1. Introduction
Antipsychotic drugs are a class of psychiatric medication primarily used to manage psychotic symptoms principally in schizophrenia, bipolar disorder and other psycho-organic disorders. Based on World Health Organization, sixty four compounds are classified as antipsychotics and for about 70% of these, analytical methods have been developed to determine them in human matrices [
1].
Typical antipsychotics, known as first-generation antipsychotics were primarily discovered at 1950s and they tend to act on D
2 and D
4 receptors in the dopamine pathways of the brain. Those drugs are used much less frequently now because they show severe side-effects. For this reason second-generation antipsychotics, or atypical antipsychotics have been developed. Those drugs have a tendency to act less to those receptors, and therefore they show less side-effects than the primarily used typical antipsychotic drugs [
2].
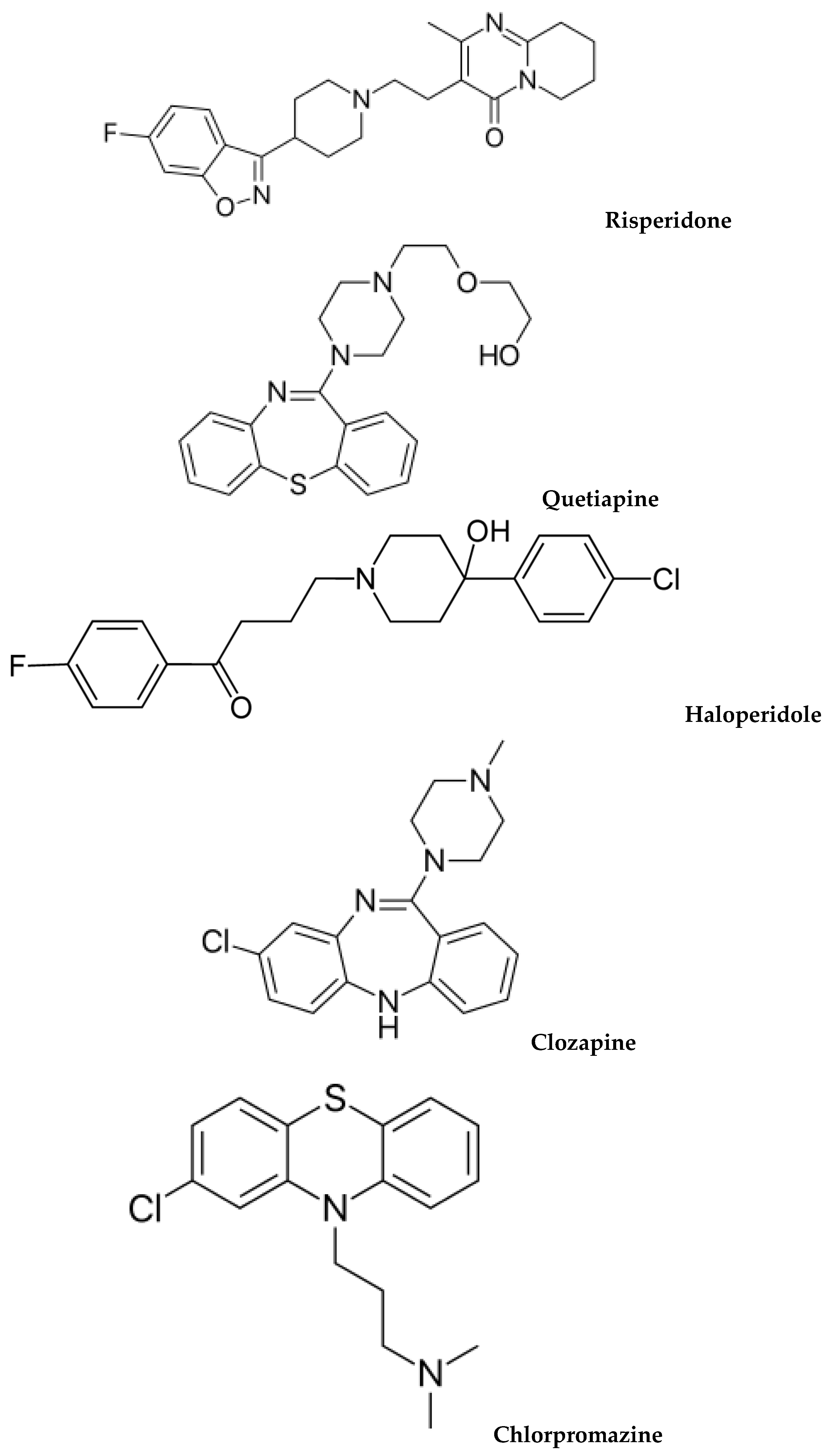
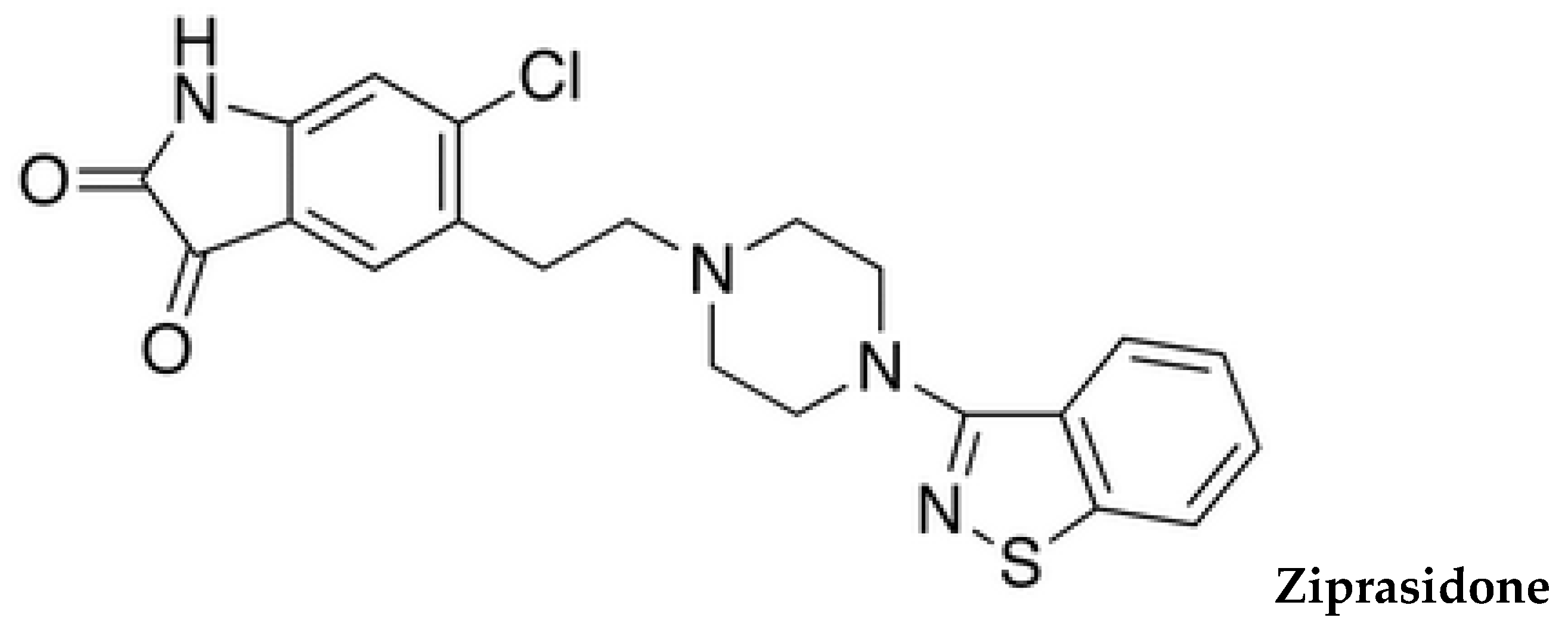
Figure 1 illustrates the chemical structures of common antipsychotics, which are currently used.
Due to the wide use of these drugs worldwide, there is a great need of analytical methods in order to analyze biological samples. The quantitative determination of antipsychotics in human matrices is of great interest both for therapeutic drug monitoring and for forensic toxicology [
1].
The modern trend in drug analysis is shifting from gas chromatography to liquid chromatography not only because of its good quantitative results, its high reproducibility, sensitivity and wide applicability, but also because most antipsychotic drugs are not volatile. For the moment ultrahigh performance liquid chromatography-tandem mass spectrometry UHPLC-MS/MS is the most preferred technique for the separation and analysis of antipsychotic drugs in biofluids.
Conventional matrices used for this purpose are serum, plasma and whole blood. However, other alternative matrices like oral fluid and urine; which are easily collected, keratinized matrices namely hair and nails; which are stable and capable of providing information for long periods of time, dry blood spots (DBS) and cerebrospinal fluid, are widely used [
1].
Due to the high complexity of biological materials, which often contain proteins, salts, organic compounds with similar properties to the analytes and other endogenous compounds that may deteriorate the performance of separation, a sample preparation procedure is required. An ideal sample preparation technique should be fast and comprise the minimum number of working steps, should be easy to learn and easy to use, should be economical and environmental friendly and should be compatible with many analytical instruments [
