Abstract
In this work, Chromabond® HLB was evaluated as an extraction sorbent of a group of seven phthalic acid esters (PAEs; i.e., dipropyl phthalate, DPP, dibutyl phthalate, DBP, diisopentyl phthalate, DIPP, di-n-pentyl phthalate, DNPP, butylbenzyl phthalate, BBP, dicyclohexyl phthalate, DCHP, and di(2-ethylhexyl)phthalate, DEHP) and one adipate (di(2-ethylhexyl) adipate, DEHA) from tap and waste water prior to gas chromatography-mass spectrometry. After the optimization of the extraction conditions (200 mg of sorbent conditioned with 10 mL of acetonitrile and 2 mL of Milli-Q water, extraction of 50 mL of water at pH 6.0, vacuum drying for 20 min and elution with 10 mL of ethyl acetate), a recovery study was developed at different concentration levels in each matrix, which revealed that most of the target analytes could be recovered between 75 and 112%, with relative standard deviation values for all of them below 20%. Matrix effect was evaluated, finding that matrix-matched calibration should be developed for most analytes in both matrices. The limits of quantification (LOQs) of the method were in the 0.82–71 ng L−1 range. The developed method was also applied to the extraction of the target PAEs in different water samples finding some of them, in particular, DNPP in tap water samples, and BBP and DCHP in waste water, but below the LOQs of the method.
1. Introduction
In the plastic industry, an important number of compounds are currently being used as additives to improve plastic properties, among which plasticizers, and, in particular, phthalic acid esters (PAEs) are one of the groups of compounds mostly used. When PAEs are present in the polymeric matrix, plastics can be more flexible and manageable. Their use with this objective began in the 1920s by replacing other natural compounds, such as camphor (highly volatile and fragrant). In the following decades, the use of PAEs was extended up to the point that phthalates accounted for 65% of the world consumption of plasticizers in 2017 [1].
The analysis of PAEs, particularly in environmental samples, is of high interest, since PAEs can be released from plastics and also because many of them have an endocrine-disrupting activity [2]. As a result, some PAEs have been forbidden or regulated by different countries, especially in plastic items. That is the case of the European Union (EU) [3], the United States (US) [4], or Canada [5], among others. At the same time, PAEs analysis in any matrix is also a challenge, in the sense that they are widely present in every laboratory, and suitable precautions and strategies should be developed to minimize, control, and correct their presence during their analysis [6,7].
As it is well-known, solid-phase extraction (SPE) [8] constituted an extremely active technique in the field of separation sciences decades ago, but it is still being highly used nowadays in sample preparation for both analyte enrichment/preconcentration, and/or clean-up [9,10]. Generally, there are four main characteristics of a chemical compound that are mainly responsible for extraction/retention in SPE: polarity, electrical charge, molecular recognition, and molecular size (the last of them not so widely used nowadays). Depending on the analyte’s characteristics and, therefore, on the main extraction/retention mechanism, the sorbent is selected. In this sense, the sorbents most widely and traditionally used in SPE are bonded-phase silica particles, C18 being the most important and one of the first to be used. The later introduction of polymeric sorbents clearly increased the applicability of the technique, since they do not have labile silanol groups, they have a superior stability in a wide pH range, and they offer the possibility of the retention of polar analytes. One of those polymeric sorbents that has also found a very good acceptance among analytical laboratories is Oasis® HLB (hydrophilic-lipophilic balance) introduced by Waters Chromatography in 1996. This sorbent is indeed a copolymer of the hydrophilic N-vinylpyrrolidone and the lipophilic divinylbenzene (no silanol groups are present). The first monomer allows the retention of polar compounds and also provides wetting properties, while the second provides reversed-phase properties. It is appropriate for acidic, basic and neutral analytes and it is stable from pH 0 to 14 [11]. According to the supplier, it has an average specific surface area of 810 m2 g−1, an average pore size diameter of 80 Å and specific pore volume of 1.3 mL g−1 [12]. The particles’ size of commercialized sorbents is also variable: 5, 15, 25, 30, and 60 µm [13]. These types of cartridges have been applied in several works for the extraction of diverse groups of PAEs from different water samples with success [14,15,16,17,18,19,20].
