Advances and Pitfalls in the Capillary Electrophoresis Analysis of Aggregates of Beta Amyloid Peptides

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Samples and Reagents Preparation
2.3. CE-LIF
3. Results
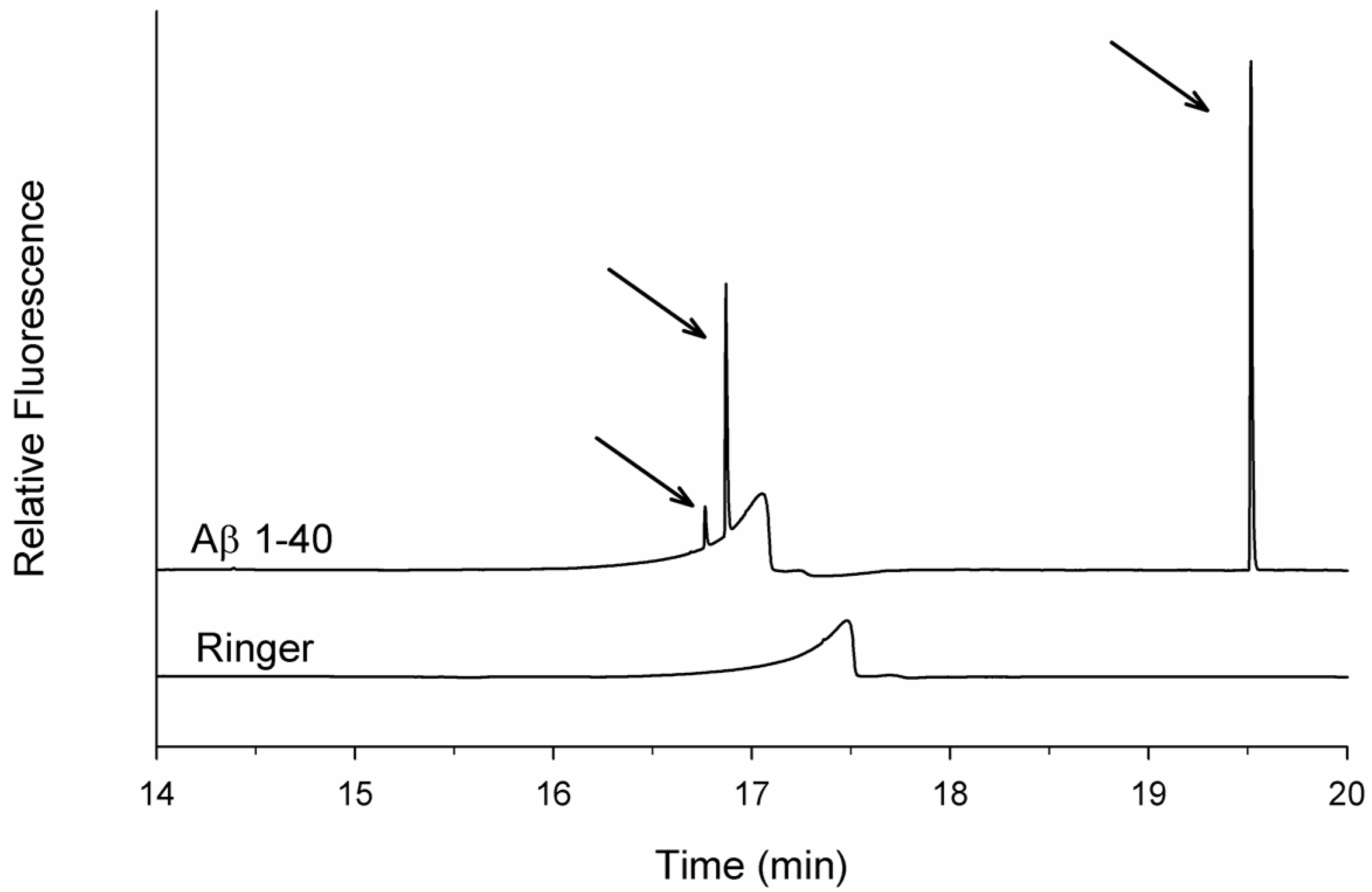
3.1. Spikes Are Present in Samples of Incubated Aβ 1-40 and Reveal Aggregates
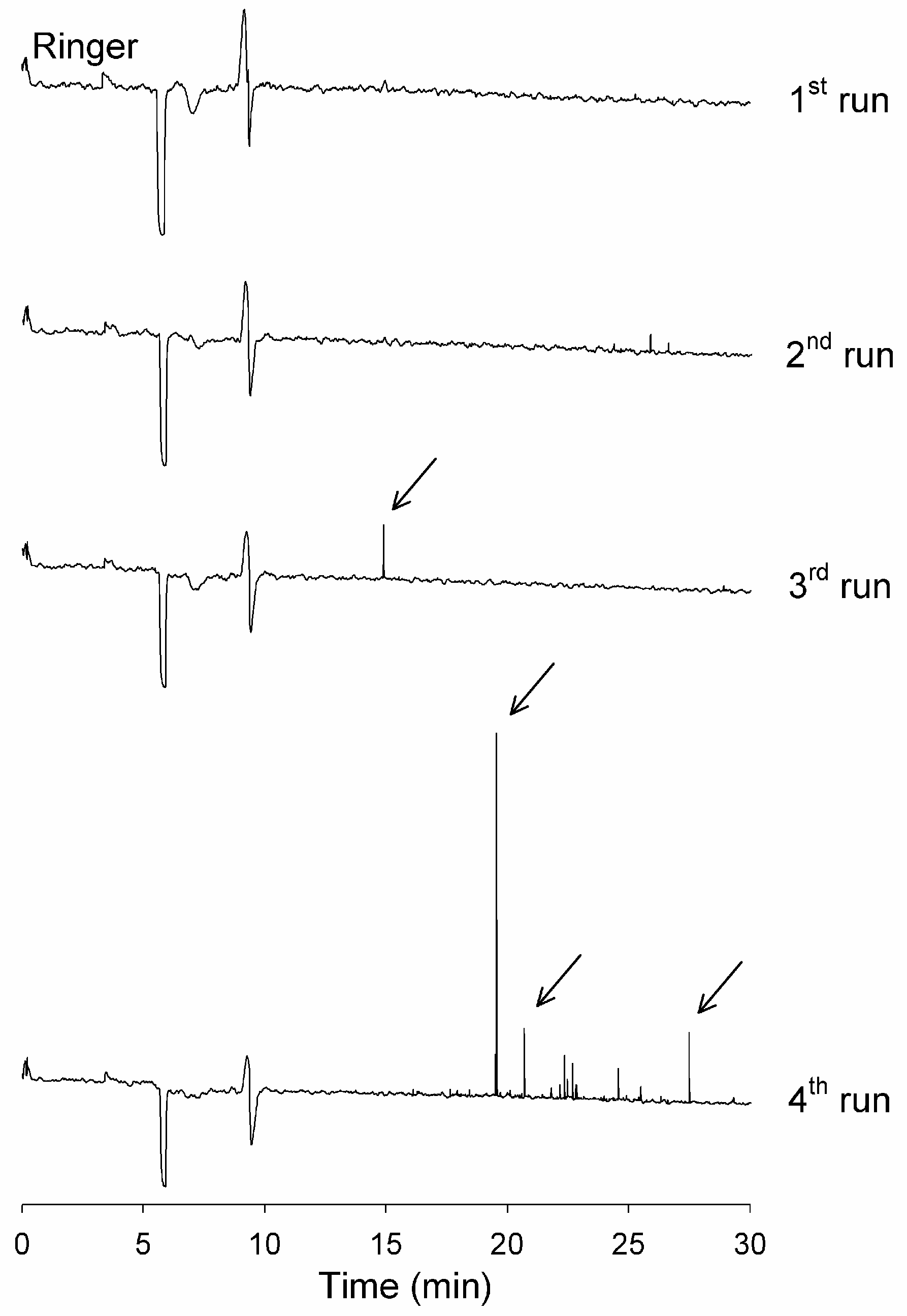
3.2. Analytical Instrument Can Generate Artefactual Spikes During the Analysis of Incubated Aβ
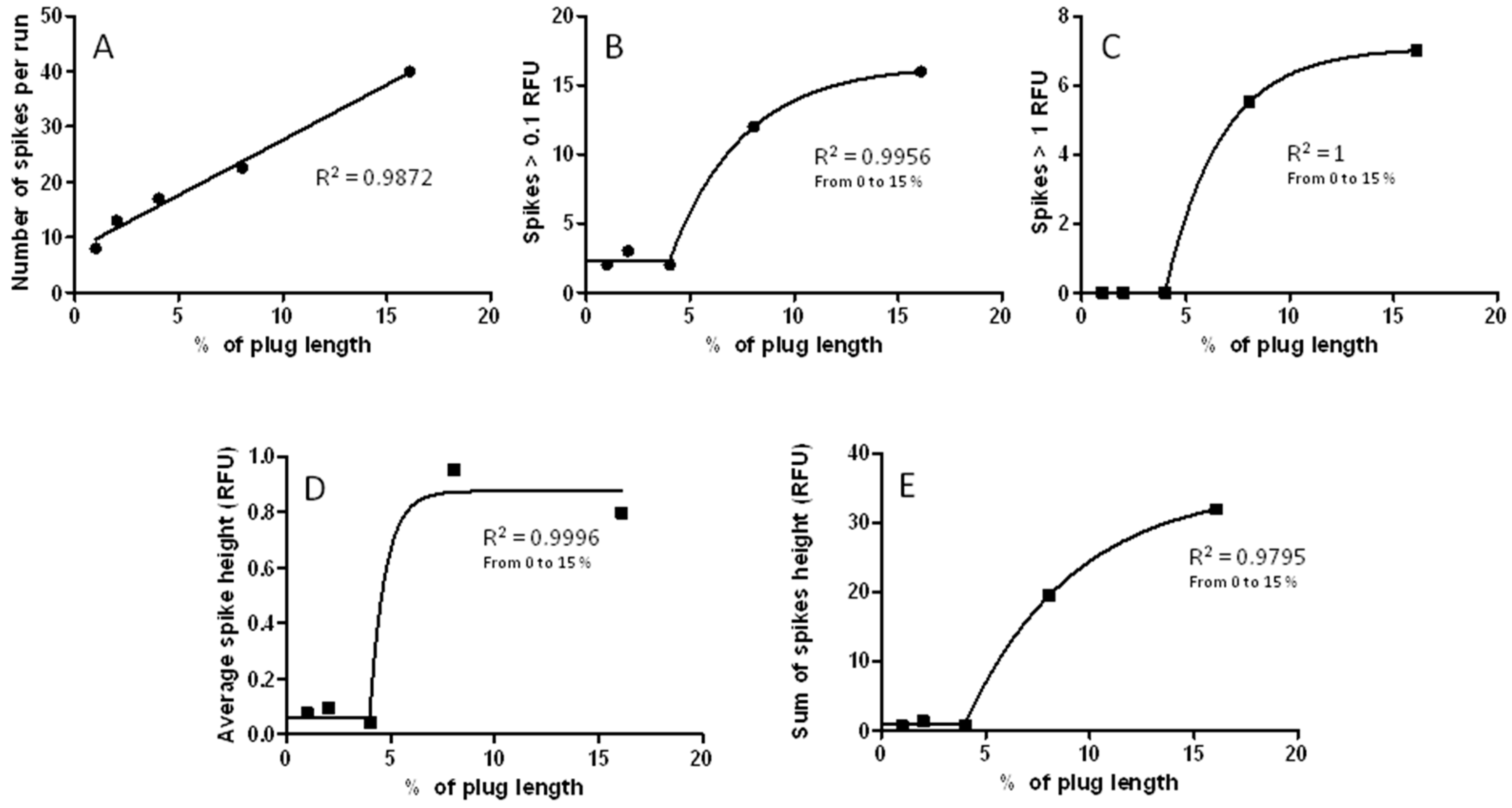
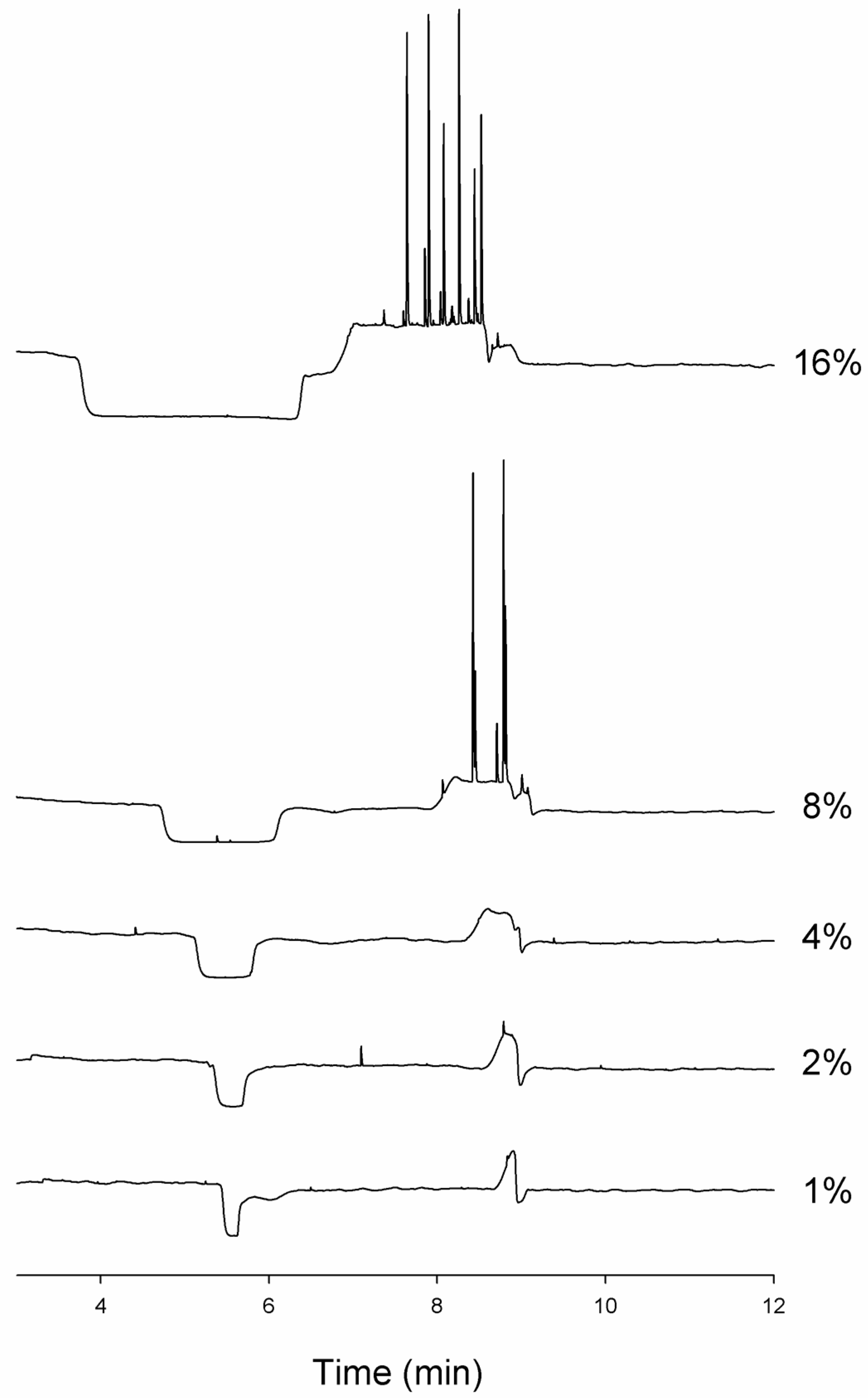
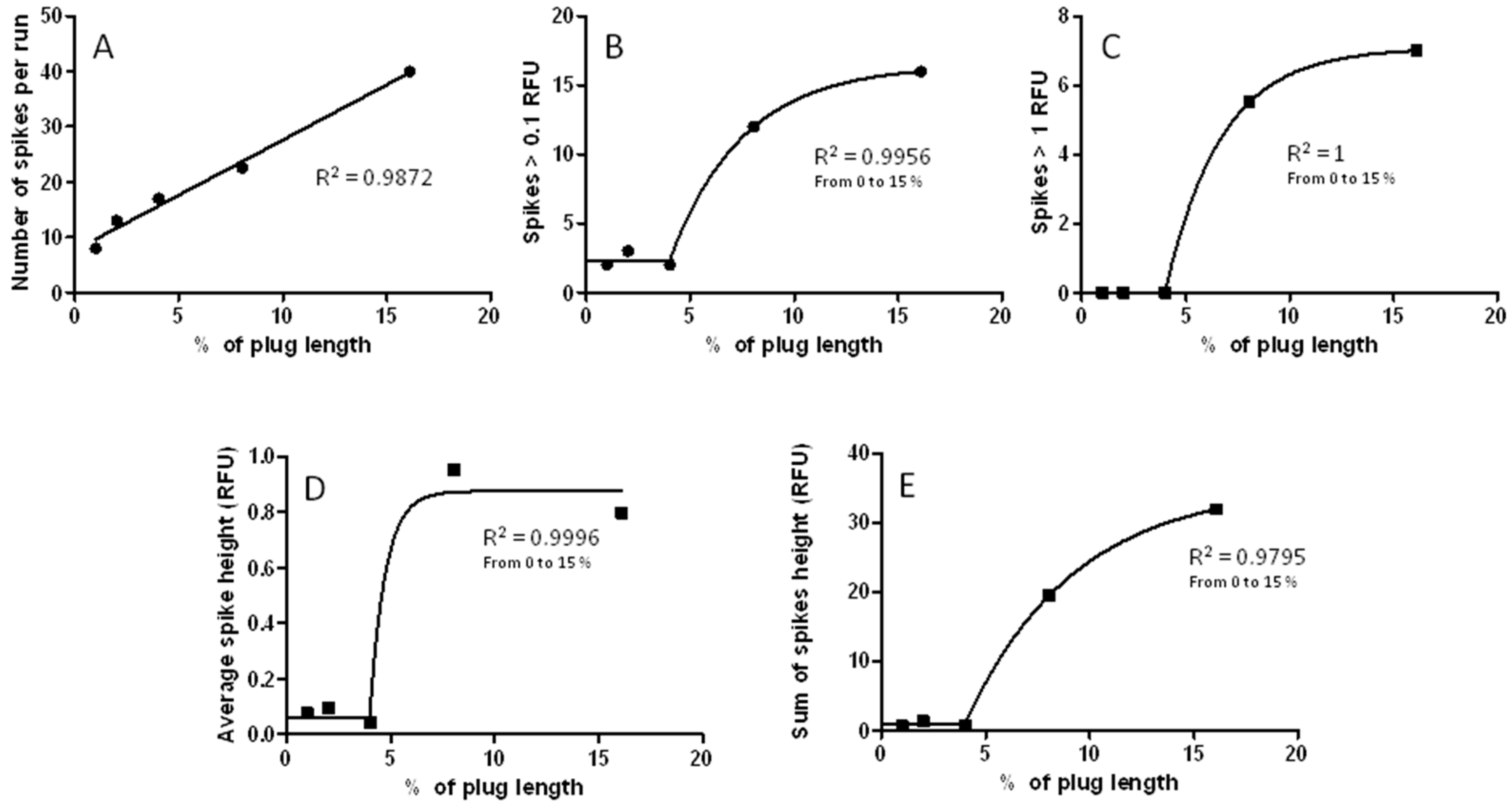
3.3. Changes in Injection Volume Reveal That Spikes of Incubated Aβ 1-40 Exhibit Particle-Like Electrophoresis Behavior
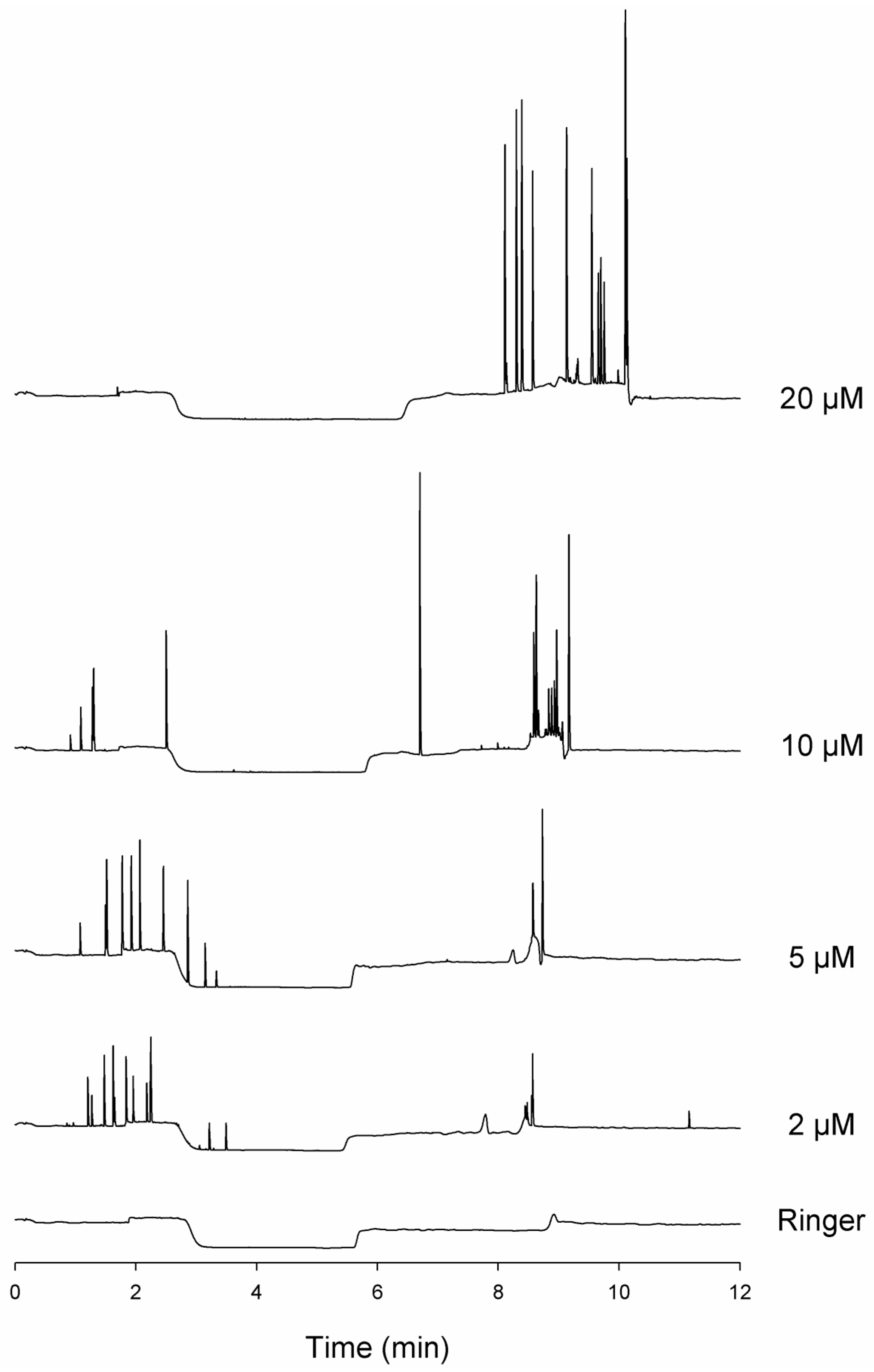
3.4. The Nature of Running Buffer Affects Spikes Detection
3.5. The Link between Spikes and Aggregates Is Supported by Ultrasound-Treatment of Samples
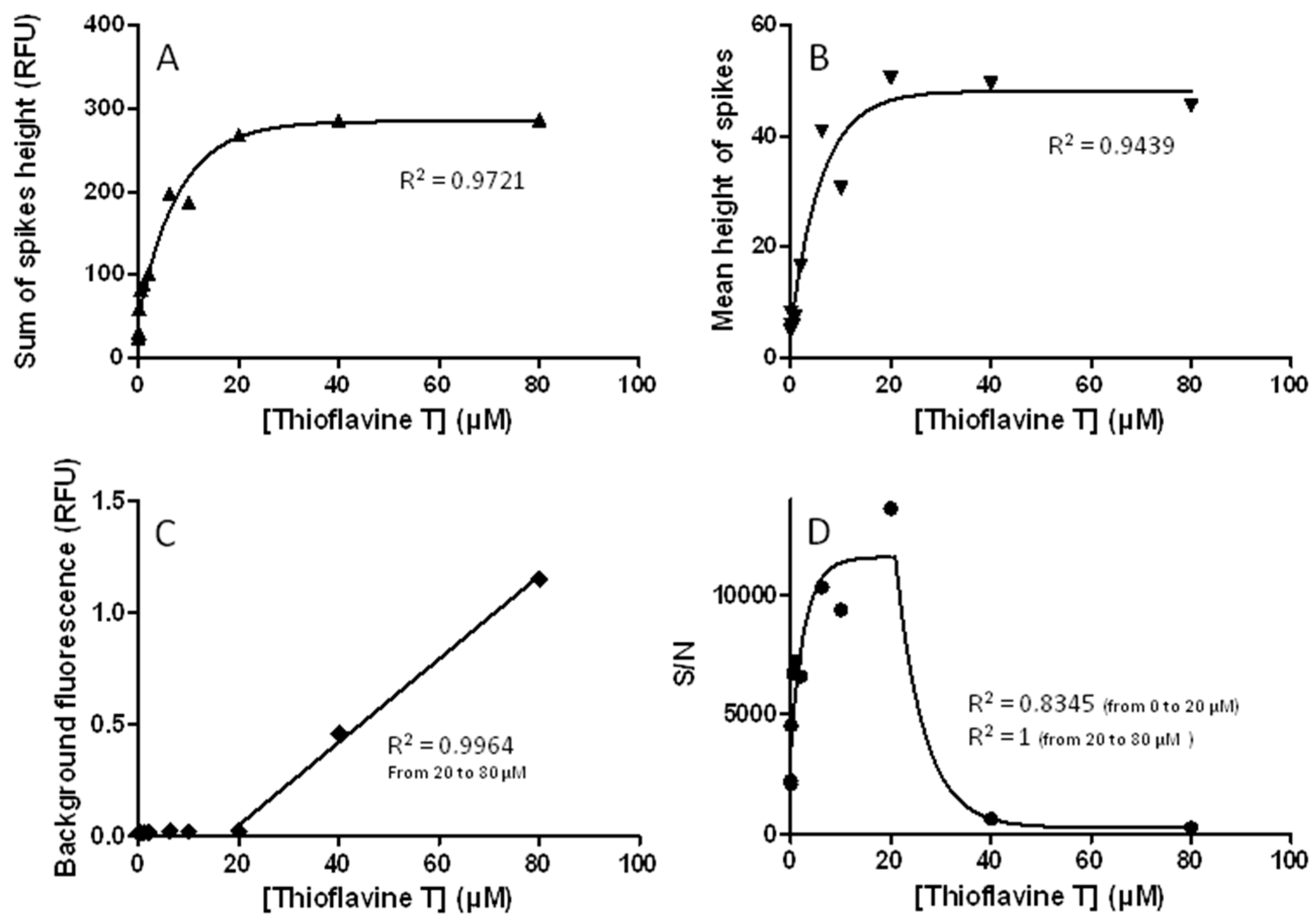
3.6. The Use of Thioflavine T in Running Buffer Must Be Carefully Controlled and Validated
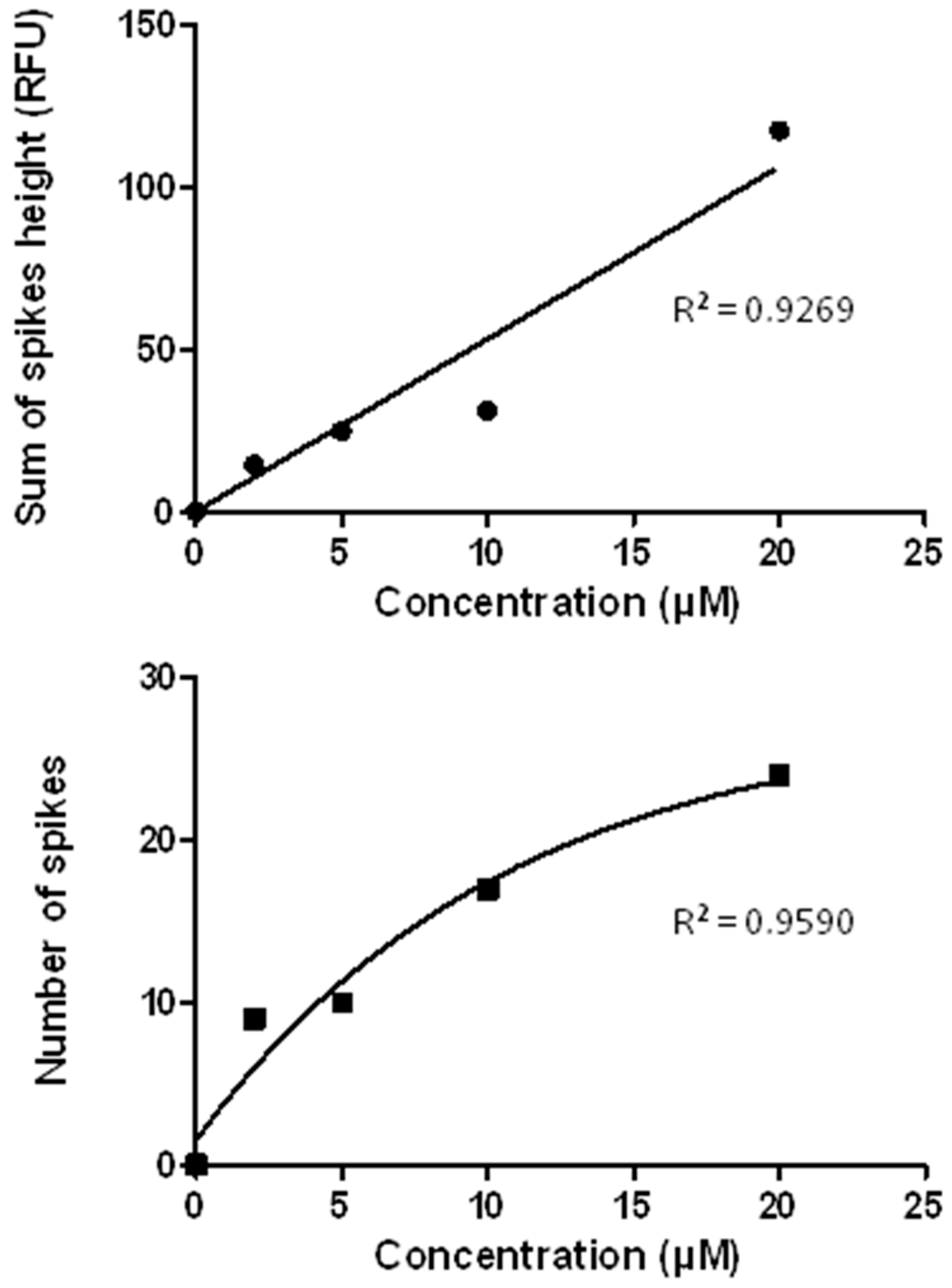
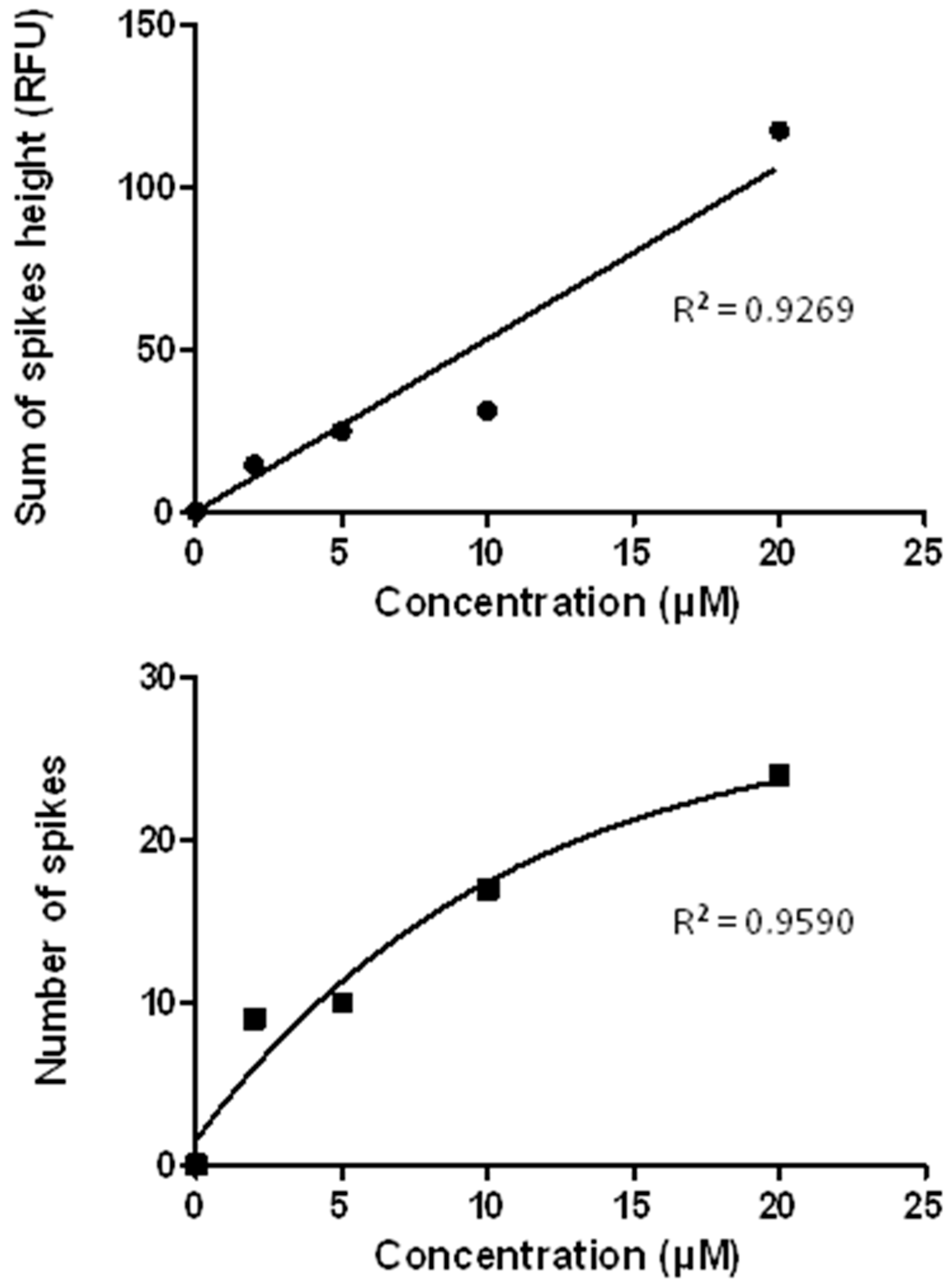
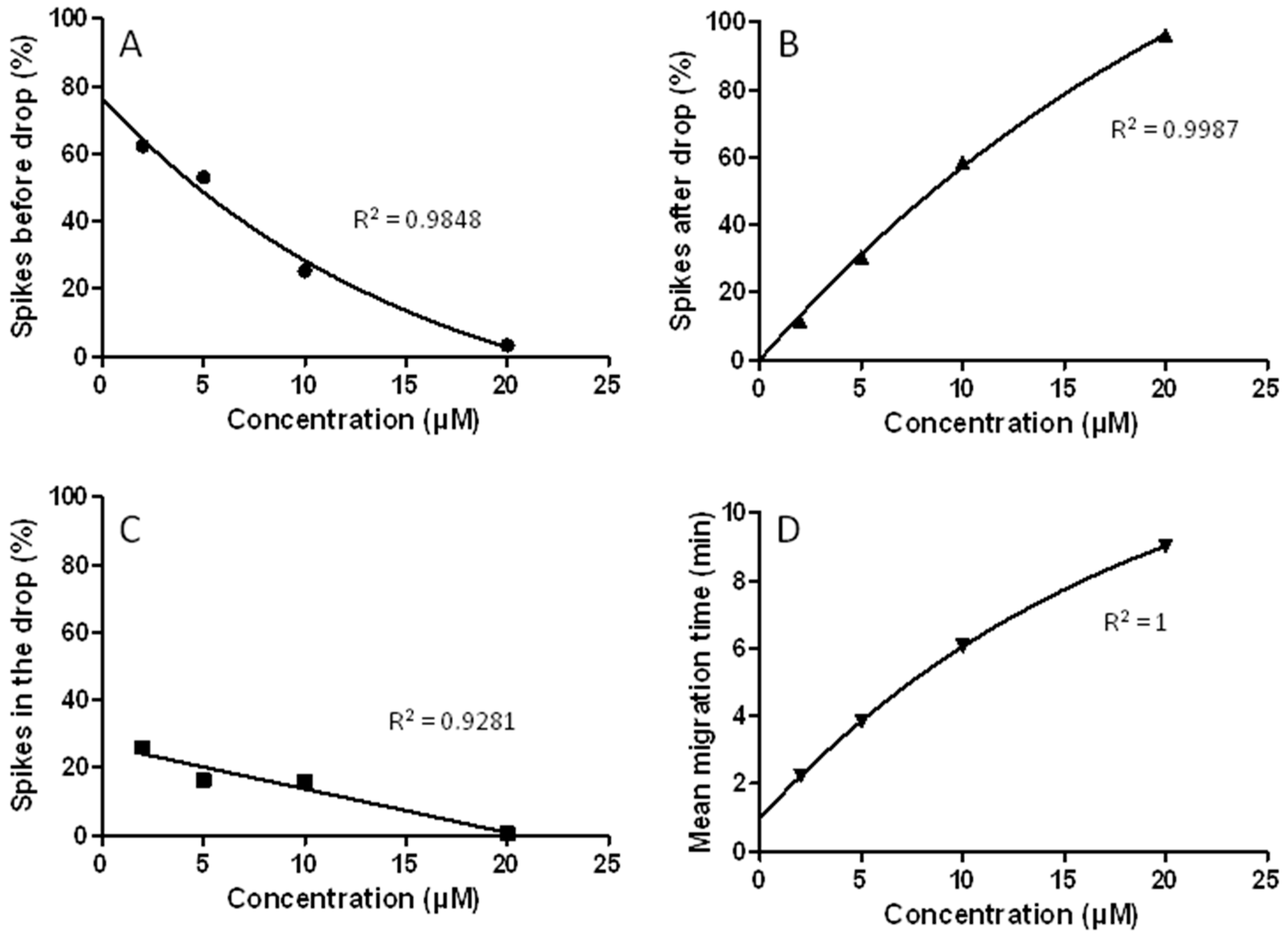
3.7. The CE-LIF Separation of Aβ Aggregates Exhibits Quantification Capabilities
3.8. This CE-LIF Method Allows Separation ofAβ Aggregates of Different Size
