MPL | Maximum permitted level |
| PD | Photometric detection |
| RSD | Relative standard deviation |
| SD | Standard deviation |
| UV–Vis | Ultraviolet-visible |
| WHO | World Health Organization |
References
- Amchova, P.; Kotolova, H.; Ruda-Kucerova, J. Health safety issues of synthetic food colorants. Regul. Toxicol. Pharmacol. 2015, 73, 914–922. [Google Scholar] [CrossRef]
- Scotter, M.J. Colour Additives for Food and Beverages; Woodhead Publishing: Cambridge, UK, 2015. [Google Scholar]
- The European Parliament and the Council of the European Union. Council Regulation (EC) 1333/2008 of 16 December 2008 on food additives. Off. J. Eur. Union 2008, L354, 16–33. [Google Scholar]
- The European Parliament and the Council of the European Union. Directive 2009/35/EC of the European Parliament and of the Council of 23 April 2009 on Colouring Matters Which May Be Added to Medical Products. Off. J. Eur. Union 2009, L109, 10–13. [Google Scholar]
- Martins, N.; Roriz, C.L.; Morales, P.; Barros, L.; Ferreira, I.C.F.R. Food colorants: Challenges, opportunities and current desires of agro-industries to ensure consumer expectations and regulatory practices. Trends Food Sci. Technol. 2016, 52, 1–15. [Google Scholar] [CrossRef]
- Delgado-Vargas, F.; Paredes-López, O. Natural Colorants for Food and Nutraceutical Uses; CRC Press: Boca Raton, FL, USA, 2002. [Google Scholar]
- Samari, F.; Hemmateenejad, B.; Shamsipur, M. Spectrophotometric determination of carminic acid in human plasma and fruit juices by second order calibration of the absorbance spectra–pH data matrices coupled with standard addition method. Anal. Chim. Acta 2010, 667, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Merinas-Amo, R.; Martínez-Jurado, M.; Jurado-Güeto, S.; Alonso-Moraga, Á.; Merinas-Amo, T. Biological effects of food coloring in in vivo and in vitro model systems. Foods 2019, 8, 176. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Food Additives and Nutrient Sources added to Food. Scientific Opinion on the re-evaluation of cochineal, carminic acid, carmines (E 120) as a food additive. EFSA J. 2015, 13. [Google Scholar] [CrossRef]
- Scotter, M.J. Methods for the determination of European Union-permitted added natural colours in foods: A review. Food Addit. Contam. Part A Chem. 2011, 28, 527–596. [Google Scholar] [CrossRef] [PubMed]
- Ntrallou, K.; Gika, H.; Tsochatzis, E. Analytical and sample preparation techniques for the determination of food colorants in food matrices. Foods 2020, 9, 58. [Google Scholar] [CrossRef]
- The European Commission. Commission Regulation (EU) No 231/2012 of 9 March 2012 laying down specifications for food additives listed in Annexes II and III to Regulation (EC) No 1333/2008 of the European Parliament and of the Council. Off. J. Eur. Union 2012, L83, 1–295. [Google Scholar]
- Heydari, R.; Hosseini, M.; Zarabi, S. A simple method for determination of carmine in food samples based on cloud point extraction and spectrophotometric detection. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 150, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, A.H.; Alshammery, H.M.; Abdalla, M.A.; Alghamdi, A.F. Determination of carmine food dye (E120) in foodstuffs by stripping voltammetry. J. AOAC Int. 2009, 92, 1454–1459. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, U.T.; Ergun, F.; Yilmaz, H. Determination of the food dye carmine in milk and candy products by differential pulse polarography. J. Food Drug Anal. 2014, 22, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Šuleková, M.; Hudák, A.; Smřcová, M. The determination of food dyes in vitamins by RP-HPLC. Molecules 2016, 21, 1368. [Google Scholar] [CrossRef]
