
In Situ Measurement of Polymer Layer Thickness in Porous Layer Open Tubular (PLOT) Columns Using Optical Absorbance in the Near-IR Range


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Theory
Optical Absorbance
3. Experimental
3.1. Reagents and Materials
3.2. Instrumentation
3.3. Procedures
3.3.1. Fabrication of monoPLOT Columns
3.3.2. Determination of monoPLOT Layer Thickness by Optical Absorbance Measurement
4. Results
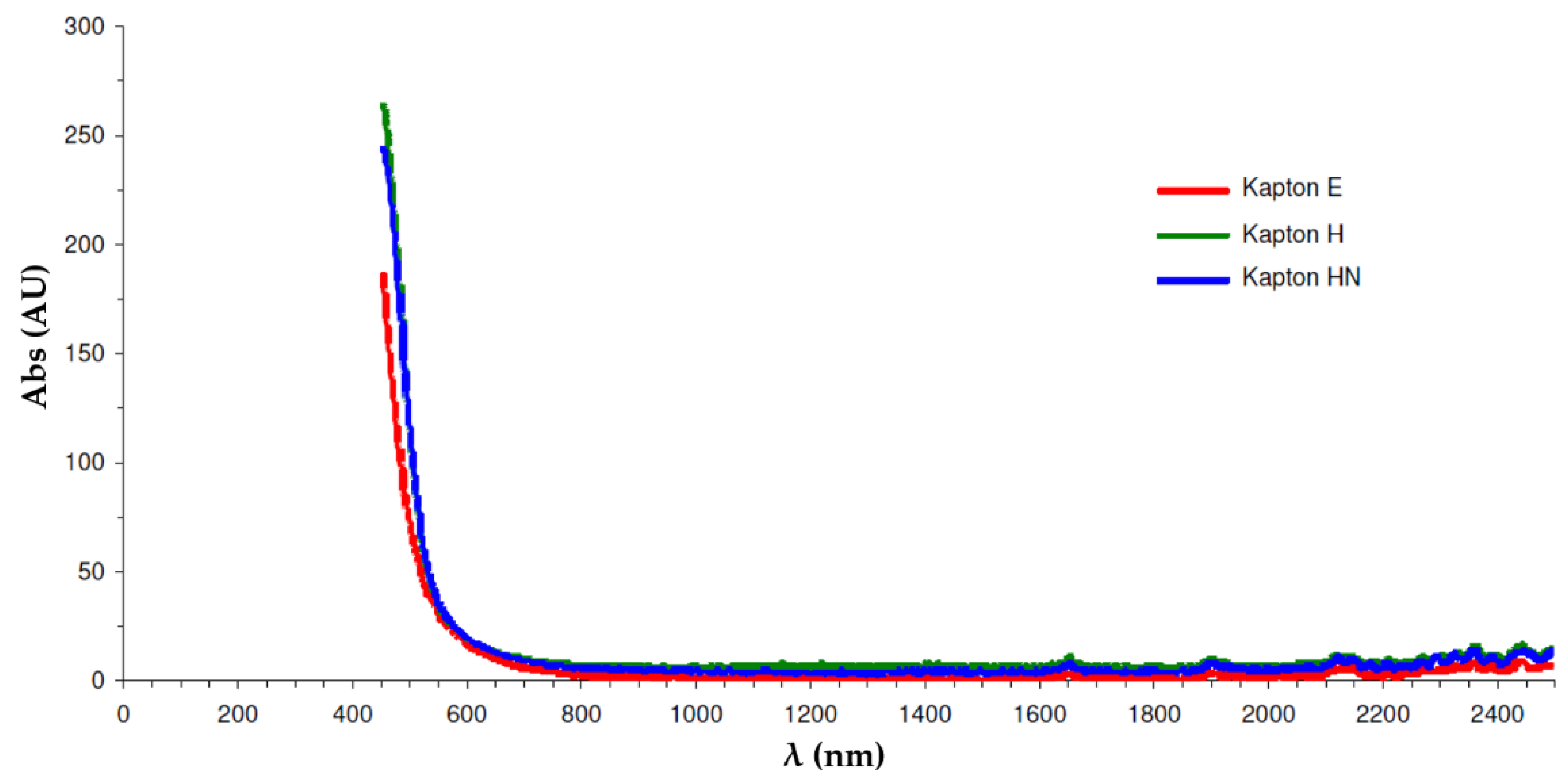
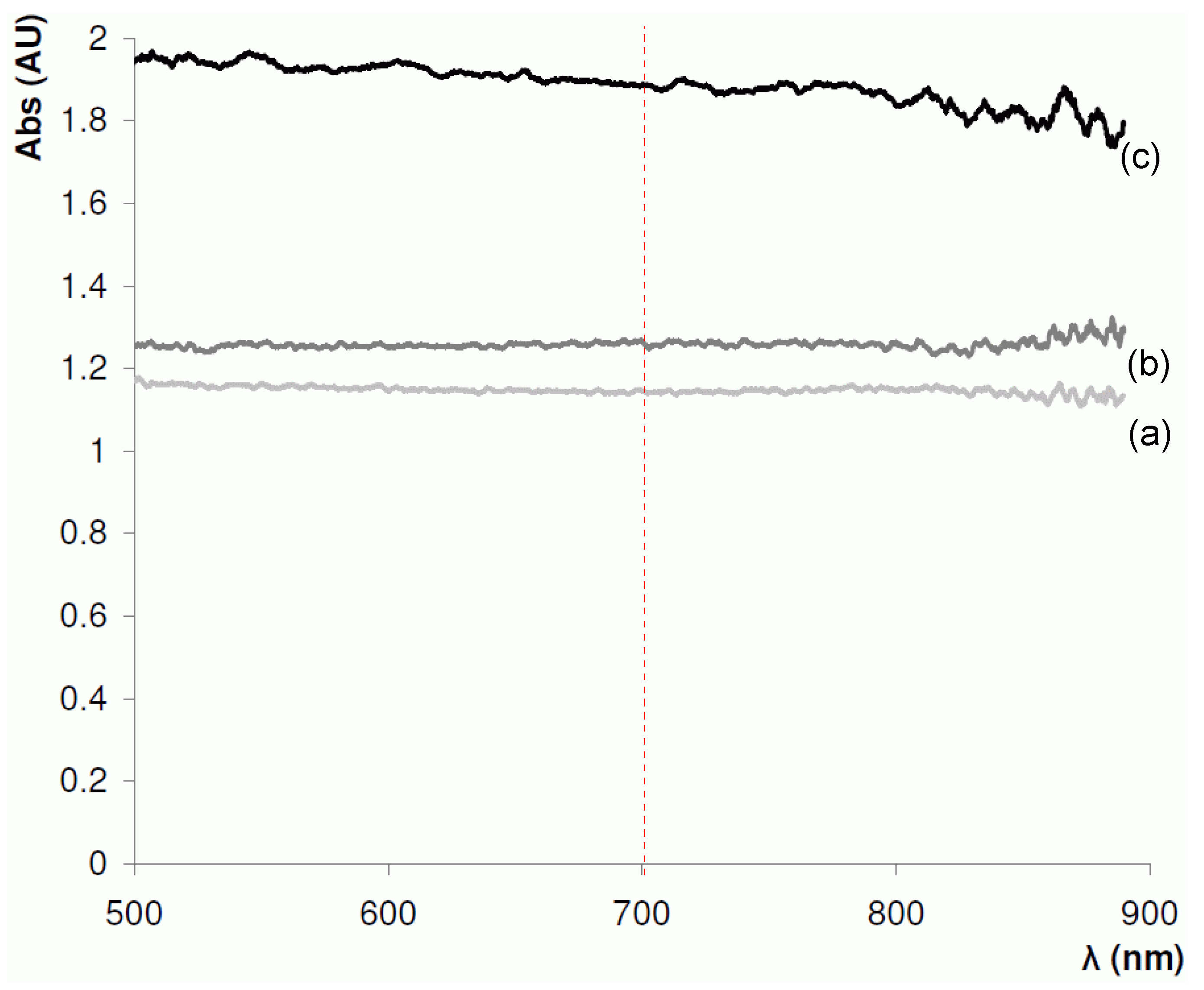
4.1. Selection of Optimal Wavelength
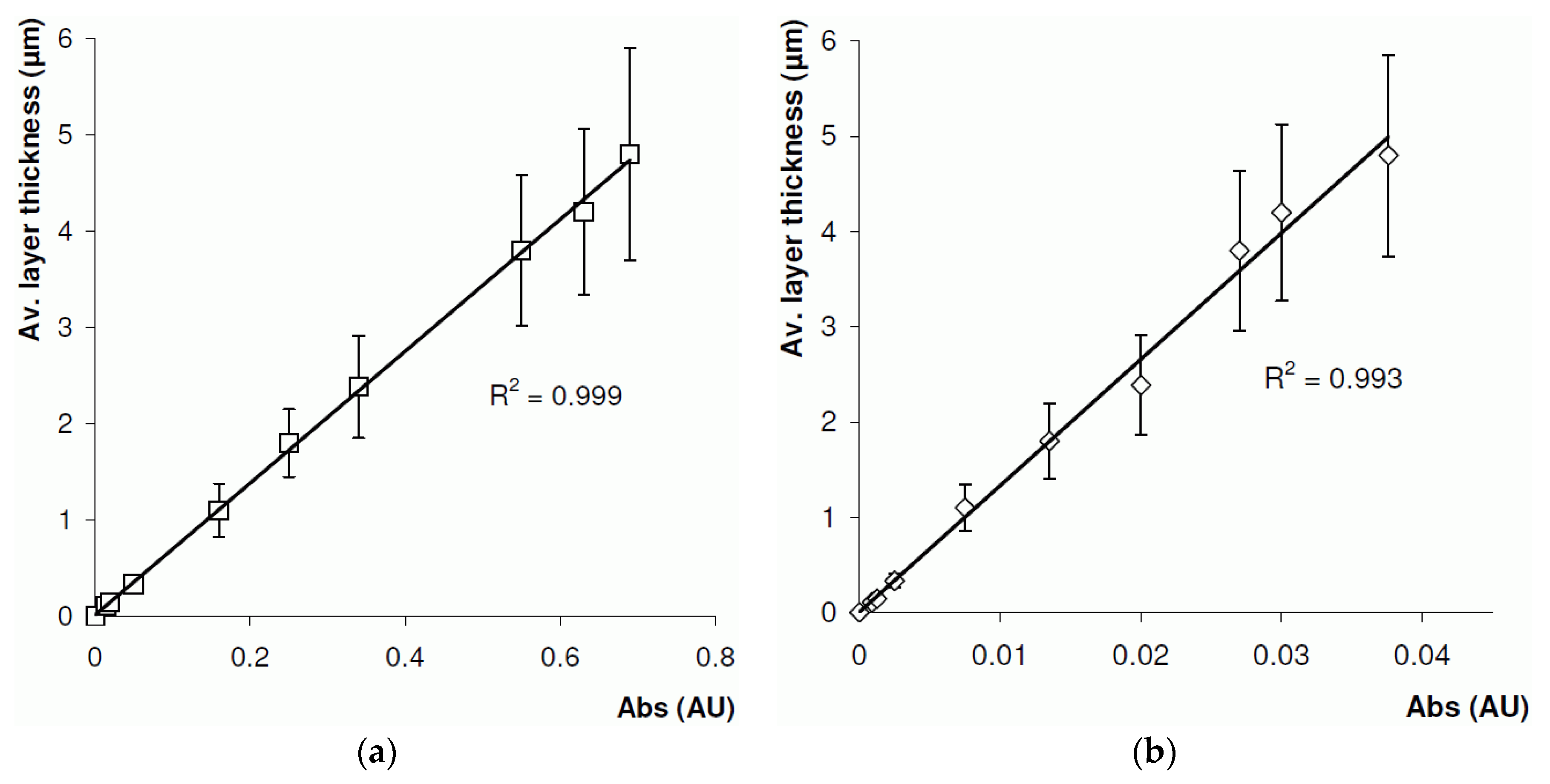
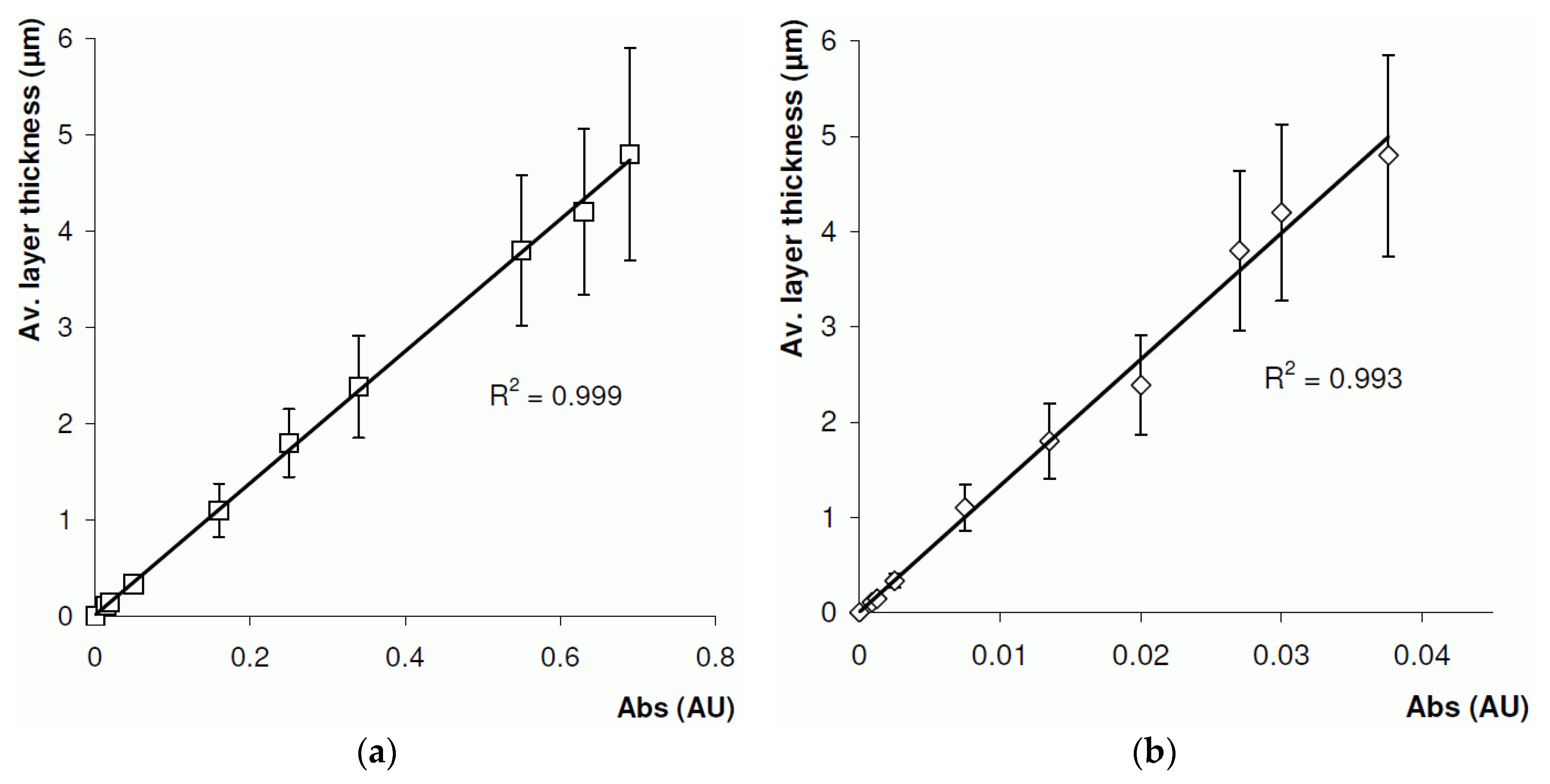
4.2. On-Line Layer Thickness Measurement
4.3. Off-Line Layer Thickness Measurement
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AFM | Atomic force microscopy |
| BuMA | butyl methacrylate |
| C4D | Capacitively coupled contactless conductivity detection |
| DVB | divinylbenzene |
| EDMA | ethylene glycol dimethacrylate |
| IR | Infrared |
| monoPLOT | monolithic porous layer open tubular |
| PLOT | porous layer open tubular |
| PS | polystyrene |
| RSD | Relative standard deviation |
| SNR | Signal to noise ratio |
| UV | Ultraviolet |
References
- Niessen, W.M.A.; Poppe, H.J. Open-tubular liquid-chromatography mass-spectrometry using direct liquid introduction. J. Chromatogr. A 1985, 323, 37–46. [Google Scholar] [CrossRef]