Recently, Macherey-Nagel has commercialized Chromabond® HLB as an alternative sorbent to Oasis® HLB (Supplementary Materials Figure S1 shows its structure). In this case, the specific surface area reported by the manufacturer is 750 m2 g−1 and the average pore size diameter is 65 Å [21]. No specific pore volume is provided, while the particle sizes available are only 30 and 60 µm [21]. The range of pH work goes from 1 to 14. As a result of the recent release of this sorbent, suitable studies should be developed to appropriately evaluate its effectivity and to compare it with that of previous sorbents. Up to now, only some technical notes of the manufacturer have been provided concerning the application of Chromabond® HLB for extraction purposes (basically for some pharmaceuticals, drugs, alkaloids or pesticides analysis) [22], being necessary more studies to evaluate its real performance. That is why the main objective of this work is to evaluate the effectiveness of Chromabond® HLB sorbent on the SPE of a group of 7 PAEs (dipropyl phthalate, DPP, dibutyl phthalate, DBP, diisopentyl phthalate, DIPP, di-n-pentyl phthalate, DNPP, butylbenzyl phthalate, BBP, dicyclohexyl phthalate, DCHP, di(2-ethylhexyl)phthalate, DEHP) and one adipate (di(2-ethylhexyl) adipate, DEHA) from tap and waste water. To the best of our knowledge, this is the first time that these analytes have been extracted with this sorbent, and also it is the first article published at the moment regarding the use of this sorbent.
2. Materials and Methods
2.1. Chemical and Materials
DPP (CAS 131-16-8), DBP (CAS 84-74-2), DCHP (CAS 84-61-7) and DEHP (CAS 117-81-7) were purchased from Sigma-Aldrich (San Luis, MO, USA) and BBP (CAS 85-68-7), DIPP (CAS 605-50-5), DNPP (CAS 131-18-0), and DEHA (CAS 103-23-1) were obtained from Dr. Ehrenstorfer (Augsburg, Germany, purity ≥ 97.3%). DBP-3,4,5,6-d4 (DBP-d4, CAS 93952-11-5) and DHP-3,4,5,6-d4 (DHP-d4, CAS 1015854-55-3), which were used as internal standards (ISs), were from Sigma-Aldrich (purity ≥ 99.1%). Supplementary Materials Figure S2 shows the structure and log KOW values of the target analytes.
Solutions of each analyte were prepared in cyclohexane at 1200 mg L−1 and at 500 mg L−1 for the ISs, storing them at −18 °C. Working solutions were prepared at different concentration levels by diluting individual stock solutions with an appropriate volume of cyclohexane and also stored at −18 °C.
Milli-Q water was obtained from a Milli-Q gradient system A10 in which previously deionized water obtained from an Elix® Essential water purification system was further purified, both from Millipore (Burlington, MA, USA). Acetonitrile (ACN) of HPLC-MS grade, methanol (MeOH) of HPLC-MS grade, and dichloromethane (DCM) of pesticide residue analysis quality were from VWR International (Radnor, PA, USA). Ethyl acetate (EtOAc) of HPLC-MS hypergrade was from Merck (Darmstadt, Germany). Hydrochloric acid (37%, w/w) from VWR International. Sodium hydroxide (purity 99.2%) was from VWR International. Chromabond® HLB polypropylene columns of 500 mg (6 mL) were purchased from Macherey-Nagel (Düren, Germany).
Volumetric glassware was cleaned with a sulfuric acid (95%, w/w, VWR International) solution of Nochromix® from Godax Laboratories (Cabin John, MD, USA) for 24 h. Non-volumetric glassware was maintained at 550 °C for 4–5 h to eliminate any organic matter.
2.2. Instrumentation
A 7820A GC system from Agilent Technologies (Santa Clara, CA, USA) equipped with an autosampler was used for analytes separation, which was coupled to an 5977B single quadrupole (Q) mass spectrometer from Agilent Technologies for analytes detection. The carrier gas was helium at a flow rate of 1.5 mL min−1. Separation was carried out in a HP-5ms Ultra Inert column ((5%-phenyl)-methylpolysiloxane, 30 m × 250 μm × 0.25 μm) from Agilent Technologies. The thermal gradient program was as follows: temperature was increased from 60 to 160 °C at 30 °C min−1, then increased to 260 °C at 3 °C min−1, and finally increased to 300 °C at 30 °C min−1 and held for 5 min, achieving a total run time of 40 min. Injection was performed in the splitless mode (after 0.75 min the split was opened with a purge flow of 40 mL min−1) at 280 °C, injecting 2 µL of sample. An electron ionization energy of −70 eV, and a temperature of the ion source and of the MS transfer line of 280 °C were set. Single ion monitoring (SIM) mode was selected. GC-MS system was controlled using the Enhanced MassHunter software from Agilent Technologies.
A FiveEasy™ Plus pH/mV meter from Mettler Toledo (Columbus, OH, USA) was used for pH and conductivity measurements. A RV8 rotary evaporator supplied with a HB thermostatic bath and a CVC 3000 vacuum pump with a vacuum controller from VWR International was used for solvent evaporation.