3].
The four major sample preparation techniques used for those matrices are liquid–liquid extraction (LLE), solid-phase extraction (SPE), protein precipitation (PP) and direct injection. However, these conventional techniques tend to have many fundamental drawbacks because they include complicated, time-consuming steps and they require large amounts of sample and organic solvents, while there are many difficulties in automation [
1,
3].
Therefore, there is a great need of developing novel, relatively simple, fast and solvent-free microextraction procedures which use smaller volumes of samples and solvents (microliter range or even smaller) and can be widely used to analyze these samples. To date, there is a big number of different microextraction techniques which are used for sample preparation of biological fluids and other biological matrices in order to enhance compatibility with modern analytical instrumentation, as well as to minimize the use of toxic chemicals and to decrease the size of biofluids or reagents’ demand.
In this review we aim to present an overview of microextraction techniques which are used prior to liquid chromatography analysis in order to analyze biological fluids and to detect and quantify antipsychotics in conventional and alternative biological matrices.
2. Solid Phase Microextraction for the Determination of Antipsychotic Drugs in Biological Samples
Solid-phase microextraction (SPME) is an efficient solvent-free sample preparation method which was first introduced in the early 1990s by Pawliszyn and co-workers. It enables automation, miniaturization and high-throughput performance. This technique uses fibers and capillary tubes coated by stationary phases and it can be applied to samples in any state of matter gaseous, liquid and solid. The technique is based on partitioning of the analytes between the sample matrix and the extraction phase which is immobilized on a fused-silica SPME fiber coated with polymers, until the equilibrium is reached and subsequent thermal desorption of the extracts into a gas chromatograph, reconstitution in the mobile phase used for a separation with a liquid chromatograph, or direct injection to an HPLC injection port using suitable interface [
4].
The type of the polymers which are used depends on the properties of the analyte. For drug analysis of biological matrices the most common coatings are the polydimethylsiloxane (PDMS) and polyacrylate (PA) while other polymers such as polypyrrole coatings, coatings based on restricted access materials, and those based on mixtures of biocompatible polymers with sorbents used for SPE have also been developed and used [
5,
6].