- Escoffier, B.H.; Parker, C.E.; Mester, T.C.; Dewit, J.S.M.; Corbin, F.T.; Jorgensen, J.W.; Tomer, K.B. Comparison of open-tubular liquid-chromatography mass-spectrometry and direct liquid introduction liquid-chromatography mass-spectrometry for the analysis of metribuzin and its metabolites in plant-tissue and water samples. J. Chromatogr. A 1989, 474, 301–316. [Google Scholar] [CrossRef]
- Swart, R.; Kraak, J.C.; Poppe, H. Recent progress in open tubular liquid chromatography. TrAC Trends Anal. Chem. 1997, 16, 332–342. [Google Scholar] [CrossRef]
- Yue, G.; Luo, Q.; Zhang, J.; Wu, S.L.; Karger, B.L. Ultratrace LC/MS proteomic analysis using 10-µm-i.d. Porous layer open tubular poly(styrene-divinylbenzene) capillary columns. Anal. Chem. 2007, 79, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Rogeberg, M.; Wilson, S.R.; Greibrokk, T.; Lundanes, E. Separation of intact proteins on porous layer open tubular (PLOT) columns. J. Chromatogr. A 2010, 1217, 2782–2786. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hincapie, M.; Rejtar, T.; Karger, B.L. Ultrasensitive characterization of site-specific glycosylation of affinity-purified haptoglobin from lung cancer patient plasma using 10 µm i.d. porous layer open tubular liquid chromatography-linear ion trap collision-induced dissociation/electron transfer dissociation mass spectrometry. Anal. Chem. 2011, 83, 2029–2037. [Google Scholar] [PubMed]
- Causon, T.J.; Shellie, R.A.; Hilder, E.F.; Desmet, G.; Eeltink, S. Kinetic optimisation of open-tubular liquid-chromatography capillaries coated with thick porous layers for increased loadability. J. Chromatogr. A 2011, 1218, 8388–8393. [Google Scholar] [CrossRef] [PubMed]