2.3. Samples
Tap water was collected at our laboratory while waste water was collected in a waste water treatment plant located at the island of Tenerife (Canary Islands, Spain). Both types of water samples were used for method validation. Additionally, two more tap water samples were collected at the towns of La Orotava and San Cristóbal de La Laguna. Tap water samples were directly extracted while waste water was first filtered through Durapore® polyvinylidene fluoride (PVDF) filter membranes with a pore size of 0.45 µm and a diameter of 47 mm from Millipore. Therefore, for waste water samples, only dissolved analytes are taken into account.
For sample spiking, the corresponding volumes of the PAEs and DEHA mixture, as well as those of the ISs, were introduced in a flask, evaporated at ambient air and temperature without the need of using a stream of nitrogen (less than 15 s were necessary since the volume was really low), 50 mL of the water sample were added using a pipette and sonicated for 2 min. After pH adjustment, the samples were passed through the SPE columns as indicated in Section 2.4. When non-spiked samples were analyzed, only both ISs were added.
2.4. SPE Procedure
Commercial Chromabond® HLB packed in plastic columns (500 mg) were opened and 200 mg of the sorbent were introduced into a glass column of 6 mL that had two PTFE frits inside, both from Sigma-Aldrich. The column was placed on a Chromabond® SPE vacuum manifold from Macherey-Nagel. Once the sorbent was introduced, a PTFE frit was located onto it to retain it. Then, the stationary phase was conditioned with 10 mL of ACN (ACN was also used to eliminate possible residual PAEs of the sorbent) and 2 mL of Milli-Q water. Afterwards, 50 mL of the water sample previously adjusted to pH 6.0 with a HCl solution, was passed through the column (one drop per second) and vacuum-dried for 20 min. Afterwards, elution was carried out using 10 mL of EtOAc which was later evaporated to dryness at 40 °C and 180 mbar in a rotary evaporator. Finally, 400 μL of cyclohexane were added. After ultrasound application for a couple of seconds, the solvent was transferred to a vial and injected in the GC system.
Since PAEs background contamination might exist (it may also slightly vary between days) and has to be assessed before the analysis of PAEs content in any sample, procedural blanks were analyzed on a daily basis by applying the entire procedure without using/adding any kind of water. Consequently, the amount of such analytes was subtracted when necessary to correctly determine its concentration in the real samples analyzed.
3. Results and Discussion
3.1. Gas Chromatography-Mass Spectrometry Analysis
Gas chromatography-mass spectrometry (GC-MS) was used for the analysis of the selected group of PAEs as well as the adipate, using the conditions selected in our previous work [23] which are summarized in the Experimental Section. The MS system was operated in the SIM mode selecting 129 m/z for DEHA, 149 m/z for all the target PAEs and 153 m/z for both ISs as well as the retention time as identification points. DBP-d4 was used for DPP, DBP, DIPP, DNPP, BBP, and DCHP analysis while DHP-d4 was used for DEHP and DEHA. Figure 1 shows a GC-MS chromatogram obtained under the SIM mode when a mixture of the analytes (75 µg L−1) and ISs (125 µg L−1) dissolved in cyclohexane was injected. Since all PAEs share the same m/z value, it is important to achieve a good separation between them.
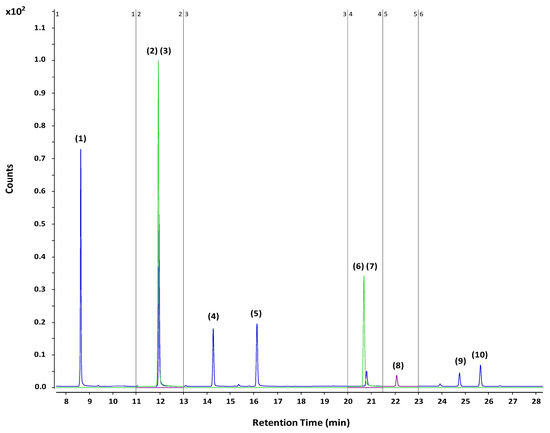
Figure 1.
GC-Q-MS chromatogram obtained under SIM mode. Concentration of all the analytes: 75 µg L−1. Concentration of the ISs: 125 µg L−1. Peak identification: DPP (1), DBP-d4 (2), DBP (3), DIPP (4), DNPP (5), DHP-d4 (6), BBP (7), DEHA (8), DCHP (9), and DEHP (10).