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Roher, A.; Wolfe, D.; Palutke, M.; KuKuruga, D. Purification, ultrastructure, and chemical analysis of Alzheimer disease amyloid plaque core protein. Proc. Natl. Acad. Sci. USA 1986, 83, 2662–2666. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.D.; Wong, S.S.; Lieber, C.M.; Lansbury, P.T. Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chem. Biol. 1997, 4, 119–125. [Google Scholar] [CrossRef]
- Walsh, D.M.; Lomakin, A.; Benedek, G.B.; Condron, M.M.; Teplow, D.B. Amyloid β-protein fibrillogenesis detection of a protofibrillar intermediate. J. Boil. Chem. 1997, 272, 22364–22372. [Google Scholar] [CrossRef]
- Fandrich, M. Oligomeric intermediates in amyloid formation: Structure determination and mechanisms of toxicity. J. Mol. Biol. 2012, 421, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Sakono, M.; Zako, T. Amyloid oligomers: Formation and toxicity of Aβ oligomers. FEBS J. 2010, 277, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, A.; Yankner, B.A. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc. Natl. Acad. Sci. USA 1994, 91, 12243–12247. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Motter, R.; Vigo-Pelfrey, C.; Kholodenko, D.; Barbour, R.; Johnson-Wood, K.; Galasko, D.; Chang, L.; Miller, B.; Clark, C.; Green, R.; et al. Reduction of β-amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer’s disease. Ann. Neurol. 1995, 38, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Kanai, M.; Matsubara, E.; Isoe, K.; Urakami, K.; Nakashima, K.; Arai, H.; Sasaki, H.; Abe, K.; Iwatsubo, T.; Kosaka, T.; et al. Longitudinal study of cerebrospinal fluid levels of tau, Aβ 1-40, and Aβ 1-42(43) in Alzheimer’s disease: A study in Japan. Ann. Neurol. 1998, 44, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Welge, V.; Fiege, O.; Lewczuk, P.; Mollenhauer, B.; Esselmann, H.; Klafki, H.W.; Wolf, S.; Trenkwalder, C.; Otto, M.; Kornhuber, J.; et al. Combined CSF tau, p-tau181 and amyloid-β 38/40/42 for diagnosing Alzheimer’s disease. J. Neural Transm. (Vienna) 2009, 116, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Verwey, N.A.; Veerhuis, R.; Twaalfhoven, H.A.; Wouters, D.; Hoozemans, J.J.; Bollen, Y.J.; Killestein, J.; Bibl, M.; Wiltfang, J.; Hack, C.E.; et al. Quantification of amyloid-beta 40 in cerebrospinal fluid. J. Immunol. Methods 2009, 348, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Wiltfang, J.; Esselmann, H.; Bibl, M.; Smirnov, A.; Otto, M.; Paul, S.; Schmidt, B.; Klafki, H.W.; Maler, M.; Dyrks, T.; et al. Highly conserved and disease-specific patterns of carboxyterminally truncated Aβ peptides 1-37/38/39 in addition to 1-40/42 in Alzheimer’s disease and in patients with chronic neuroinflammation. J. Neurochem. 