
Multi-Template Molecularly Imprinted Polymers Coupled with a Solid-Phase Extraction System in the Selective Determination of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) in Environmental Water Samples

 , , , ,
, , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. mt-MIP Synthesis
2.3. Instruments
2.4. Capillary Electrophoresis
2.5. Adsorption Experiments
2.6. Sample Treatment
3. Results and Discussion
3.1. MIP Composition Effects on ACE, DCF, IBP, and NPX Extraction
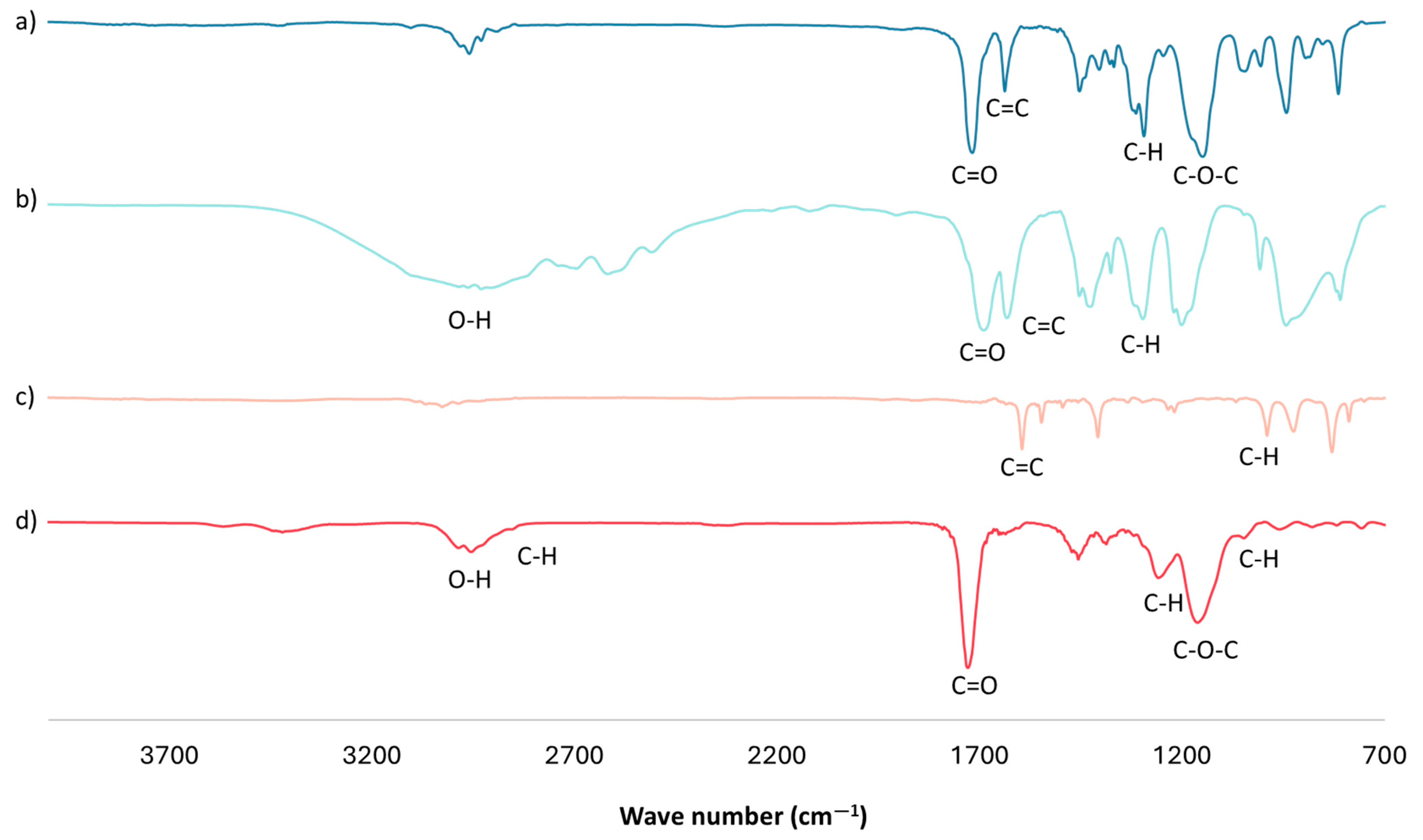
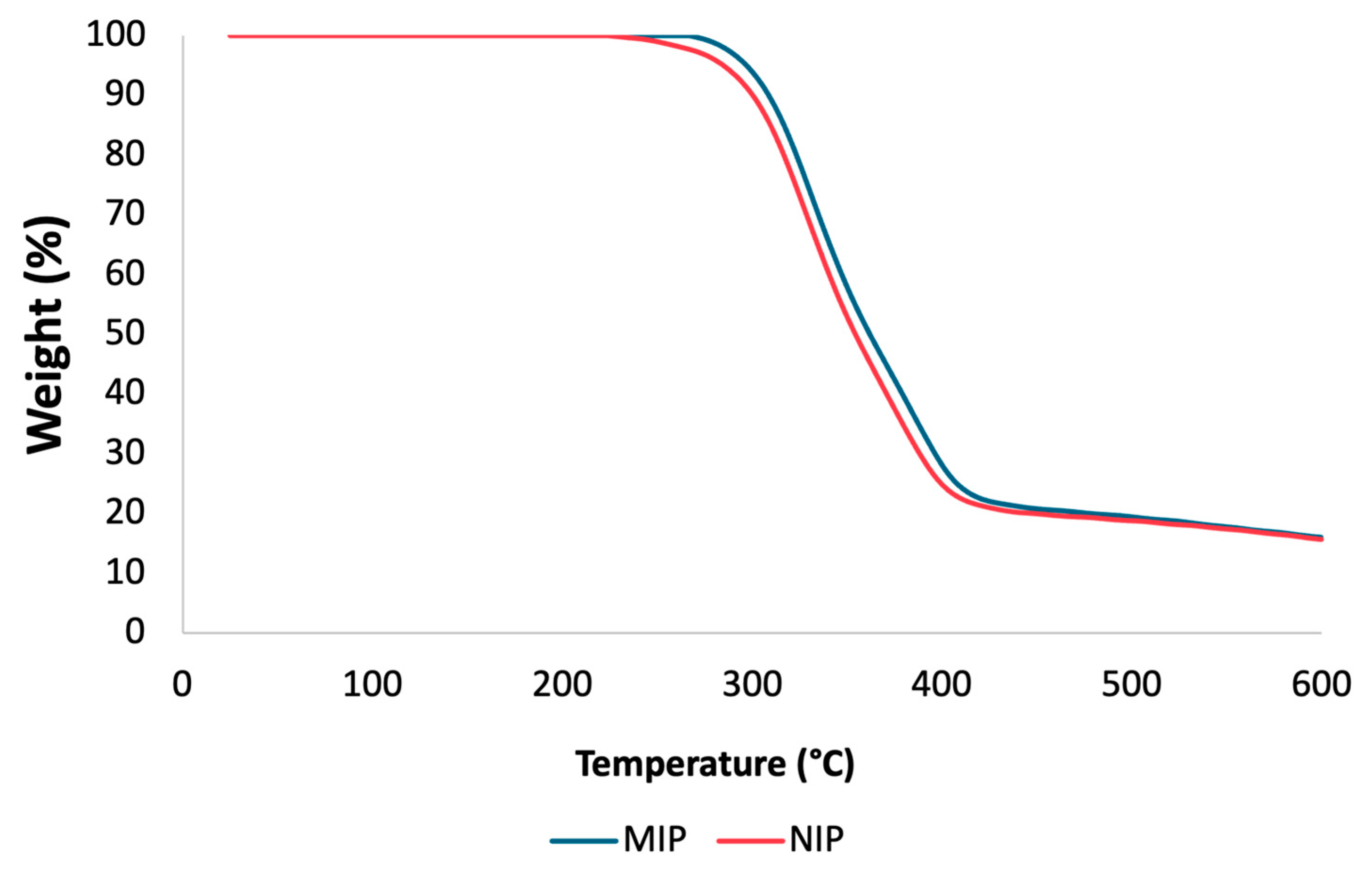
3.2. Characterization of Optimal mt-MIP
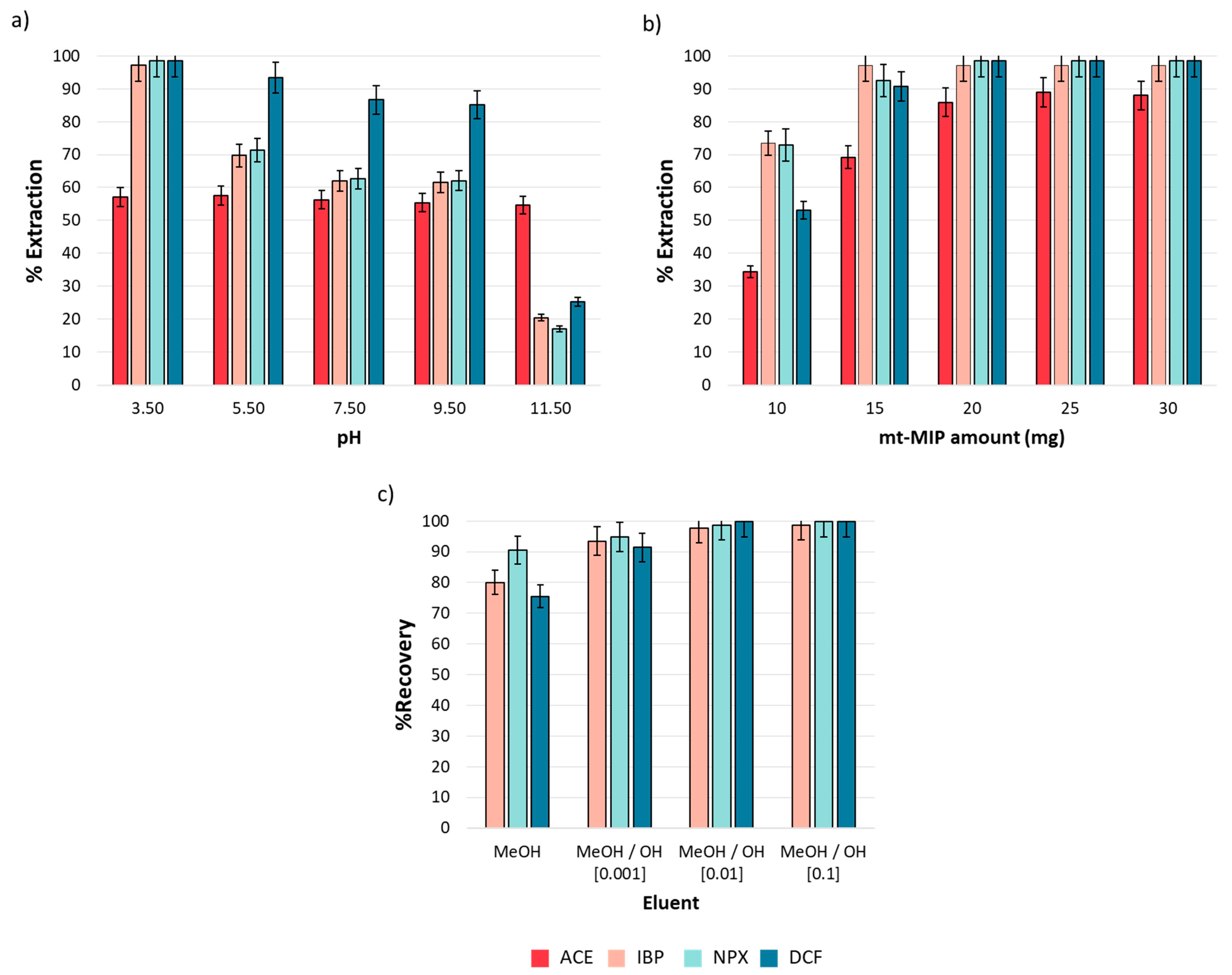
3.3. pH Effect on Adsorption Efficiency
3.4. Optimization of SPE Process
3.4.1. mt-MIP Dosage Effect
3.4.2. Eluent Composition
3.5. Analytical Parameters
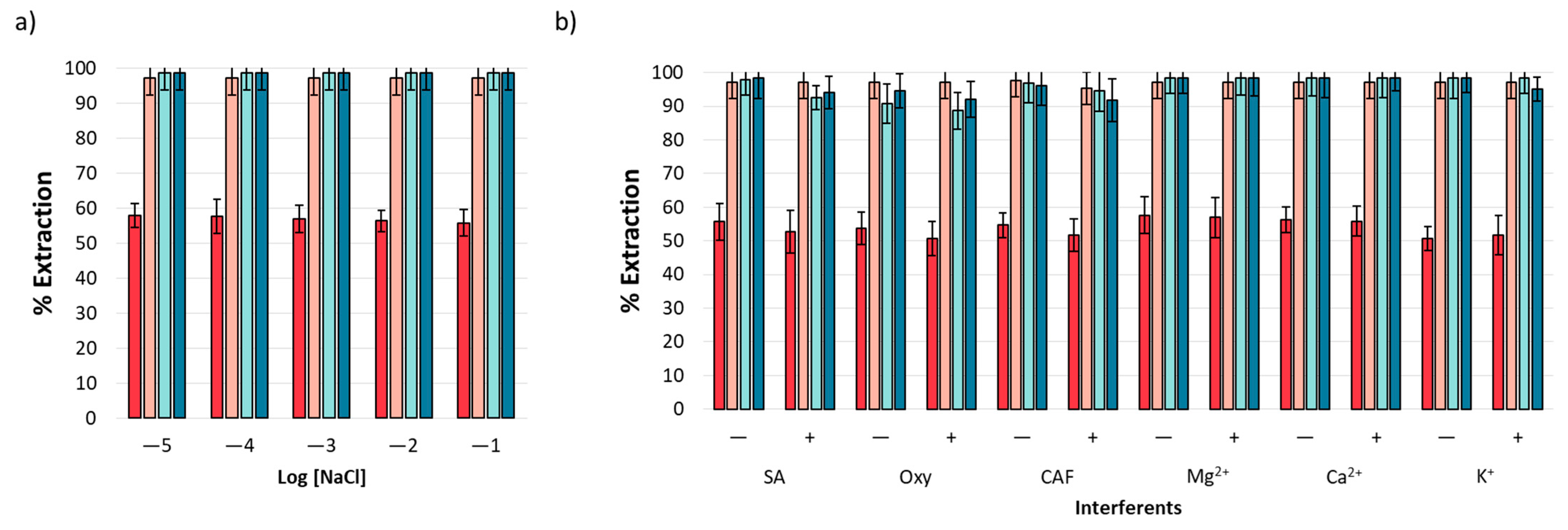
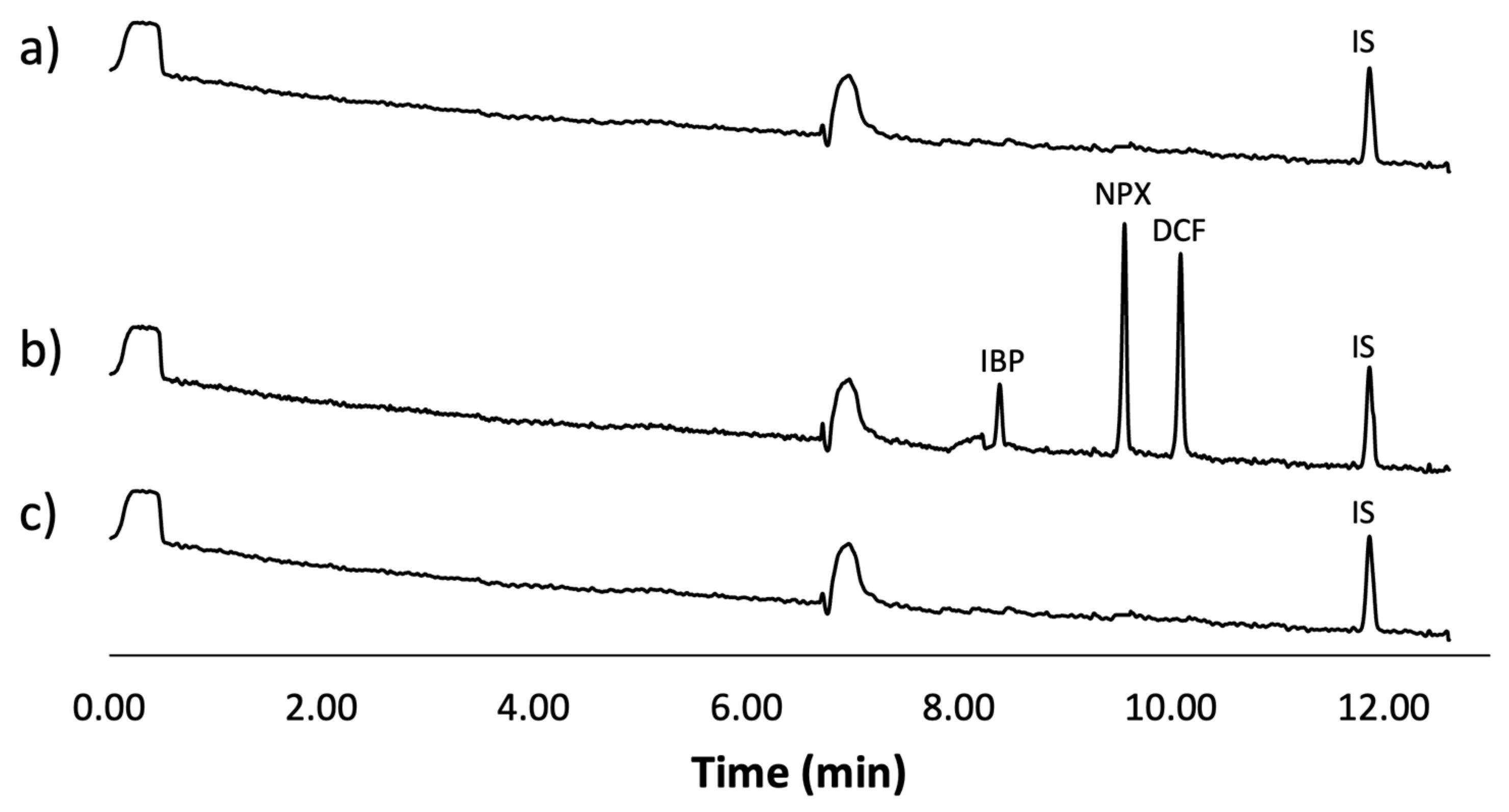
3.6. Interferent Analysis
3.7. Real Sample Analysis
3.8. Comparison of Proposed SPE Method
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rivera-Utrilla, J.; Sánchez-Polo, M.; Ferro-García, M.Á.; Prados-Joya, G.; Ocampo-Pérez, R. Pharmaceuticals as emerging contaminants and their removal from water. A review. Chemosphere 2013, 93, 1268–1287. [Google Scholar] [CrossRef]
- Białk-Bielińska, A.; Kumirska, J.; Borecka, M.; Caban, M.; Paszkiewicz, M.; Pazdro, K.; Stepnowski, P. Selected analytical challenges in the determination of pharmaceuticals in drinking/marine waters and soil/sediment samples. J. Pharm. Biomed. Anal. 2016, 121, 271–296. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, P.; Shukla, P.; Giri, B.S.; Chowdhary, P.; Chandra, R.; Gupta, P.; Pandey, A. Prevalence and hazardous impact of pharmaceutical and personal care products and antibiotics in environment: A review on emerging contaminants. Environ. Res. 2021, 194, 110664–110685. [Google Scholar] [CrossRef] [PubMed]
- Valdez-Carrillo, M.; Abrell, L.; Ramírez-Hernández, J.; Reyes-López, J.A.; Carreón-Diazconti, C. Pharmaceuticals as emerging contaminants in the aquatic environment of Latin America: A review. Environ. Sci. Pollut. Res. 2020, 27, 44863–44891. [Google Scholar] [CrossRef]