Linearity of the response of the detection system was tested by the injection in triplicate of solutions of the target analytes in cyclohexane of 9 different concentration levels. ISs were also included at a concentration of 125 µg L−1. Information regarding the quality parameters of the calibration curves (slope and intercept as well as their confidence intervals, error of the estimate, determination coefficients (R2) and studied linear range) are shown in Supplementary Materials Table S1 (calibration curves were built considering the analytes’ relative peak areas). Since a good sensitivity was obtained, the lowest calibration levels (LCLs) shown in the table (and which ranged between 0.1 and 5 µg L−1) were established. In all cases, R2 values were higher than 0.990. The analysis of the residual plot reflected a random distribution around zero, with no appreciable trend.
3.2. Solid-Phase Extraction Optimization
In order to develop the SPE procedure, 200 mg of the sorbent were introduced in empty glass columns and retained using PTFE frits, as indicated in the Experimental Section. Such amount of sorbent was used in order to facilitate the comparison of the results, since for most applications, 200 or 500 mg of sorbents are used. Concerning the activation procedure, 10 mL of ACN were initially passed through the cartridge followed by 2 mL of Milli-Q water. ACN was used, as recommended by the manufacturer [24] though at a higher volume (10 mL instead of 5 mL) since some interferences coming from the sorbent at very low amounts were detected and further washing was also required, probably due to the fact that it is commercialized in polypropylene columns. After activation (no PAEs peaks were observed even at higher volumes of ACN), spiked Milli-Q water at 2 µg L−1 (final concentration in the extract of 250 µg L−1) was passed through the stationary phase. Spiked Milli-Q water was selected as a sample to avoid the influence of matrix effects. Absolute recovery values were calculated by comparing the peak areas obtained for spiked samples (spiked before the SPE procedure) and matrix-matched standards (spiked at the theoretical expected final concentration when the final extract was reconstituted with cyclohexane).
Though PAEs are not ionized in aqueous solution and pH should not affect their extraction as previously reported [25], Milli-Q water was adjusted at different pH values (4, 6, and 10) by adding small amounts of HCl or NaOH in order to reach the desired pH value. For these experiments, elution was carried out with 10 mL of ACN (extractions were developed in duplicate) after vacuum drying the cartridge for 20 min. It was observed that it was not necessary to adjust the pH, though, for further experiments, and to minimize the possible irreproducibility of the extraction, pH was adjusted to 6.0 in all cases since it is close to the pH value of environmental water samples (pH 6–8).
Regarding the elution solvent, ACN, MeOH, DCM and EtOAc were tested by carrying out duplicate extractions. A matrix-matched standard of the expected final concentration was also analyzed in each case to compare the peak areas and to calculate absolute recovery values. Elution was performed with each solvent using a volume of 10 mL plus 2 × 5 mL more (a total of 20 mL) in order to simultaneously study the elution volume. After the evaporation of the three consecutive eluates separately (10, 5, and 5 mL) in a rotary evaporator at 40 °C and at the ideal/recommended vacuum pressure (180 mbar for ACN, 200 mbar for EtOAc, 220 mbar for MeOH and 700 mbar for DCM), 400 µL of cyclohexane were added and sonicated. Finally, 2 µL were injected in triplicate in the GC-MS system (n = 3). As shown in Figure 2, 10 mL of DCM or EtOAc provided similar results and the highest absolute recovery values for most of the analytes. In particular, absolute recovery percentages above 75% were obtained for DPP, DBP, DIPP, DNPP, BBP, and DCHP with both of them. Recovery of DEHA (which is not a PAE) was nearly 60% while DEHP recovery was close to 50%. On the contrary, ACN and MeOH provided the worst results, except for DEHP with MeOH which recovery values were higher. It was also observed that above 10 mL of each solvent, analyte recovery did not improve. In addition, the reduction of the elution solvent volume was also tested below 10 mL, but it was observed that elution was incomplete. Among EtOAc and DCM, the first was selected since it is not a chlorinated solvent like the second.
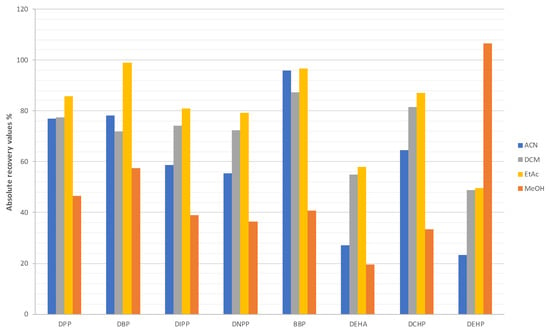
Figure 2.
Absolute recovery values obtained with ACN, DCM, EtOAc, and MeOH during extraction procedure optimization.
The optimized procedure was later applied to the extraction of tap and waste water samples as it will be shown below, in order to fully demonstrate its applicability.