- Lim, H.S.; Choi, J.C.; Song, S.B.; Kim, M. Quantitative determination of carmine in foods by high-performance liquid chromatography. Food Chem. 2014, 158, 521–526. [Google Scholar] [CrossRef]
- Ordoudi, S.A.; Staikidou, C.; Kyriakoudi, A.; Tsimidou, M.Z. A stepwise approach for the detection of carminic acid in saffron with regard to religious food certification. Food Chem. 2018, 267, 410–419. [Google Scholar] [CrossRef]
- Carvalho, P.R.N.; Collins, C.H. HPLC determination of carminic acid in foodstuffs and beverages using diode array and fluorescence detection. Chromatographia 1997, 45, 63–66. [Google Scholar] [CrossRef]
- Feng, F.; Zhao, Y.; Yong, W.; Sun, L.; Jiang, G.; Chu, X. Highly sensitive and accurate screening of 40 dyes in soft drinks by liquid chromatography-electrospray tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 1813–1818. [Google Scholar] [CrossRef]
- Huang, H.Y.; Chiu, C.W.; Sue, S.L.; Cheng, C.F. Analysis of food colorants by capillary electrophoresis with large-volume sample stacking. J. Chromatogr. A 2003, 995, 29–36. [Google Scholar] [CrossRef]
- Nevado, J.J.B.; Cabanillas, C.G.; Salcedo, A.M.C. Method development and validation for the simultaneous determination of dyes in foodstuffs by capillary zone electrophoresis. Anal. Chim. Acta 1999, 378, 63–71. [Google Scholar] [CrossRef]
- Huang, H.Y.; Shih, Y.C.; Chen, Y.C. Determining eight colorants in milk beverages by capillary electrophoresis. J. Chromatogr. A 2002, 959, 317–325. [Google Scholar] [CrossRef]
- Huang, H.Y.; Chuang, C.L.; Chiu, C.W.; Chung, M.C. Determination of food colorants by microemulsion electrokinetic chromatography. Electrophoresis 2005, 26, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.-J.; Liu, C.-T.; Li, W.; Tang, A.-N. Dispersive solid-phase microextraction and capillary electrophoresis separation of food colorants in beverages using diamino moiety functionalized silica nanoparticles as both extractant and pseudostationary phase. Talanta 2015, 132, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Musso, N.; Grasso, M.; Costantino, A.; Lazzarino, G.; Tascedda, F.; Gulisano, M.; Lunte, S.M.; Caraci, F. Microfluidics as a novel tool for biological and toxicological assays in drug discovery processes: Focus on microchip electrophoresis. Micromachines 2020, 11, 593. [Google Scholar] [CrossRef]
- Guzman, N.A.; Guzman, D.E. A two-dimensional affinity capture and separation mini-platform for the isolation, enrichment, and quantification of biomarkers and its potential use for liquid biopsy. Biomedicines 2020, 8, 255. [Google Scholar] [CrossRef]
- Kaniansky, D.; Masár, M.; Bodor, R.; Žúborová, M.; Ölvecká, M.; Jöhnck, M.; Stanislawski, B. Electrophoretic separations on chips with hydrodynamically closed separation systems. Electrophoresis 2003, 24, 2208–2227. [Google Scholar] [CrossRef]
- Rasimas, J.P.; Berglund, K.A.; Blanchard, G.J. A molecular lock-and-key approach to detecting solution phase self-assembly. A fluorescence and absorption study of carminic acid in aqueous glucose solutions. J. Phys. Chem. 1996, 100, 7220–7229. [Google Scholar] [CrossRef]
- International Conference on Harmonisation of Technical Requirements of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline: Validation of Analytical Procedures: Text and Methodology Q2(R1). Available online: https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf (accessed on 20 October 2020).
- ISO. International Standard ISO 3632-2: Spices—Saffron (Crocus sativus L.). Part 2: Test methods; ISO: Geneva, Switzerland, 2010. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).