- Bruno, A.; Tacconelli, S.; Contursi, A.; Ballerini, P.; Patrignani, P. Cyclooxygenases and platelet functions. Adv. Pharmacol. 2023, 97, 133–165. [Google Scholar] [CrossRef] [PubMed]
- Farré, M.; Petrovic, M.; Barceló, D. Recently developed GC/MS and LC/MS methods for determining NSAIDs in water samples. Anal. Bioanal. Chem. 2007, 387, 1203–1214. [Google Scholar] [CrossRef]
- Zhou, Y.; Lin, J.Y.; Bian, Y.; Ren, C.J.; Xiao-Li, N.; Yang, C.Y.; Feng, X.S. Non-steroidal anti-inflammatory drugs (NSAIDs) in the environment: Updates on pretreatment and determination methods. Ecotoxicol. Environ. Saf. 2023, 267, 115624. [Google Scholar] [CrossRef]
- Petrie, B.; Camacho-Muñoz, D. Analysis, fate and toxicity of chiral non-steroidal anti-inflammatory drugs in wastewaters and the environment: A review. Environ. Chem. Lett. 2021, 19, 43–75. [Google Scholar] [CrossRef]
- Tanwar, S.; Di Carro, M.; Magi, E. Innovative sampling and extraction methods for the determination of nonsteroidal anti-inflammatory drugs in water. J. Pharm. Biomed. Anal. 2015, 106, 100–106. [Google Scholar] [CrossRef]
- Martinez-Sena, T.; Armenta, S.; De la Guardia, M.; Esteve-Turrillas, F.A. Determination of non-steroidal anti-inflammatory drugs in water and urine using selective molecular imprinted polymer extraction and liquid chromatography. J. Pharm. Biomed. Anal. 2016, 131, 48–53. [Google Scholar] [CrossRef]
- Paíga, P.; Lolić, A.; Hellebuyck, F.; Santos, L.H.; Correia, M.; Delerue-Matos, C. Development of a SPE-UHPLC-MS/MS methodology for the determination of non-steroidal anti-inflammatory and analgesic pharmaceuticals in seawater. J. Pharm. Biomed. Anal. 2015, 106, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.M.; Huang, C.P.; Chen, S.K.; Chen, C.W.; Dong, C.D. Electrochemical analysis of naproxen in water using poly (L-serine)-modified glassy carbon electrode. Chemosphere 2020, 254, 126686–126694. [Google Scholar] [CrossRef]
- Kovacs, E.D.; Silaghi-Dumitrescu, L.; Kovacs, M.H.; Roman, C. Determination of the uptake of ibuprofen, ketoprofen, and diclofenac by tomatoes, radishes, and lettuce by gas chromatography–mass spectrometry (GC-MS). Anal. Lett. 2021, 54, 314–330. [Google Scholar] [CrossRef]
- Mynul Hassan, M.; Nam, S.W. High-performance liquid chromatography for determining a mixture of nonsteroidal anti-inflammatory drugs. Electron. Mater. Lett. 2021, 17, 414–420. [Google Scholar] [CrossRef]