The most widely used technique is fiber-SPME. In this technique the analyte is directly extracted onto the coating of the fiber which is usually is inside a needle in a device with an assembly holder. For the procedure of SPME the sample is placed in a capped vial with a septum which is pierced by the needle of the device followed by the extension of the fiber either to the vapor above the sample (Head-Space SPME) for volatile analytes or directly to the sample (Direct Immersion-SPME) for the extraction of non-volatile analytes until equilibrium is reached. An alternative microextraction technique is in-tube SPME that uses a fused-silica capillary column. The extraction of the analytes takes place either onto the inner coating of the fiber or onto a sorbent bed. Compared to fiber SPME, in-tube SPME is more mechanically stable and can be used with on line coupling with HPLC or LC/MS instruments [
7].
Various SPME methods have been developed for the analysis of antipsychotic drugs in biological matrices prior to liquid chromatography analysis.
Theodoridis et al. developed a method for the determination of a typical antipsychotic; haloperidol, together with other four drugs: quinine, naproxen, ciprofloxacin and paclitaxel in urine. Each analyte was studied independently. Haloperidol was determined using an Analyticals Erbasil Symmetry C
18 column, with a mixture of 0.05 M aqueous ammonium acetate and acetonitrile (35:65
v/
v) as a mobile phase, while the detection was accomplished with a UV detector at 210 nm. For the SPME procedure, a PDMS 100 μm fiber was conditioned for 30 min in a GC injector operating at 250 °C. Then, 4 mL of a solution of each pharmaceutical in buffer (20 μg/mL in 0.9% NaCl, pH 9) was transferred in a glass vial containing a magnetic stirring bar, which was then capped and the sample was agitated at 700 rpm. Multiple SPME was also applied, but it did not provide yield enhancement for haloperidol, despite that this technique is supposed to be an excellent way to increase extraction yields. At the end of the extraction, the analyte was desorbed in 200 μL of methanol and an aliquot of 80 μL was injected to the HPLC. This method overcomes the problems that exist in conventional sample preparation techniques and can be applied to real urine samples [
5].
In 2012, Bocato et al. published a method for the analysis of paliperidone after stereoselective fungal biotransformation of the atypical antipsychotic drug risperidone with SPME extraction prior to HPLC-MS/MS analysis. Paliperidone or 9-hydroxyrisperidone (9-RispOH) is characterized by the same pharmacologic activity of the parent drug risperidone. Another metabolite of risperidone, 7-hydroxyrisperidone, was also included in this study. The chromatographic separation was achieved using a Chiralcel OJ-H column with a mixture of methanol: ethanol (50:50,
v/
v) plus 0.2% triethylamine as a mobile phase at a flow rate of 0.8 mL·min
−1. Firstly, the SPME C
18 fiber probe 45 μm which was selected, was conditioned for 30 min with methanol and water (50:50,
v/
v). Extraction was performed by immersing the fiber in the fungal sample, the pH of which was controlled with a phosphate buffer (pH 7) and also 20% NaCl (
w/
v) was added. The addition of the electrolyte reduces its solubility of the organic analyte and increases its extraction yield. The adsorption lasted 30 min at room temperature with 600 rpm stirring speed following desorption in a 120 μL glass vial filled with the mobile phase. After 5 min, the fiber was withdrawn into the needle and an aliquot of 20 μL was injected into the HPLC–MS/MS system with no further treatment. To avoid carryover, after each desorption step the fiber was washed for 30 min with methanol. The obtained SPME recoveries were 28% for risperidone, 16% for 9-RispOH and 11% for 7-RispOH [
6].
Kumazawa et al. have successfully developed an HPLC-MS/MS method to determine eleven phenothiazine derivatives (clospirazine, fluphenazine, perazine, thiethylperazine, thioridazine, flupentixol, thioproperazine, trifluoperazine, perphenazine, prochlorperazine and propericiazine) in human whole blood and urine using solid-phase microextraction (SPME) with a polyacrylate-coated fiber. The pH of the samples was adjusted to about 8 with KOH solution and the vial was sealed with a silicone-rubber septum cap. The syringe needle of the SPME device was passed through the septum and the polyacrylate fiber was pushed out from the needle and immersed directly in the sample solution in the vial at 40 °C and the extraction lasted 60 min at continuous stirring at 250 rpm. The fiber was then injected into the desorption chamber of the SPME-HPLC interface. This was filled with distilled water containing 10 mM ammonium acetate plus 0.1% formic acid—100% acetonitrile (70:30,
v/
v). The desorption time was 10 min. Subsequently the entire contents of the desorption chamber were flushed directly on to the HPLC column by means of the mobile phase flow at 0.2 mL/min. This method was effectively applied to real samples after oral administration and it can be recommended for use in both therapeutic monitoring and clinical or forensic toxicology [
8].
3. Microextraction by Packed Sorbent for the Determination of Antipsychotic Drugs in Biological Samples
Another microextraction technique developed in the last decade is microextraction by packed sorbent (MEPS). This novel technique is based on the same general principle of solid phase extraction (SPE), but with MEPS the packing is integrated directly into the syringe, in a very small barrel (BIN) which sets up the needle assembly of an HPLC syringe and not in a separate SPE cartridge. The sorbents that are used in MEPS are usually the same as conventional SPE columns. Most of the applied sorbents include silica-based sorbents (C
2, C
8 and C
18). When the biological sample passes through the solid support of the syringe, the analytes are adsorbed onto the sorbent which is packed in the BIN. Because MEPS and SPE build on the same principles there is the option of transferring a method from conventional SPE to MEPS relatively straight forward [
9,
10,
11,
12].