3.3. Matrix-Matched Calibration
The presence of matrix components can suppress or enhance the chromatographic signal [26], as it is well-known. In GC, signal enhancement is very frequent as a result of the partial occupation/deactivation of active surfaces of the system (silanol and metal ion active sites of the injector, column, or detector) by matrix components. Such fact may improve mass transfer of the analytes to the detector which results in the enhancement of the peak symmetry as well as in the improvement of detection and quantification limits [26]. However, signal suppression may also occur when an unspecific adsorption of the analytes on non-volatile components retained in the active sites takes place [26].
To evaluate and correct the matrix effect (ME), matrix-matched calibration curves were obtained by spiking the final extracts of nine tap and waste water aliquots of 50 mL with the ISs and the analytes (spiking was developed once the extract was evaporated). Non-spiked samples (only the ISs were added before the SPE procedure) were also analyzed in order to detect and/or correct the possible presence of the selected analytes. A new liner was used before the injection of the calibration curves (injections were developed in triplicate). Table 1 compiles the calibration curves, including the standard deviation of the slope and intercept, the R2 values, the standard deviation of the estimate (sy/x), the studied linear range and the ME percentage (%ME), which was calculated in the following way: %ME = ((slope of matrix-matched calibration - slope of pure solvent calibration)/(slope of solvent calibration)) × 100 [27,28]. As can be seen, good R2 values, beyond 0.991, were obtained for all analytes and matrices. In this case, also the analysis of the residual plot reflected a random distribution around zero, with no appreciable trend.

Table 1.
Matrix-matched calibration data and matrix effect percentage of the target analytes.
Concerning the ME for tap water, a negligible effect (%ME values between −20 and 20%) was found for three analytes: DPP, DBP, and DEHP. A soft signal enhancement effect (between 20 and 50%) was found for DIPP, DNPP, and DEHA while also a strong signal enhancement effect (higher than 50%) was found for BPP and DCHP [29]. Regarding waste water, a negligible effect was found for four PAEs: DIPP, DNPP, DEHA, and DEHP. A soft signal suppression effect was found for DPP and DBP while also a strong signal enhancement effect was found again for BBP and DCHP. These results suggest the need of developing a matrix-matched calibration for most analytes in both matrices to compensate such effects.
3.4. Recovery Study
The trueness of the method was determined by developing a recovery study at 0.4 and 1.6 µg L−1 in the sample (final concentration of the analytes in the vial of 50 and 200 µg L−1, respectively). The ISs were also added at a concentration of 1 µg L−1 in the sample (final concentration of 125 µg L−1). Four extractions were carried out at each level. Relative recovery values were calculated by comparing the peak area/IS peak area ratios obtained for spiked samples (spiked before the SPE procedure) and matrix-matched standards (spiking at the theoretical expected final concentration when the final extract was reconstituted with cyclohexane).
Results are shown in Table 2. As can be seen, relative recovery values ranged between 75 and 112% for tap water (except for DPP at the highest concentration level, which was 64%), with RSD values ≤ 19%, and in the range 87–110% (except for BBP and DCHP at the lowest concentration level, which were 126 and 123%, respectively) for waste water, with RSD values ≤ 10%. Although, the recovery values of those three analytes (DPP, BBP, and DCHP) were out of the 70–120% range at one of the concentration levels and samples, the mean values of all levels were in the acceptable range 70–120% and always with RSD values below 20% [30]. Such recovery values are also similar to the ones reported by previous works in which Oasis® HLB cartridges were used for the extraction of some of these analytes from water samples [16,18,19] except for DIPP, DNPP, and DCHP, which were not analyzed in those cases. Such data, which can be seen in Supplementary Materials Table S2, suggests that Chromabond® HLB stationary phase can perfectly be used as substitute of Oasis® HLB for the extraction of the selected PAEs and DEHA from tap and waste water samples. Further work to demonstrate its application to other samples and analytes should still be demonstrated.
Table 2.
Relative recovery and RSD (between parenthesis) values of the target analytes in water samples (n = 3 at each spiking level).
Concerning the LOQs of the method, they ranged between 1.1 and 71 ng L−1 for tap water and between 0.82 and 46 ng L−1 for waste water. Such values were assessed contemplating the absolute recovery values, the enrichment factors and the LCL of the matrix-matched calibration curves. These LOQs are analogous to those reported by previous works in which PAEs have been analyzed in tap and waste water using other extraction procedures [31,32], also using Oasis® HLB cartridges [17]. Since PAEs appear at extremely low concentrations in environmental water samples [16,33,34], it is important to achieve LOQs in the low ng L−1 level. It should be highlighted that among the existing PAEs, only DEHP is currently included in the EU list of priority substances to be monitored in environmental waters [35] being the annual average-environmental quality standard of 1.3 µg L−1. Our method is able to quantify such compound at much lower concentrations, down to 1.5 ng L−1 in tap water and 9.4 ng L−1 in waste water.