- Othman, W.M.; Al-Zoman, N.Z.; Darwish, I.A.; Almomen, A.; Saad, S.S.; Abdallah, F.F.; Farid, N.F. Development of an eco-friendly capillary electrophoresis method for the simultaneous determination of piperacillin, tazobactam and ibuprofen in plasma samples: Application to a pharmacokinetic study in rats. RSC Adv. 2024, 14, 23378–23391. [Google Scholar] [CrossRef]
- Jickells, S. Sample Preparation. In Analytical Techniques in Forensic Science; John Wiley & Sons: Hoboken, NJ, USA, 2021; pp. 71–108. [Google Scholar] [CrossRef]
- Badawy, M.E.; El-Nouby, M.A.; Kimani, P.K.; Lim, L.W.; Rabea, E.I. A review of the modern principles and applications of solid-phase extraction techniques in chromatographic analysis. Anal. Sci. 2022, 38, 1457–1487. [Google Scholar] [CrossRef]
- Wan, Q.; Liu, H.; Deng, Z.; Bu, J.; Li, T.; Yang, Y.; Zhong, S. A critical review of molecularly imprinted solid phase extraction technology. J. Polym. Res. 2021, 28, 401. [Google Scholar] [CrossRef]
- Jiang, Q.; Zhang, S.; Sun, M. Recent advances on graphene and graphene oxide as extraction materials in solid-phase (micro) extraction. TrAC. Trends Anal. Chem. 2023, 168, 117283–117301. [Google Scholar] [CrossRef]
- Karaket, R.; Khattiya, A.; Lerdpiriyaskulkij, N.; Mathaweesansurn, A.; Detsri, E. Covalent organic frameworks blended cellulose nanocrystal for in-needle syringe solid phase extraction of polycyclic aromatic hydrocarbons in dark roasted coffee. Microchem. J. 2024, 206, 111566–111588. [Google Scholar] [CrossRef]
- Wen, A.; Li, G.; Wu, D.; Yu, Y.; Yang, Y.; Hu, N.; Wu, Y. Sulphonate functionalized covalent organic framework-based magnetic sorbent for effective solid phase extraction and determination of fluoroquinolones. J. Chromatogr. A 2020, 1612, 460651–460689. [Google Scholar] [CrossRef]
- Aguilar, J.F.; Miranda, J.M.; Rodriguez, J.A.; Paez-Hernandez, M.E.; Ibarra, I.S. Selective removal of tetracycline residue in milk samples using a molecularly imprinted polymer. J. Polym. Res. 2020, 27, 176–188. [Google Scholar] [CrossRef]
- Murdaya, N.; Triadenda, A.L.; Rahayu, D.; Hasanah, A.N. A review: Using multiple templates for molecular imprinted polymer: Is it good? Polymers 2022, 14, 4441. [Google Scholar] [CrossRef]