MEPS holds the high selectivity, the good sample purifying efficiency and extraction yields of SPE. Compared to traditional sample preparation techniques like LLE and SPE, MEPS procedure is faster, simpler, cheaper, more feasible, more environmental friendly, more user-friendly and uses both small amounts of biological sample (10 μL of plasma, urine or water) and large volumes (1000 μL). Compared to protein precipitation, this microextraction procedure is much more efficient. In general, MEPS can reduce sample volume and time necessary for the analysis. Moreover it can be fully automated and it can be connected to liquid chromatography (LC), gas chromatography (GC) or capillary electrochromatography (CEC) [
9,
13].
The most important factors in MEPS performance, which should be optimized before the sample analysis are conditioning, loading, washing and eluting solvents, sample flow rate, washing solution and the type and volume of the elution, which should be suitable for injection into LC or GC systems. Also, the volume of the sample should be optimized leading to the best equilibrium between a good analytical performance and a good extraction methodology. The optimum conditions will be contingent to the nature of the matrix being used and the retention capacity and specificity of the sorbent in order to obtain the highest recovery of the analytes [
9].
To date, there are several MEPS methods that have been developed for the determination of antipsychotic drugs in biological matrices prior to liquid chromatography analysis.
In 2010 Saracino et al. aimed to develop an analytical method for the determination of an atypical antipschycotic drug; risperidone and its main active metabolite 9-hydroxyrisperidone in human plasma and saliva based on HPLC with coulometric detection and an innovative MEPS procedure. Those two analytes were also studied with a SPME procedure prior to HPLC-MS/MS analysis. The microextraction procedure was carried out using a BIN containing 4 mg of solid-phase material silica-C
8, after being activated with 100 μL of methanol for three times and conditioning with 100 μL of water for another three times, at a flow rate of 20 μL/s. For the extraction of the analytes, the samples were drawn up and down through the syringe 15 times (at a flow rate of 5 μL/s) without discarding and a washing step once with water (100 μL) and once with a mixture of water and methanol (95:5,
v/
v) took place, in order to remove biological interference from the samples. Then, the analytes were eluted with 250 μL of methanol and they were subsequently separated on a reversed phase C
18 column, using a mobile phase composed of acetonitrile (26%) and a pH 6.5 phosphate buffer (74%).After extraction, the sorbent was cleaned similarly to the activating and conditioning step in order to decrease memory effects and to condition for the next extraction. The same sorbent was used for about 50 extractions. The limit of quantitation for the two compounds was 0.5 ng/mL, while the limit of detection was 0.17 ng/mL. Extraction yields were higher than 90.1% and intra-day and inter-day precision results were good. As a result, this method was successfully applied to real saliva and blood samples from patients [
13].
For the determination of the same analytes in plasma, urine and saliva, there is also a more recent MEPS-HPLC-UV method. The column which was used was a Chromsep C
8 reversed-phase (150 × 4.6 mm i.d., 5 μm) and the mobile phase consisted of a mixture of acetonitrile (27%,
v/
v) and a pH 3.0, 30 mM phosphate buffer containing 0.23% (
v/
v) triethylamine (73%,
v/
v) with a gradient elution program. The UV detector was set at 238 nm and diphenhydramine was used as the internal standard. The C
8 MEPS cartridges were activated and conditioned with 300 μL of methanol and then with 300 μL of water. For the loading step the samples were drawn into the syringe and discharged back 10 times. The cartridge was then washed with 200 μL of water and then with 200 μL of a water/methanol mixture. For the elution step, 500 μL of methanol were used. The eluate was dried and redissolved in mobile phase and 50 μL of the solution was injected into the HPLC system. For the MEPS procedure, extraction efficiencies were higher than 90%, while relative standard deviation (RSD) for precision was always lower than 7.9% for the two compounds. In the biological samples limits of quantification were lower than 4 ng/mL for risperidone and lower than 6 ng/mL for 9-hydroxyrisperidone. Finally, the developed method was successfully applied to the analysis of biological samples from patients and seems suitable for therapeutic drug monitoring [
14].
In 2014, Mercolini et al. developed an HPLC method for the determination of a recent atypical antipsychotic; ziprasidone in plasma samples, using MEPS procedure. The analytes were separated on a RP C
18 column, with a mobile phase which was a mixture of acetonitrile (30%,
v/
v) and a pH 2.5, 50 mM phosphate buffer containing 0.2% (
v/
v) diethylamine (70%,
v/
v) that was delivered isocratically and the detection was performed at 320 nm. For the microextration, the C
2 sorbent which was chosen was conditioned with 200 μL of methanol and equilibrated with 200 μL of water. The sample was loaded and discarded back 10 times. Washing of the sorbent took place with 100 μL of water and 100 μL of a water/methanol mixture (90/10,
v/
v). Finally, the elution was done by drawing and discharging 500 μL of methanol. The eluate was dried under vacuum, redissolved in 100 μL of mobile4 phase and injected in the HPLC-UV system. Extraction yields were higher than 90% while limit of quantitation was 1 ng/mL. The sensitivity and the selectivity of the method was also good. The developed method was compared to a SPE procedure, using C
2 cartridges and the results were satisfactory. As a result, this procedure was successfully applied to real plasma samples from patients who were using ziprasidone and can be used for therapeutic drug monitoring of patients undergoing treatment with ziprasidone [
15].