Supplementary Materials Table S2 shows a comparison among different previous works in which Oasis® HLB cartridges have been applied for the extraction of PAEs and/or DEHA from water samples. In general, more complicated and longer extraction procedures were carried out, using several solvents for the sorbent activation as well as the elution of the analytes, even making use of chlorinated solvents in some cases [17,18,19,20]. As can be seen, the methodology proposed in this work presents in many cases a simpler and faster alternative for the extraction of PAEs from water samples, maintaining an excellent extraction efficiency and, in most cases, a comparable sensitivity, despite a much lower volume of sample is analyzed compared to the works previously published using this kind of sorbent.
3.5. Analysis of Real Samples
Three different tap water samples collected at different towns of Tenerife, including our laboratory and one waste water were also analyzed to demonstrate the applicability of the method (see Supplementary Materials Table S3 for more details). For this purpose, only the ISs were added before the extraction procedure. Only DNPP was found in some of the tap water samples below the LOQ of the method. Concerning the waste water, only BBP and DCHP were found, but also at a concentration below the LOQ of the method. As it was previously mentioned, PAEs have been found in different environmental water samples, being BBP one of the most common, at concentrations in the ng L−1 or even µg L−1 order [16,36]. Analyzing these findings carefully, it is not strange that these PAEs appear in the environment at the mentioned levels, since many of them (including BBP) are highly used in the manufacture of PVC products or even as part of adhesives, inks, or paints [37]. Definitely, the current problematic of plastic contamination is not only a problem related to the polymeric particles themselves, but also to the contaminants they release to the environment and can be even more dangerous.
4. Conclusions
Chromabond® HLB was found a suitable and alternative sorbent for the SPE of most of the group of PAEs selected in this work from tap and waste water, as a result of the acceptable recovery (75–112%) and RSD (≤ 19%) values that were achieved. Since PAEs are additives (plasticizers) of many plastics, it is necessary to develop the extraction in glass columns and to suitably wash the sorbent before its use. The proposed procedure is able to provide LOQs in the low ng L−1 level, which are adequate for the determination of the target compounds in environmental samples. The application of the method to the analysis of several water samples, revealed the presence of some of the target analytes below the LOQs of the method. In general terms, the methodology developed in this work has shown to be comparable or even a better alternative to the previously published ones for the analysis of PAEs using Oasis® HLB sorbent.
Supplementary Materials
The following are available online at https://www.mdpi.com/2297-8739/7/2/21/s1, Table S1. Instrumental calibration data of the target analytes, Table S2. Comparison of previous works in which Oasis® HLB cartridges have been used to extract PAEs and/or DEHA from water samples. Table S3. Results of the analysis of tap and waste water samples after the SPE-GC-MS procedure, Figure S1. Chemical structure of Chromabond® HLB sorbent, Figure S2. Chemical structures of the target analytes and log KOW values.
Author Contributions
Conceptualization: J.G.-S. and J.H.-B.; data curation: C.O.-Z., D.A.V.-M., and M.Á.G.-C.; formal analysis: C.O.-Z.; investigation: C.O.-Z., D.A.V.-M., and M.Á.G.-C.; methodology: C.O.-Z., J.G.-S., and J.H.-B.; project administration: J.G.-S. and J.H.-B.; resources: J.G.-S. and J.H.-B.; supervision: J.G.-S. and J.H.-B.; writing—original draft: C.O.-Z. and J.H.-B.; writing—review and editing: J.G.-S., M.Á.G.-C., and J.H.-B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Fundación CajaCanarias grant number 2016TUR07 and Alumni ULL Pratronage Program 20018-2019.
Acknowledgments
Javier González-Sálamo thanks “Cabildo de Tenerife” for the Agustín de Betancourt contract at the Universidad de La Laguna. The authors thank Verónica Pino and Juan Ayala from GIN-QAAMA group of the Universidad de La Laguna, for instrumental facilities.
Conflicts of Interest
Authors declare no conflict of interest.
References
- IHS Markit Plasticizers. Chemical Economics Handbook. Available online: https://ihsmarkit.com/products/plasticizers-chemical-economics-handbook.html (accessed on 20 January 2020).
- Tsatsakis, A.M.; Katsikantami, I.; Kalantzi, O.-I.; Sevim, Ç.; Tsarouhas, K.; Sarigiannis, D.; Tzatzarakis, M.N.; Rizos, A.K. Phthalates: Exposure and Health Effects; Elsevier: Oxford, UK, 2019; pp. 163–173. ISBN 978-0-444-63952-3. [Google Scholar]
- Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), establishing a European Chemicals Agency, amending Directive 1999/4. Off. J. Eur. J. 2006, L396, 1–520.