- Tlili, A.; Attia, G.; Khaoulani, S.; Mazouz, Z.; Zerrouki, C.; Yaakoubi, N.; Fourati, N. Contribution to the understanding of the interaction between a polydopamine molecular imprint and a protein model: Ionic strength and pH effect investigation. Sensors 2021, 21, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Ansari, A.; Li, Z.; Mazumdar, H.; Siddiqui, M.A.; Khan, A.; Kumar, P. Point-of-Care Health Diagnostics and Food Quality Monitoring by Molecularly Imprinted Polymers-Based Histamine Sensors. Adv. Sens. Res. 2025, 4, 2400132–2400157. [Google Scholar] [CrossRef]
- Azizi, A.; Shahhoseini, F.; Langille, E.A.; Akhoondi, R.; Bottaro, C.S. Micro-gel thin film molecularly imprinted polymer coating for extraction of organophosphorus pesticides from water and beverage samples. Anal. Chim. Acta 2021, 1187, 339135–339152. [Google Scholar] [CrossRef]
- Gerdan, Z.; Saylan, Y.; Uğur, M.; Denizli, A. Ion-imprinted polymer-on-a-sensor for copper detection. Biosensors 2022, 12, 91–105. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, L.; Zeng, J.; Hu, X.; Wang, X.; Yu, L.; Zhang, Z. Multi-templates molecularly imprinted polymers for simultaneous recognition of multiple targets: From academy to application. TrAC Trends Anal. Chem. 2023, 166, 117173–117197. [Google Scholar] [CrossRef]
- Aurelio-Soria, D.; Canales, X.H.; Vázquez-Garrido, I.; Islas, G.; Álvarez-Romero, G.A.; Ibarra, I.S. Design of Selective Nanoparticles of Layered Double Hydroxide (Mg/Al-LDH) for the Analysis of Anti-Inflammatory Non-Steroidal Agents in Environmental Samples, Coupled with Solid-Phase Extraction and Capillary Electrophoresis. Separations 2024, 11, 259–273. [Google Scholar] [CrossRef]
- Cormack, P.A.; Elorza, A.Z. Molecularly imprinted polymers: Synthesis and characterisation. J. Chromatogr. B 2004, 804, 173–182. [Google Scholar] [CrossRef]
- Hasanah, A.N.; Safitri, N.; Zulfa, A.; Neli, N.; Rahayu, D. Factors affecting preparation of molecularly imprinted polymer and methods on finding template-monomer interaction as the key of selective properties of the materials. Molecules 2021, 26, 5612–5633. [Google Scholar] [CrossRef]