In 2015, Souza et al. synthesized hybrid silica monoliths which were functionalized with aminopropyl- or cyanopropyl- groups by sol-gel process and used the mass selective stationary phase for MEPS to determine five antipsychotics, namely: olanzapine, quetiapine, clozapine, haloperidol and chlorpromazine) simultaneously with seven antidepressants, two anti-convulsants and two anxiolytics in plasma using UPLC-MS/MS. Due to the higher selectivity of the cyanopropyl hybrid silica for most of the drugs and its good mechanical strength, it was finally selected as the stationary MEPS phase. For the MEPS procedure, the stationary phase was conditioned with 4 × 200 μL of a methanol and acetonitrile mixture (50:50
v/
v) and 4 × 200 μL of water. Then, 4 × 100 μL of plasma samples diluted with ammonium acetate solution (pH 10) was manually drawn. Then, the sorbent was washed with 150 μL of water and desorption took place using 100 μL of a 50:50 (
v/
v) mixture of methanol and acetonitrile. The extract was dried, and reconstituted with 50 μL of the mobile phase, which consisted of ammonium acetate solution 5 mmol/L (with 0.1% formic acid) and acetonitrile and then injected into a XSelects CSH C
18 (2.5 μm, 2.1 × 100 mm) column for analysis with liquid chromatography. The linearity of the method ranged from 0.05 to 1.00 ng/mL (limit of quantification) to 40–10,500 ng/mL. The absolute recoveries, the precision and the accuracy were good, so the developed method can be applied to the therapeutic drug monitoring of patients [
16].
Clozapine and its metabolites were also determined in dried blood spots on filter paper with a HPLC method coupled with a coulometric detection, after being extracted with phosphate buffer and cleaned-up with MEPS procedure. The use of this matrix has many advantages because it eliminates the blood withdrawal, it has low cost and low biohazard risk and it is easy to use and to store. For the microextraction procedure, the sorbent which was 4 mg of solid phase silica-C
8 material, inserted into a syringe was activated using 3 × 100 μL of methanol and subsequently conditioned with 3 × 100 μL of water. For the clean-up 10 × 150 μL of the extract from DBS was drawn up and down, followed by a washing step first with 100 μL of water and second with a mixture of water and methanol (95:5,
v/
v). The elution step took place using 150 μL of the mobile phase and the liquid was injected into the HPLC system. For the HPLC analysis a reversed phase C
18 column was used with a mobile phase composed of methanol, acetonitrile and phosphate buffer. All MEPS steps namely: activation, loading, washing and elution were carried out in manual mode. The extraction yields were higher than 90%, the method validation gave satisfactory results for accuracy, precision, sensitivity and selectivity. The developed method was successfully applied to real samples obtained from patients. Therefore, this developed method is suitable for therapeutic drug monitoring for patients undergoing treatment with clozapine [
17].
Hendrickx et al. developed a capillary UHPLC-UV method in combination with MEPS as a sample clean-up procedure, in order to determine chlorpromazine, olanzapine and their flavin-containing monooxygenase mediated N-oxides in rat brain microdialysates. The analysis was carried out with an Acclaim Pepmap RP C
18 capillary column. For the MEPS procedure 4 mg of a mixed solid phase M1 (80% C
8, 20% SCX) cartridge was selected. Firstly, the sorbent was activated using first 100 μL of a solution consisting of 5% ammonia in 80% methanol (
v/
v) and second 100 μL of methanol and then conditioned with a 1:3 mixture (
v/
v) of Ringer's solution and phosphate buffer (pH 2.5). For the adsorption 3 × 50 μL microdialysate sample diluted with the same buffer was drawn through the syringe and ejected. Then the sorbent was washed first with 100 μL 5% acetic acid (
v/
v) and second with 100 μL of a mixture of methanol and water (10:90,
v/
v). The elution step took place with 50 μL of a solution containing 5% ammonia in 80% methanol (
v/
v) and the extracts were diluted with 150 μL of mobile phase which consisted of 10 mM ammonium acetate with 0.05% triethylamine, adjusted to pH 3.00 using formic acid and acetonitrile and injected into the UHPLC system. For the examined analytes MEPS recoveries were higher than 92% and intra- and inter-day variabilities were below 15%. The applicability of the method was checked by analyzing real samples from patients, thus proving that it can be used for therapeutic drug monitoring [
18].
4. Liquid Phase Microextraction for the Determination of Antipsychotic Drugs in Biological Samples
Liquid Phase Microextraction (LPME) is a miniaturized form of liquid-liquid extraction, which was firstly introduced at 1990s, when Dasgupta [
19] and Jeannot and Cantwell [
20] suggested almost at the same time the use of extraction solvents in the low microliter range. It is considered as a simple, rapid and cheap sample preparation technique, which requires only several microliters of organic solvents in contrast to traditional LLE, which requires several hundred of milliliters.
Based on hydrodynamic features, this technique can be classified into static LPME and dynamic LPME. In the static LPME, a solvent is used as an extractant and it is suspended in the sample. As a result transference of the target compounds to the extractant is carried out. On the other hand, in the dynamic mode, the exractant solvent forms a microfilm inside of an extraction unit, such as a microsyringe and the mass transfer of the analytes takes place between the sample and the microfilm [
21].
The main forms of LPME are (1) single drop microextraction; the oldest form of LPME, which is less frequently used today compared to more recently developed techniques, because it was based on a droplet of solvent hanging at a needle of a syringe and it was not considered very robust, (2) Dispersive LPME (DLLME) and (3) Hollow fiber LPME (HF-LPME). Hollow fiber LPME is a recently developed technique, which is based on immobilized organic solvents inside pores of hollow fibers. This technique partly consists of (1) a donor phase, which is the aqueous sample containing the target compounds, (2) the porous fiber with the organic solvent trapped inside and (3) a receptor phase inside the hollow fiber lumen.
Prior to analysis, the organic solvent is immobilized in the fiber’s pores by dipping the hollow fiber in a vial containing the solvent in order to form a layer. Next, the lumen is filled with the acceptor phase, which could be an organic, an acidic or a basic solution. For the analysis, the fiber is inserted in the sample, which is the donor phase containing the analytes and extraction takes place into the immobilized organic solvent [
22].