2002, 81, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Verpillot, R.; Esselmann, H.; Mohamadi, M.R.; Klafki, H.; Poirier, F.; Lehnert, S.; Otto, M.; Wiltfang, J.; Jean-Louis, V.; Taverna, M. Analysis of amyloid-β peptides in cerebrospinal fluid samples by capillary electrophoresis coupled with LIF detection. Anal. Chem. 2011, 83, 1696–1703. [Google Scholar] [CrossRef] [PubMed]
- Naiki, H.; Higuchi, K.; Hosokawa, M.; Takeda, T. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavinT. Anal. Biochem. 1989, 177, 244–249. [Google Scholar] [CrossRef]
- Bartolini, M.; Naldi, M.; Fiori, J.; Valle, F.; Biscarini, F.; Nicolau, D.V.; Andrisano, V. Kinetic characterization of amyloid-beta 1-42 aggregation with a multimethodological approach. Anal. Biochem. 2011, 414, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Verpillot, R.; Otto, M.; Klafki, H.; Taverna, M. Simultaneous analysis by capillary electrophoresis of five amyloid peptides as potential biomarkers of Alzheimer’s disease. J. Chromatogr. A 2008, 1214, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Picou, R.; Moses, J.P.; Wellman, A.D.; Kheterpal, I.; Gilman, S.D. Analysis of monomeric Aβ (1-40) peptide by capillary electrophoresis. Analyst 2010, 135, 1631–1635. [Google Scholar] [CrossRef] [PubMed]