- Mueller, A. A note about crosslinking density in imprinting polymerization. Molecules 2021, 26, 5139–5149. [Google Scholar] [CrossRef]
- Sikiti, P.; Msagati, T.A.; Mamba, B.B.; Mishra, A.K. Synthesis and characterization of molecularly imprinted polymers for the remediation of PCBs and dioxins in aqueous environments. J. Environ. Health Sci. Eng. 2014, 12, 82. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; She, X.; Wang, L.; Fan, H.; Zhou, Q.; Huang, X.; Tang, J.Z. Preparation, characterization and application of a molecularly imprinted polymer for selective recognition of sulpiride. Materials 2017, 10, 475–491. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Pham, H.Q.; Nguyen, D.A.S.; Nguyen, L.T.; Huynh, K.P.H.; Le Tran, H.; Mai, P.T.; Nguyen, H.T.; Truong, T.T. 10-(pyren-1-yl)-10h-phenothiazine and pyrene as organic catalysts for photoinitiated ATRP of 4-vinylpyridine. Polímeros 2021, 31, 2021001–2021008. [Google Scholar] [CrossRef]
- Flores-Aguilar, J.F.; Islas, G.; Rodríguez, J.A.; Paez-Hernandez, M.E.; Galán-Vidal, C.A.; Ibarra, I.S. Selective Pb (II)-imprinted polymer for solid phase extraction in the trace determination of lead in infant formula by capillary electrophoresis. J. Mex. Chem. Soc. 2022, 66, 221–236. [Google Scholar] [CrossRef]
- Dada, A.O.; Lekan, A.P.; Olatunya, A.M.; Dada, O. Langmuir, Freundlich, Temkin and Dubinin-Radushkevich isotherms studies of equilibrium sorption of Zn2+ unto phosphoric acid modified rice husk. J. Appl. Chem. 2012, 3, 38–45. [Google Scholar] [CrossRef]
- Yamin, M.; Ghouri, Z.K.; Rohman, N.; Syed, J.A.; Skelton, A.; Ahmed, K. Unravelling pH/pKa influence on pH-responsive drug carriers: Insights from ibuprofen-silica interactions and comparative analysis with carbon nanotubes, sulfasalazine, and alendronate. J. Mol. Graph. Model. 2024, 128, 108720–108736. [Google Scholar] [CrossRef]
- Madikizela, L.M.; Chimuka, L. Synthesis, adsorption and selectivity studies of a polymer imprinted with naproxen, ibuprofen and diclofenac. J. Environ. Chem. Eng. 2016, 4, 4029–4037. [Google Scholar] [CrossRef]
- Currie, L.A. Nomenclature in evaluation of analytical methods including detection and quantification capabilities (IUPAC Recommendations 1995). Pure Appl. Chem. 1995, 67, 1699–1723. [Google Scholar] [CrossRef]