Depending on the acceptor phase that is used HF-LPME can be classified into (1) two phase HF-LPME, in which the organic solvent, which is immobilized in the hollow fiber and the receptor phase, which is inside the lumen of the fiber are the same solvent. In this case the solution that is used as a receptor phase can be directly injected to the gas chromatography system, whereas, for liquid chromatography and capillary electroapothesis, evaporation of the solvent and reconstitution in an aqueous solution are mandatory, so that the sample is compatible with the analytical apparatus.
Another form of HF-LPME is the three-phase HF-LPME, in which the acceptor phase is an acidic or basic aqueous solution. In this case, extraction of the analytes takes place from the aqueous sample primarily into the immobilized organic solvent. After that, back extraction from the organic solvent takes place in the final receptor solution, which is the aqueous solution placed into the lumen of the hollow fiber. This extraction mode is limited to basic or acidic analytes that can be ionized [
22].
Hollow fiber-LPME can also be classified as static HF-LPME, which includes magnetic stirring of the solution and dynamic HF-LPME, in which small volumes of the sample are repeatedly pulled in and out of the fiber in order to increase the extraction speed [
21].
During the development of an ideal HF-LPME process, many parameters should be optimized in order to achieve the best results. These parameters are the material of the fiber, the type of organic solvent, the pH of the sample and the acceptor phase, the volume of sample and of solvent, the time, the temperature, the ionic strength and the stirring speed [
22].
Dispersive LLPME is a recent novel approach of liquid-phase microextraction, introduced by Assadi and their co-workers in 2006 [
23]. This technique is based on a ternary solvent system consisting of an extraction solvent, a disperser solvent and an aqueous sample. A mixture consisting of the organic and the disperser solvent is rapidly and vigorously injected in the aqueous sample, which contains the target analytes. For this purpose a syringe is used [
24]. As a result, a cloudy solution is formed, which is supposed to be stable for a specific time. As a next step, phase separation takes place by gently shaking and centrifuging the mixture. If the density of the organic solvent is higher than this of water, the solvent goes to the bottom of the tube and it can be removed by a microsyringe, after discarding the aqueous solution. The crucial parameters in this procedure are the type and the volume of extraction and disperser solvents, the extraction time after the formation of the cloudy solution, the pH of the sample and its ionic strength. As for the extraction solvent, it should be miscible with the disperser solvent and it should be able to extract the target analytes. Moreover, its high density and low solubility in water assist the centrifugation step. As for the disperser solvent, it has to be soluble in the organic solvent and miscible in water in order to enable the organic solvent to be dispersed in the sample and to form a cloudy solution. The most common disperser solutions are acetone, methanol and acetonitrile [
22].
4.1. DLLME for the Determination of Antipsychotic Drugs in Biological Samples
Several LPME methods have been developed for the determination of antipsychotic drugs in biological matrices prior to liquid chromatography analysis using either HF-LPME or DLLME.
Cruz-Vera et al. published an almost solvent-less HPLC-UV method for the determination of seven phenothiazine derivatives in urine using the dynamic liquid-phase microextraction (dLPME) procedure. The whole process took place under dynamic conditions in an automatic flow system. The extraction unit was consisted of a syringe pump and a 1 mL syringe connected to a Pasteur pipette. For the microextraction, 100 μL of a mixture of ionic liquid 1-butyl-3-methyl imidazolium hexafluorophosphate and acetonitrile (50:50,
v/
v) were picked up in the pipette, which was then inserted into a vial containing the sample, the pH of which was primarily fixed at 8. A volume of 10 mL was drawn with a flow rate of 0.5 mL·min
−1. When the extraction was completed, 50 μL of the ionic liquid were drawn out at a flow rate of 0.05 mL·min
−1 and recovered in a vial containing 50 μL of acetonitrile. Finally, 20 μL of the mixtures were injected into tandem LiChrosorb C
8 (4.6 mm × 150 mm)–LiChrosorb C
18 (4.6 mm × 150 mm) cartridge columns and determined with a mobile phase consisting of acetonitrile/water/acetic acid/trimethylamine 40/40/20/2 (
v/
v/
v/
v). A new pipette was used for each extraction so there is no carry-over effect. The recovery values was between 72% and 98%, the limits of detection were between 21 ng/mL and 60 ng/mL and the repeatability expressed as RSD varied between 2.2% and 3.9% and the method was successfully validated [
22].
Xiong et al. developed a HPLC-UV method for the separation and quantitative determination of three psychotropic drugs (amitryptiline, clomipramine and thioridazine) in urine, using DLLME as a sample preparation technique. For the microextraction, 5 mL of the sample, the pH of which was adjusted to 10 using NaOH, was placed in a test tube, in which 0.50 mL of acetonitrile (as a disperser solvent) containing 20 μL of carbon tetrachloride (as an extraction solvent) were rapidly and vigorously injected, in order to form a cloudy solution. Accordingly, the mixture was shaken and then the separation of the phases took place by centrifugation at 4000 rpm for 3 min. The extraction time was fixed at 3 min. After slowly discarding the aqueous solution, the resulting droplet and the lipidic solid were dissolved in 200 μL acetonitrile, filtered and injected into the HPLC system for analysis with a C
8column and a mixture of ammonium acetate (0.03 mol/L, pH 5.5)–acetonitrile (60:40,
v/
v) as a mobile phase. The absolute recoveries were between 96% and 101%, the limit of detection was 3 ng/mL, while the limit of quantification was 10 ng/mL. The developed method was efficiently applied to urine samples obtained from patients to estimate and personalize the drug dose [
24].