- Collins, D.; Nesterenko, E.; Paull, B. Porous layer open tubular columns in capillary liquid chromatography. Analyst 2014, 139, 1292–1302. [Google Scholar] [CrossRef] [PubMed]
- Knob, R.; Kulsing, C.; Boysend, R.I.; Macka, M.; Hearn, M.T.W. Surface-area expansion with monolithic open tubular columns. TrAC Trends Anal. Chem. 2015, 67, 16–25. [Google Scholar] [CrossRef]
- Eeltink, S.; Svec, F.; Frechet, J.M.J. Open-tubular capillary columns with a porous layer of monolithic polymer for highly efficient and fast separations in electrochromatography. Electrophoresis 2006, 27, 4249–4256. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Svec, F.; Frechet, J.M.J. Towards stationary phases for chromatography on a microchip: Molded porous polymer monoliths prepared in capillaries by photoinitiated in situ polymerization as separation media for electrochromatography. Electrophoresis 2000, 21, 120–127. [Google Scholar] [CrossRef]
- Zaidi, A.; Cheong, W.J. Long open tubular molecule imprinted polymer capillary columns with excellent separation efficiencies in chiral and non-chiral separation by capillary electrochromatography. Electrophoresis 2009, 30, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.L.; Lin, Y.C. Succinyl methacrylate polymerized in porous-layered phases for open-tubular capillary electrochromatography: Comparison with silica hydride monolayered phases. J. Chromatogr. A 2010, 1217, 4328–4336. [Google Scholar] [CrossRef] [PubMed]