- Burlingame, G.A.; Dietrich, A.M.; Whelton, A.J. Understanding the basics of tap water taste. J. Am. Water Works Assoc. 2007, 99, 100–111. [Google Scholar] [CrossRef]
- Akhter, F.; Nag, A.; Alahi, M.E.E.; Liu, H.; Mukhopadhyay, S.C. Electrochemical detection of calcium and magnesium in water bodies. Sens. Actuators A Phys. 2020, 305, 111949–111959. [Google Scholar] [CrossRef]
- Costa Junior, I.L.; Machado, C.S.; Pletsch, A.L.; Torres, Y.R. Simultaneous HPLC-PDA determination of commonly prescribed antidepressants and caffeine in sludge from sewage treatment plants and river sediments in the Itaipu reservoir region, Paraná, Brazil. Int. J. Environ. Anal. Chem. 2020, 100, 1004–1020. [Google Scholar] [CrossRef]
- McCarthy, B.; Apori, S.O.; Giltrap, M.; Bhat, A.; Curtin, J.; Tian, F. Hospital effluents and wastewater treatment plants: A source of oxytetracycline and antimicrobial-resistant bacteria in seafood. Sustainability 2021, 13, 13967–13989. [Google Scholar] [CrossRef]
- Payán, M.R.; López, M.Á.B.; Torres, R.F.; Navarro, M.V.; Mochón, M.C. Electromembrane extraction (EME) and HPLC determination of non-steroidal anti-inflammatory drugs (NSAIDs) in wastewater samples. Talanta 2011, 85, 394–399. [Google Scholar] [CrossRef]
- Dahane, S.; Galera, M.M.; Marchionni, M.E.; Viciana, M.S.; Derdour, A.; García, M.G. Mesoporous silica based MCM-41 as solid-phase extraction sorbent combined with micro-liquid chromatography–quadrupole-mass spectrometry for the analysis of pharmaceuticals in waters. Talanta 2016, 152, 378–391. [Google Scholar] [CrossRef]
- Toledo-Neira, C.; Álvarez-Lueje, A. Ionic liquids for improving the extraction of NSAIDs in water samples using dispersive liquid–liquid microextraction by high performance liquid chromatography-diode array–fluorescence detection. Talanta 2015, 134, 619–626. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coded (Uncoded) | %Extraction | Output Variable | ||||||
|---|---|---|---|---|---|---|---|---|
| Exp | A | B | C | ACE | IBP | NPX | DCF | Average |
| 1 | 0.00 (0.20) | 0.00 (4.00) | 1.00 (40.00) | 38.00 | 97.15 | 98.50 | 98.50 | 83.04 |
| 2 | 0.50 (0.50) | 0.00 (4.00) | 0.50 (30.00) | 41.00 | 97.15 | 98.50 | 98.50 | 83.79 |
| 3 | 0.33 (0.40) | 0.33 (5.33) | 0.33 (26.66) | 47.32 | 97.15 | 98.50 | 98.50 | 85.37 |