In 2011, Chen et al. developed a DLLME-HPLC-UV method for the determination of two antipsychotic drugs; clozapine and chlorpromazine in urine. For the DLLME procedure 10 mL of the sample was placed in a test tube after adjusting the pH to 10 with NaOH and 200 μL of ethanol (as a disperser solvent) containing 40 μL CCl
4 (as an extraction solvent) was fast and vigorously injected, in order to form a cloudy solution, which was then shaken and centrifuged for 2 min at 4000 rpm to achieve phase separation. After that, the precipitate was dissolved by 0.5 mL methanol after careful removal of the supernatant solution, the extract was filtered and injected into the HPLC. For the separation, a Symmetry
® C
18 column packed with 5.0 μm particle size of dimethyloctylsilyl bounded amorphous silica was used with a mixture of CH
3COONH
4 (0.03 g/mL, pH 5.5)-CH
3CN (60:40,
v/
v) as a mobile phase. With these conditions, the limits of detection were lower than 6 ng/mL, and the limits of quantification lower than 39 ng/mL. The absolute extraction efficiencies were higher than 97%. The method was successfully applied to the analysis of real samples obtained from patients [
25].
In 2011, Zhang et al. developed a DLLME-HPLC-UV method for the determination of tetrahydropalmatine and tetrahydroberberine in rat urine; two active components in
Rhizoma corydalis, which possess strong antipsychotic actions. For the DLLME procedure 1.00 mL of the sample solution was placed in a glass tube and 100 mL of 1 mol/L of NaOH solution were added. The mixture was vortexed and 100 μL of methanol (dispersive solvent) containing 37 μL of chloroform (extraction solvent) was injected rapidly and vigorously in order to form a cloudy solution which was then centrifuged for 3 min at 4000 rpm. After discarding the upper layer solution, the sedimented phase was evaporated to dryness and the dry residue was reconstituted in 50 μL of a acetonitrile-0.1% phosphoric acid solution which was used as a mobile phase and a 20 μL aliquot was injected into a Ultimate XB-C
18 (150 × 4.6 mm i.d., packed with 5 mm particles) column. The extraction recoveries with this technique were higher than 69% and this method was efficiently applied in real urine samples [
26].
In 2015, Fisichella et al. developed a DLLME method for the determination of many different classes of drugs including main drugs of abuse (cocaine and metabolites, amphetamines and analogues, LSD, ketamine, opiates, methadone and fentanyl and analogues), Z-compounds and 44 benzodiazepines and antipsychotic drugs in blood samples followed by analysis with liquid UHLC-MS/MS. For the microextraction procedure, 100 μL of chloroform (extraction solvent) and 250 μL of methanol (disperser solvent) were rapidly and vigorously injected into the blood which was primarily deproteinized with 500 μL of methanol and its pH was fixed at 9 with the use of 0.2 g of NaCl and 100 μL of saturated carbonate buffer. A cloudy solution was formed, which was then shaken for 1 min using an ultrasonic water bath and centrifuged at 4000 rpm for 5 min. About 50 μL of the organic phase was evaporated to dryness and reconstituted in 100 μL of mobile phase. Then, 10 μL was then injected into a superficially porous Kinetex Biphenyl column (2.6 μm, 100 × 2.1 mm) with UHPLC-MS/MS instrument. Τhe mobile phase consisted of H
2O with 0.1% HCOOH MeOH with 0.1% HCOOH and a gradient elution program was chosen. The limits of detection were lower than 2 ng/mL and limits of quantification were lower than 10 ng/mL, with satisfactory accuracy and precision. The developed method was subsequently applied to the analysis of 50 blood samples from forensic cases [
27].
4.2. HF-LPME for the Determination of Antipsychotic Drugs in Biological Samples
For trace amounts of chlorpromazine in biological fluids, a hollow fiber liquid phase microextraction HF-LPME-HPLC-UV method was also developed. The drug was extracted from 11 mL of sample into an organic phase which was n-dodecane trapped in the pores of the fiber followed by the back-extraction into a receiving aqueous solution consisting of 0.01 M phosphate buffer (pH 2.0), located inside the lumen of the hollow fiber. For the extraction 11 mL of the aqueous sample solution was placed into a glass vial with a stirring bar and the vials were put on a magnetic stirrer. The stirring speed was 1000 rpm. Then, 20 μL of the receiving phase were injected into the polypropylene fiber, which was placed into the organic solution for 5 s and then into water for 5 s to remove the extra organic solution from its surface. After that, the fiber was bent and placed into the sample for 60 min and at the end of the extraction the fiber was removed, the receiving phase was withdrawn into the syringe and 10 μL of the receiving phase was injected into the HPLC. The whole procedure was carried out in absence of salt. The detection limit for chlorpromazine was 0.5 μg/L and intra-day and inter-day assay (RSD %) were lower than 10.3%.The method was successfully applied to drug level monitoring in biological fluids (urine and serum) of patients and gave satisfactory results [
28].
5. Novel LPME and SPME Techniques for the Determination of Antipsychotic Drugs in Biological Samples
Except for the conventional DLLME process, which was described above, many DLLME variations are widely used nowadays. Firstly, in-situ DLLME is also gaining attention, while temperature assisted-DLLME, UV-assisted-DLLME, Microwave-assisted-DLLME and vortex-assisted DLLME are becoming more and more popular as observed in the literature. Additionally, DLLME variations which use deep eutectic solvents (DES’s), surfactants, or ionic liquis (ILs) in order to form emulsions are also reported through scientific papers. Some of these processes are described below.
These procedures tend also to avoid the main problems existed in conventional sample preparation techniques such as LLE, SPE and PP which have been widely used to extract antipsychotic drugs from biological fluids.