- Tarangoy, F.M., Jr.; Haddad, P.R.; Boysen, R.I.; Hearn, M.T.W.; Quirino, J. Open tubular-capillary electrochromatography: Developments and applications from 2013 to 2015. Electrophoresis 2016, 37, 66–85. [Google Scholar] [CrossRef] [PubMed]
- Bakry, R.; Gjerde, D.; Bonn, G.K. Derivatized nanoparticle coated capillaries for purification and micro-extraction of proteins and peptides. J. Proteome Res. 2006, 5, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, Y.; Ohmichi, M.; Tatsumoto, H. Simple and rapid determination of golf course pesticides by in-tube solid-phase microextraction coupled with liquid chromatography. J. Health Sci. 2005, 51, 526–532. [Google Scholar] [CrossRef]
- de Zeeuw, J. The Development and Applications of PLOT Columns in Gas-Solid Chromatography. LC-GC Eur. 2011, 24, 38–45. [Google Scholar]
- Nesterenko, E.; Burke, M.; de Bosset, C.; Pessutto, P.; Malafosse, C.; Collins, D. Monolithic porous layer open tubular (monoPLOT) capillary columns for gas chromatography. RSC Adv. 2015, 5, 7890–7896. [Google Scholar] [CrossRef]
- Collins, D.; Nesterenko, E.; Paull, B. Infrared photo-initiated fabrication of monolithic porous layer open tubular (monoPLOT) capillary columns for chromatographic applications. RSC Adv. 2014, 4, 28165–28170. [Google Scholar] [CrossRef]