| 4 | 0.16 (0.30) | 0.16 (4.67) | 0.66 (33.33) | 46.00 | 97.15 | 98.50 | 98.50 | 85.04 |
| 5 | 1.00 (0.80) | 0.00 (4.00) | 0.00 (20.00) | 43.00 | 97.15 | 98.50 | 98.50 | 84.29 |
| 6 | 0.16 (0.30) | 0.66 (6.66) | 0.16 (23.33) | 50.00 | 97.15 | 98.50 | 98.50 | 86.04 |
| 7 | 0.67 (0.60) | 0.16 (4.67) | 0.16 (23.33) | 52.00 | 97.15 | 98.50 | 98.50 | 86.54 |
| 8 | 0.50 (0.50) | 0.50 (6.00) | 0.00 (20.00) | 46.00 | 97.15 | 98.50 | 98.50 | 85.04 |
| 9 | 0.00 (0.20) | 0.50 (6.00) | 0.50 (30.00) | 46.00 | 97.15 | 98.50 | 98.50 | 85.04 |
| 10 | 0.00 (0.20) | 1.00 (8.00) | 0.00 (20.00) | 46.00 | 97.15 | 98.50 | 98.50 | 85.04 |
| Source | D.F | ADJ SS | ADJ MS | F-Value | p-Value |
|---|---|---|---|---|---|
| Model | 5 | 6.26 | 1.25 | 1.62 | 0.33 |
| Linear | 2 | 3.34 | 1.19 | 1.54 | 0.32 |
| Square | 3 | 2.92 | 0.97 | 1.26 | 0.40 |
| A*B | 1 | 0.76 | 0.78 | 1.01 | 0.37 |
| A*C | 1 | 0.44 | 0.45 | 0.58 | 0.49 |
| B*C | 1 | 1.72 | 1.72 | 2.24 | 0.21 |
| Error | 4 | 3.09 | 0.77 | ||
| Total | 9 | 9.35 |
| Langmuir | Dubinin–Radushkevich | ||||
|---|---|---|---|---|---|
| Qmax (mg g−1) | RL | r2 | E (kJ mol−1) | r2 | |
| ACE | 2.71 | 0.38 | 0.99 | 12.91 | 0.98 |
| IBP | 2.97 | 0.39 | 0.99 | 11.95 | 0.99 |
| NPX | 5.46 | 0.17 | 0.96 | 14.43 | 0.96 |
| DCF | 5.73 | 0.67 | 0.99 | 8.33 | 1.00 |
| IBP | NPX | DCF | ||
|---|---|---|---|---|
| Correlation coefficient, r2 | 0.9989 | 0.9999 | 0.9999 | |
| Intercept | ||||
| b0 ± ts(b0) | 0.3796 ± 0.1284 | 0.4705 ± 0.1929 | 0.3558 ± 0.0938 | |
| Slope | ||||
| b1 ± ts(b1) | 21.7960 ± 1.1198 | 79.4721 ± 2.0442 | 52.7654 ± 0.7321 | |
| Inter-day repeatability (RSD %, n = 3) | 50.00 µg L−1 | 4.68 | 5.78 | 6.56 |
| 100.00 µg L−1 | 3.97 | 2.89 | 4.83 | |
| Intra-day repeatability (RSD %, n = 3) | 50.00 µg L−1 | 8.56 | 9.45 | 8.12 |
| 100.00 µg L−1 | 6.13 | 6.21 | 7.44 | |
| Linearity range (µg L−1) | 36.00–200.00 | 12.00–150.00 | 10.00–200.00 | |
| Limit of detection (µg L−1) | 12.00 | 4.00 | 3.00 | |
| Limit of quantification (µg L−1) | 36.00 | 12.00 | 10.00 |
| Added (μg L−1) | IBP | NPX | DCF | |||
|---|---|---|---|---|---|---|
| % Recovery | % RSD | % Recovery | % RSD | % Recovery | % RSD | |
| 0 | ND | - | ND | - | ND | - |
| 50 | 97.02 | 7.21 | 96.31 | 8.01 | 95.52 | 7.34 |