- Sabella, S.; Quaglia, M.; Lanni, C.; Racchi, M.; Govoni, S.; Caccialanza, G.; Calligaro, A.; Bellotti, V.; De Lorenzi, E. Capillary electrophoresis studies on the aggregation process of β-amyloid 1-42 and 1-40 peptides. Electrophoresis 2004, 25, 3186–3194. [Google Scholar] [CrossRef] [PubMed]
- Colombo, R.; Carotti, A.; Catto, M.; Racchi, M.; Lanni, C.; Verga, L.; Caccialanza, G.; De Lorenzi, E. CE can identify small molecules that selectively target soluble oligomers of amyloid β protein and display antifibrillogenic activity. Electrophoresis 2009, 30, 1418–1429. [Google Scholar] [CrossRef] [PubMed]
- Brinet, D.; Kaffy, J.; Oukacine, F.; Glumm, S.; Ongeri, S.; Taverna, M. An improved capillary electrophoresis method for in vitro monitoring of the challenging early steps of Aβ 1-42 peptide oligomerization: Application to anti-Alzheimer’s drug discovery. Electrophoresis 2014, 35, 3302–3309. [Google Scholar] [CrossRef] [PubMed]
- Paracha, S.; Hestekin, C. Field amplified sample stacking of amyloid beta (1-42) oligomers using capillary electrophoresis. Biomicrofluidics 2016, 10, 033105. [Google Scholar] [CrossRef] [PubMed]