- Collins, D.; Nesterenko, E.; Brabazon, D.; Paull, B. Fabrication of bonded monolithic porous layer open tubular (monoPLOT) columns in wide bore capillary by laminar flow thermal initiation. Chromatographia 2013, 76, 581–589. [Google Scholar] [CrossRef]
- Collins, D.; Nesterenko, E.; Brabazon, D.; Paull, B. Controlled ultraviolet (UV) photoinitiated fabrication of monolithic porous layer open tubular (monoPLOT) capillary columns for chromatographic applications. Anal. Chem. 2012, 84, 3465–3472. [Google Scholar] [CrossRef] [PubMed]
- Collins, D.; Nesterenko, E.; Brabazon, D.; Paull, B. In-process phase growth measurement technique in the fabrication of monolithic porous layer open tubular (monoPLOT) columns using capacitively coupled contactless conductivity. Analyst 2013, 138, 2540–2545. [Google Scholar] [CrossRef] [PubMed]
- Svec, F.; Tennikova, T.B.; Deyl, Z. Monolithic Materials: Preparation, Properties and Applications, 1st ed.; Elsevier Science: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Veerkamp, T.H.; De Kock, R.J.; Veermans, A.; Lardinoye, M.H. Thickness measurement of polymer films for infrared spectrometry by beta-ray absorption analysis of ethane-propene copolymers. Anal. Chem. 1964, 36, 2277–2278. [Google Scholar] [CrossRef]
- Toney, M.; Mate, M.; Leach, A.; Pocker, D. Thichness measurements of thin perfluoropolyether polymer films on silicon and amorphous-hydrogenated carbon with X-ray reflectivity, ESCA and optical ellipsometry. J. Colloid Interface Sci. 2000, 225, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Macomber, C.; Eastman, M.; Porter, T.L.; Manygoats, K.; Delinger, W. Chemical