- Oppt Phthalates ActionPlan; US Environmental Protection Agency: Washington, DC, USA, 2012.
- Hazardous Products Act. Phthalates Regulations; Government of Canada: Ottawa, ON, Canada, 2010.
- González-Sálamo, J.; Socas-Rodríguez, B.; Hernández-Borges, J. Analytical methods for the determination of phthalates in food. Curr. Opin. Food Sci. 2018, 22, 122–136. [Google Scholar] [CrossRef]
- Fankhauser-Noti, A.; Grob, K. Blank problems in trace analysis of diethylhexyl and dibutyl phthalate: Investigation of the sources, tips and tricks. Anal. Chim. Acta 2007, 582, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Fritz, J.S. Analytical Solid-Phase Extraction, 1st ed.; Wiley-VCH Verlag: New York, NY, USA, 1999; ISBN 978-0-471-24667-1. [Google Scholar]
- Andrade-Eiroa, A.; Canle, M.; Leroy-Cancellieri, V.; Cerdà, V. Solid-phase extraction of organic compounds: A critical review (Part I). TrAC Trends Anal. Chem. 2016, 80, 641–654. [Google Scholar] [CrossRef]
- Andrade-Eiroa, A.; Canle, M.; Leroy-Cancellieri, V.; Cerdà, V. Solid-phase extraction of organic compounds: A critical review. part ii. TrAC Trends Anal. Chem. 2016, 80, 655–667. [Google Scholar] [CrossRef]
- Trikas, E.D.; Papi, R.M.; Kyriakidis, D.A.; Zachariadis, G.A. Evaluation of Ion Exchange and Sorbing Materials for Their Adsorption/Desorption Performane towards Anthocyanins, Total Phenolics, and Sugars from a Grape Pomace Extract. Separations 2017, 4, 9. [Google Scholar] [CrossRef]
- Waters Sample Preparation Catalogue; Waters Corporation: Mildford, MA, USA, 2019.
- Waters Oasis Sample Extraction Products Catalogue; Waters Corporation: Mildford, MA, USA, 2019.
- Xiaoyan, T.; Suyu, W.; Yang, Y.; Ran, T.; Yunv, D.; Dan, A.; Li, L. Removal of six phthalic acid esters (PAEs) from domestic sewage by constructed wetlands. Chem. Eng. J. 2015, 275, 198–205. [Google Scholar] [CrossRef]
- Liu, X.; Shi, J.; Bo, T.; Zhang, H.; Wu, W.; Chen, Q.; Zhan, X. Occurrence of phthalic acid esters in source waters: A nationwide survey in China during the period of 2009–2012. Environ. Pollut. 2014, 184, 262–270. [Google Scholar] [CrossRef]
- Paluselli, A.; Aminot, Y.; Galgani, F.; Net, S. Occurrence of phthalate acid esters (PAEs) in the northwestern Mediterranean Sea and the Rhone River. Prog. Oceanogr. 2018, 163, 221–231. [Google Scholar] [CrossRef]
- Domínguez-Morueco, N.; González-Alonso, S.; Valcárcel, Y. Phthalate occurrence in rivers and tap water from central Spain. Sci. Total Environ. 2014, 500–501, 139–146. [Google Scholar] [CrossRef]
- Liang, Y.; Li, Z.; Shi, P.; Ling, C.; Chen, X.; Zhou, Q.; Li, A. Performance of a novel magnetic solid-phase-extraction microsphere and its application in the detection of organic micropollutants in the Huai River, China. Environ. Pollut. 2019, 252, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Guart, A.; Bono-Blay, F.; Borrell, A.; Lacorte, S. Migration of plasticizersphthalates, bisphenol A and alkylphenols from plastic containers and evaluation of risk. Food Addit. Contam. Part A 2011, 28, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Fauvelle, V.; Castro-Jiménez, J.; Schmidt, N.; Carlez, B.; Panagiotopoulos, C.; Sempéré, R. One-Single Extraction Procedure for the Simultaneous Determination of a Wide Range of Polar and Nonpolar Organic Contaminants in Seawater. Front. Mar. Sci. 2018, 5, 295. [Google Scholar] [CrossRef]
- Macherey-Nagel Chromabond® HLB. Available online: https://www.mn-net.com/spe-columns-chromabond-hlb-30-m-6-mL/150-mg-730944p30 (accessed on 20 January 2020).
- Macherey-Nagel Macherey-Nagel Application Database. Available online: https://ftp.mn-net.com/english/Flyer_Catalogs/Chromatography/SPE/KATEN200184_Brochure_Modern_polymeric_SPE_phases_www.pdf (accessed on 20 January 2020).