- Pryor, N.E.; Moss, M.A.; Hestekin, C.N. Capillary electrophoresis for the analysis of the effect of sample preparation on early stages of Aβ 1-40 aggregation. Electrophoresis 2014, 35, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Picou, R.A.; Kheterpal, I.; Wellman, A.D.; Minnamreddy, M.; Ku, G.; Gilman, S.D. Analysis of Aβ (1-40) and Aβ (1-42) monomer and fibrils by capillary electrophoresis. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Kinoshita, H.; Enokita, M.; Hori, Y.; Hashimoto, T.; Iwatsubo, T.; Toyo’oka, T. Analytical method for β-amyloid fibrils using CE-laser-induced fluorescence and its application to screening for inhibitors of β-amyloid protein aggregation. Anal. Chem. 2007, 79, 4887–4891. [Google Scholar] [CrossRef] [PubMed]
- Picou, R.A.; Schrum, D.P.; Ku, G.; Cerqua, R.A.; Kheterpal, I.; Gilman, S.D. Separation and detection of individual Aβ aggregates by capillary electrophoresis with laser-induced fluorescence detection. Anal. Biochem. 2012, 425, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Duffy, C.F.; McEathron, A.A.; Arriaga, E.A. Determination of individual microsphere properties by capillary electrophoresis with laser-induced fluorescence detection. Electrophoresis 2002, 23, 2040–2047. [Google Scholar] [CrossRef]