sensing through measurement of thickness/impedance characteristics of ion-conducting polymer films. J. Electrochem. Soc. 2003, 150, H172–H177. [Google Scholar] [CrossRef]
- Fitt, A.D.; Owen, J.R. Simultaneous measurements of conductivity and thickness for polymer electrolyte films: A simulation study. J. Electroanal. Chem. 2002, 538–539, 13–23. [Google Scholar] [CrossRef]
- Brand, U.; Beckert, E.; Beutler, A.; Dai, G.; Stelzer, C.; Hertwig, A.; Klapetek, P.; Koglin, J.; Thelen, R.; Tutsch, R. Comparison of optical and tactile layer thickness measurements of polymers and metals on silicon or SiO2. Meas. Sci. Technol. 2011, 22, 094021. [Google Scholar] [CrossRef]
- Hong, X.; Gan, Y.; Wang, Y. Facile measurement of polymer film thickness ranging from nanometer to micrometer scale using atomic force microscopy. Surf. Interface Anal. 2011, 43, 1299–1303. [Google Scholar] [CrossRef]
- Lobo, R.; Pereira-da-Silva, M.; Raposo, M.; Faria, R.; Oliveira, O. In situ thickness measurements of ultra-thin multilayer polymer films by atomic force microscopy. Nanotechnology 1999, 10, 389–393. [Google Scholar] [CrossRef]
- Maev, R.G. Acoustic Microscopy: Fundamentals and Applications; John Wiley & Sons: New York, NY, USA, 2008. [Google Scholar]
- Grunlan, J.; Mehrabi, A.; Ly, T. High-throughput measurement of polymer film thickness using optical dyes. Meas. Sci. Technol. 2005, 16, 153–161. [Google Scholar] [CrossRef]