| 75 | 98.38 | 4.37 | 99.63 | 5.24 | 97.82 | 5.06 |
| Method | Analytes | Instrument | Matrix | LOD (μg L−1) | REF |
|---|---|---|---|---|---|
| Derivatization | IBP, ACE, NPX, and DCF | GC-MS | Human serum | 6–414 | [45] |
| EME | SAC, KTR, KTP, NPX, DCF, and IBP | HPLC-FLD | Water | 0.009–9.0 | [38] |
| SPE | IPB, NPX, DCF, and KTP | μ-LC-MS/MS | Water | 0.23–3.85 | [46] |
| DLLME | KTP, IBP, and DCF | HPLC-FLC | Water | 17–95 | [47] |
| SPE | IBP, NPX, and DCF | CE-UV | Water | 3–12 | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aurelio-Soria, D.; Alvarez-Romero, G.A.; Paez-Hernandez, M.E.; Perez-Silva, I.; Franco-Guzman, M.; Islas, G.; Ibarra, I.S. Multi-Template Molecularly Imprinted Polymers Coupled with a Solid-Phase Extraction System in the Selective Determination of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) in Environmental Water Samples. Separations 2025, 12, 140. https://doi.org/10.3390/separations12060140
Aurelio-Soria D, Alvarez-Romero GA, Paez-Hernandez ME, Perez-Silva I, Franco-Guzman M, Islas G, Ibarra IS. Multi-Template Molecularly Imprinted Polymers Coupled with a Solid-Phase Extraction System in the Selective Determination of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) in Environmental Water Samples. Separations. 2025; 12(6):140. https://doi.org/10.3390/separations12060140
Chicago/Turabian StyleAurelio-Soria, David, Giaan A. Alvarez-Romero, Maria E. Paez-Hernandez, I. Perez-Silva, Miriam Franco-Guzman, Gabriela Islas, and Israel S. Ibarra. 2025. "Multi-Template Molecularly Imprinted Polymers Coupled with a Solid-Phase Extraction System in the Selective Determination of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) in Environmental Water Samples" Separations 12, no. 6: 140. https://doi.org/10.3390/separations12060140
APA StyleAurelio-Soria, D., Alvarez-Romero, G. A., Paez-Hernandez, M. E., Perez-Silva, I., Franco-Guzman, M., Islas, G., & Ibarra, I. S. (2025). Multi-Template Molecularly Imprinted Polymers Coupled with a Solid-Phase Extraction System in the Selective Determination of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) in Environmental Water Samples. Separations, 12(6), 140. https://doi.org/10.3390/separations12060140