In 2013 Fisher et al. developed an HPLC-MS/MS method for the determination of amisulpride, aripiprazole, dehydroaripiprazole, clozapine, norclozapine, olanzapine, quetiapine, risperidone, 9-hydroxyrisperidone, and sulpiride in small volumes of plasma or serum and they also investigated its ability to be applied to haemolysed whole blood as well as to oral fluid. The extraction took place in glass test tubes, where 200 μL of the sample and 100 μL of tris solution (2 mol/L pH 10.6) was added. For the extraction, 100 μL of butyl acetate:butanol (9 + 1,
v/
v) was added and the mixture was firstly vortexed for 30 s and then centrifuged (13,600×
g, 4 min). After taking the upper layer an aliquot of 200 μL was injected into a Waters Spherisorb S5SCX sulfopropyl-modified silica column for HPLC analysis, using 50 mmol/L methanolic ammonium acetate, (pH 6.0) as a mobile phase. The limits of quantification varied between were 1–5 μg/L depending on the analyte. Recoveries varied between 16% and 107%and the reproducibility of the method was good. Thus, the developed method can be applied in the therapeutic drug monitoring of patients who undergo treatment with the examined drugs [
29].
Also, in 2013, Ebrahimzadeh et al. developed a high-performance liquid chromatography coupled to photodiode array detector (HPLC-DAD) method to preconcentrate and determine the antipsychotic drug (haloperidol) in biological samples using ultrasound-assisted emulsification microextraction. The use of ultrasonic radiation helps to speed up different procedures such as homogenization, mass transfer and emulsion formation. In this microextraction procedure, a small volume of a solvent which is not miscible with water is injected into an aqueous sample solution and an emulsion is formed using sonication without any need for a dispersive solvent. When the microextraction is over the mixture is centrifuged and the two different phases are separated. In this case, 30 μL of 1-undecanol was injected into a glass-centrifuge tube containing 4 mL of the sample solution after fixing its pH to 10 (NaCl 4%
w/
v). The mixture was put in an ultrasonic water bath for 20 min at 25 °C in order to form an emulsion, which was then centrifuged at 4000 rpm for 5 min. The droplets of the extraction solvent floated at the top of the tubes and they were solidified after being cooled in an ice bath. Finally, they were removed and they were melt again at room temperature and analyzed with HPLC. An ODS-H C
18 (250 × 4.6 mm i.d., 5 μm) column was used, using a mixture of methanol and monobasic potassium phosphate solution (0.02 mol/L, pH 4) (60:40,
v/
v) as a mobile phase. The extraction yields were higher than 90% and the limits of quantification varied between 4 and 8 μg/L. As a result, the method can be successfully applied in real plasma and urine samples obtained from patients who undergo treatment with haloperidol [
30].
In 2015, Zare et al. developed an ionic-liquid-based surfactant-emulsified microextraction procedure accelerated by ultrasound radiation followed by HPLC analysis for the determination of antidepressant and antipsychotic drugs; doxepine and perphenazine in urine. This procedure is based on the replacement of less green solvents with ionic liquids, which pose some unique properties such as tunability of their viscosity and surface tension and are widely used in green chemistry. For the microextraction of the drugs, 4 mg of the lipophilic SDS surfactant was poured into 6 mL of the sample, followed by the addition of 50 μL of the ionic liquid 1-hexyl-3-methylimidazolium hexafluorophosphate, which was used as an extracting solvent. The mixture was vortexed for 10 min and sonicated for another 10 min and a cloudy solution was formed. After centrifugation for 5 min at 5000 rpm the aqueous phase was removed and the phase containing the analytes was dissolved in methanol and injected into the HPLC system. For the analysis a Zorbax SB-C
8 (250 mm × 4.6 mm, 5 μm) column was used with a mixture of acetate buffer (pH 4)/acetonitrile (70:30,
v/
v) as a mobile phase. For the examined drugs extraction recoveries were between 89% and 98%. The performance of the developed method was compared with two other methods, one including dispersive liquid–liquid microextraction and one including ultrasound-assisted surfactant-based emulsification microextraction. The novel microextraction procedure showed some significant advantages and can be used for preconcentration, separation and determination of above mentioned drugs in real urine samples [
31].
In 2016 Li et al., prepared novel magnetic octadecylsilane (ODS)—polyacrylonitrile (PAN) thin-films for microextraction of the antipsychotic drugs; quetiapine and clozapine in plasma and urine samples followed by HPLC-UV analysis. Thin-film microextraction (TFME) is a novel form of solid-phase microextraction for preconcentration and clean-up of analytes in biofluids. Thus, those films are supposed to be superior to conventional SPME because of their high extraction rate, their sensitivity and their low extraction time. These parameters can be optimized by using novel film coatings. In this work, the thin firms were made magnetic by adding superparamagnetic SiO
2@Fe
3O
4 nanoparticles to the films. For the drug microextraction procedure, the films were preconditioned with methanol and water and then they were added into the sample, the pH of which was adjusted to 9.5 by adding a 0.1 mol/L NaOH solution. The mixture was mechanically shaken for 50 min and then the film was removed by a strong magnet and cleaned with 3 mL of water. For the desorption of the analytes the film was again mechanically shaken in 1 mL of methanol for 5 min and the obtained solution was evaporated and redissolved in 100 μL of methanol and injected into a C
18 reversed-phase column for HPLC-UV analysis. The repeatability of the method was good, the films were reusable up to 15 times, extraction recoveries were higher than 99% and detection limits were lower than 0.015 μg/mL. The method was successfully applied to real plasma and urine samples and it can be used for therapeutic drug monitoring of patients [
32].
All of the developed methods that are mentioned in the following paragraphs are summarized in
Table 1.





{kind=link}
{kind=link}