- Jiménez-Skrzypek, G.; González-Sálamo, J.; Varela-Martínez, D.A.; González-Curbelo, M.Á.; Hernández-Borges, J. Analysis of phthalic acid esters in sea water and sea sand using polymer-coated magnetic nanoparticles as extraction sorbent. J. Chromatogr. A 2019, 1611, 460620. [Google Scholar] [CrossRef]
- Macherey-Nagel Standard SPE Procedure for Chromabond® HLB. Available online: https://ftp.mn-net.com/english/Flyer_Catalogs/Chromatography/SPE/KATEN200184_Brochure_Modern_polymeric_SPE_phases_www.pdf (accessed on 20 January 2020).
- González-Sálamo, J.; González-Curbelo, M.Á.; Socas-Rodríguez, B.; Hernández-Borges, J.; Rodríguez-Delgado, M.Á. Determination of phthalic acid esters in water samples by hollow fiber liquid-phase microextraction prior to gas chromatography tandem mass spectrometry. Chemosphere 2018, 201, 254–261. [Google Scholar] [CrossRef]
- Rahman, M.M.; Abd El-Aty, A.M.; Shim, J.-H. Matrix enhancement effect: A blessing or a curse for gas chromatography?—A review. Anal. Chim. Acta 2013, 801, 14–21. [Google Scholar] [CrossRef]
- Kwon, H.; Lehotay, S.J.; Geis-Asteggiante, L. Variability of matrix effects in liquid and gas chromatography-mass spectrometry analysis of pesticide residues after QuEChERS sample preparation of different food crops. J. Chromatogr. A 2012. [Google Scholar] [CrossRef]
- Varela-Martínez, D.A.; González-Curbelo, M.Á.; González-Sálamo, J.; Hernández-Borges, J. High-throughput analysis of pesticides in minor tropical fruits from Colombia. Food Chem. 2019, 280, 221–230. [Google Scholar] [CrossRef]
- Kmellár, B.; Fodor, P.; Pareja, L.; Ferrer, C.; Martínez-Uroz, M.A.; Valverde, A.; Fernandez-Alba, A.R. Validation and uncertainty study of a comprehensive list of 160 pesticide residues in multi-class vegetables by liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2008, 1215, 37–50. [Google Scholar] [CrossRef]
- SANTE/11813/2017 Guidance document on analytical quality control and validation procedures for pesticide residues analysis in food and feed. Eur. Comm. Heal. Consum. Prot. Dir. 2017.
- González-Sálamo, J.; González-Curbelo, M.Á.; Hernández-Borges, J.; Rodríguez-Delgado, M.Á. Use of Basolite ® F300 metal-organic framework for the dispersive solid-phase extraction of phthalic acid esters from water samples prior to LC-MS determination. Talanta 2019, 195, 236–244. [Google Scholar] [CrossRef] [PubMed]
- González-Sálamo, J.; Socas-Rodríguez, B.; Hernández-Borges, J.; Rodríguez-Delgado, M.Á. Determination of phthalic acid esters in water samples using core-shell poly(dopamine) magnetic nanoparticles and gas chromatography tandem mass spectrometry. J. Chromatogr. A 2017, 1530, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Malem, F.; Soonthondecha, P.; Khawmodjod, P.; Chunhakorn, V.; Whitlow, H.J.; Chienthavorn, O. Occurrence of phthalate esters in the eastern coast of Thailand. Environ. Monit. Assess. 2019, 191, 627. [Google Scholar] [CrossRef] [PubMed]
- Paluselli, A.; Fauvelle, V.; Schmidt, N.; Galgani, F.; Net, S.; Sempéré, R. Distribution of phthalates in Marseille Bay (NW Mediterranean Sea). Sci. Total Environ. 2018, 621, 578–587. [Google Scholar] [CrossRef]
- Directive 2013/39/EU Of The European Parliament And Of The Council of 12 August 2013 amending Directives 2000/60/EC and 2008/105/EC as regards priority substances in the field of water policy. Off. J. Eur. J. 2013, L226, 1–17.
- Gao, D.; Li, Z.; Wen, Z.; Ren, N. Occurrence and fate of phthalate esters in full-scale domestic wastewater treatment plants and their impact on receiving waters along the Songhua River in China. Chemosphere 2014, 95, 24–32. [Google Scholar] [CrossRef]
- Katsikantami, I.; Sifakis, S.; Tzatzarakis, M.N.; Vakonaki, E.; Kalantzi, O.-I.; Tsatsakis, A.M.; Rizos, A.K. A global assessment of phthalates burden and related links to health effects. Environ. Int. 2016, 97, 212–236. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).