- Gunasekera, N.; Musier-Forsyth, K.; Arriaga, E. Electrophoretic behavior of individual nuclear species as determined by capillary electrophoresis with laser-induced fluorescence detection. Electrophoresis 2002, 23, 2110–2116. [Google Scholar] [CrossRef]
- Fuller, K.M.; Arriaga, E.A. Analysis of individual acidic organelles by capillary electrophoresis with laser-induced fluorescence detection facilitated by the endocytosis of fluorescently labeled microspheres. Anal. Chem. 2003, 75, 2123–2130. [Google Scholar] [CrossRef] [PubMed]
- Dziubakiewicz, E.; Buszewski, B. Capillary electrophoresis of microbial aggregates. Electrophoresis 2014, 35, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Breadmore, M.C. Recent advances in enhancing the sensitivity of electrophoresis and electrochromatography in capillaries and microchips. Electrophoresis 2007, 28, 254–281. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, F.; Otsuka, K. Recent applications of on-line sample preconcentration techniques in capillary electrophoresis. J. Chromatogr. A 2014, 1335, 43–60. [Google Scholar] [CrossRef] [PubMed]
- Svobodova, Z.; Reza Mohamadi, M.; Jankovicova, B.; Esselmann, H.; Verpillot, R.; Otto, M.; Taverna, M.; Wiltfang, J.; Viovy, J.L.; Bilkova, Z. Development of a magnetic immunosorbent for on-chip preconcentration of amyloid β isoforms: Representatives of Alzheimer’s disease biomarkers. Biomicrofluidics 2012, 6. [Google Scholar] [CrossRef] [PubMed]