- Harris, D. Quantitative Chemical Analysis, 6th ed.; W.H. Freeman & Company: New York, NY, USA, 2003. [Google Scholar]
- Florea, L.; Hennart, A.; Diamond, D.; Benito-Lopez, F. Synthesis and characterisation of spiropyran-polymer brushes in micro-capillaries: Towards an integrated optical sensor for continuous flow analysis. Sens. Actuators B 2012, 175, 92–99. [Google Scholar] [CrossRef]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Collins, D.; Nesterenko, E.; Paull, B. In Situ Measurement of Polymer Layer Thickness in Porous Layer Open Tubular (PLOT) Columns Using Optical Absorbance in the Near-IR Range. Separations 2016, 3, 34. https://doi.org/10.3390/separations3040034
Collins D, Nesterenko E, Paull B. In Situ Measurement of Polymer Layer Thickness in Porous Layer Open Tubular (PLOT) Columns Using Optical Absorbance in the Near-IR Range. Separations. 2016; 3(4):34. https://doi.org/10.3390/separations3040034
Chicago/Turabian StyleCollins, David, Ekaterina Nesterenko, and Brett Paull. 2016. "In Situ Measurement of Polymer Layer Thickness in Porous Layer Open Tubular (PLOT) Columns Using Optical Absorbance in the Near-IR Range" Separations 3, no. 4: 34. https://doi.org/10.3390/separations3040034
APA StyleCollins, D., Nesterenko, E., & Paull, B. (2016). In Situ Measurement of Polymer Layer Thickness in Porous Layer Open Tubular (PLOT) Columns Using Optical Absorbance in the Near-IR Range. Separations, 3(4), 34. https://doi.org/10.3390/separations3040034