- Mai, T.D.; Pereiro, I.; Hiraoui, M.; Viovy, J.L.; Descroix, S.; Taverna, M.; Smadja, C. Magneto-immunocapture with on-bead fluorescent labeling of amyloid-β peptides: Towards a microfluidized-bed-based operation. Analyst 2015, 140, 5891–5900. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Denoroy, L.; Parrot, S. Advances and Pitfalls in the Capillary Electrophoresis Analysis of Aggregates of Beta Amyloid Peptides. Separations 2018, 5, 2. https://doi.org/10.3390/separations5010002
Denoroy L, Parrot S. Advances and Pitfalls in the Capillary Electrophoresis Analysis of Aggregates of Beta Amyloid Peptides. Separations. 2018; 5(1):2. https://doi.org/10.3390/separations5010002
Chicago/Turabian StyleDenoroy, Luc, and Sandrine Parrot. 2018. "Advances and Pitfalls in the Capillary Electrophoresis Analysis of Aggregates of Beta Amyloid Peptides" Separations 5, no. 1: 2. https://doi.org/10.3390/separations5010002
APA StyleDenoroy, L., & Parrot, S. (2018). Advances and Pitfalls in the Capillary Electrophoresis Analysis of Aggregates of Beta Amyloid Peptides. Separations, 5(1), 2. https://doi.org/10.3390/separations5010002