Abstract
This study researches the impact of diclofenac (DCF) oxidation via UV/H2O2 on water quality, focusing on aromaticity and color changes. The process effectively degrades DCF and its intermediates through hydroxyl radical attack on the aromatic structure, leading to the formation of oxidized by-products. Initially, chromophoric compounds such as quinones and conjugated intermediates cause a yellow coloration, which diminishes as mineralization progresses. Turbidity remains below 1 NTU, aligning with European water quality standards. Aromaticity initially increases due to the stable intermediates (e.g., catechols and hydroquinones) but decreases as advanced oxidation cleaves aromatic rings. Kinetic modeling shows that DCF degradation follows first-order kinetics, while aromatic intermediates degrade via fractional-order kinetics (~0.3), indicating a non-linear relationship with concentration. The formation of chromophore compounds follows first-order kinetics, whereas their degradation transitions to zero-order kinetics when hydroxyl radicals are abundant. The study highlights the environmental relevance of these transformations, as aromatic intermediates like anilines and phenols, which contribute to water toxicity, are ultimately converted into less hazardous compounds (e.g., carboxylic acids and inorganic ions). Experimental validation confirms that degradation kinetics depend on hydrogen peroxide concentration, underscoring the potential of UV/H2O2 for water purification and pollutant removal.
1. Introduction
Recently, increasing attention has been paid to the presence of pharmaceutically active compounds (PhACs) in the environment, as they represent one of the most significant groups of emerging aquatic contaminants (ECs). As global population increases, the consumption of pharmaceuticals also increases, resulting in higher concentrations of these compounds being present in the environment, which in turn negatively impacts various organisms [1]. Additionally, widely used pharmaceuticals have an increasing impact on human health. Due to their low removal efficiency, they are persistently detected in water bodies, even in the most advanced wastewater treatment plants. One of the most commonly detected drugs in natural waters is diclofenac (2-(2-(2,6-dichlorophenylamino)phenyl) acetic acid, or DCF). This compound is a nonsteroidal anti-inflammatory drug (NSAID) that is commonly used as an analgesic, antiarthritic, and antirheumatic agent [2,3]. DCF can interact with other inorganic contaminants, such as metals, as well as with other organic contaminants or even with its own metabolites. This interaction may lead to the formation of additional emerging contaminants [4].
Annual sales of DCF can reach up to 100 tons in some countries [5], and in 2012, it was ranked as the twelfth best-selling generic molecule worldwide by Fierce Pharma [4]. The European Geosciences Union estimated that global annual DCF consumption in 2018 was approximately 2400 tons [6], with Asia and Europe accounting for 40% and 29% of the total DCF consumption, respectively [7,8]. Although controlling DCF at the source is the most effective measure, the application of an appropriate water treatment remains crucial for reducing its toxicity [5].
Traditional treatment technologies (sedimentation, coagulation, and biological processes) are inadequate for PhAC removal, given their low concentrations and high chemical stability [1,9,10]. Conventional treatments are not always sufficient to meet the growing demands regarding water quality, especially when faced with persistent and difficult-to-remove contaminants, such as pharmaceuticals. For this reason, new technological alternatives, including Advanced Oxidation Processes (AOPs), have been developed and have become areas of intense research. AOPs, particularly UV/H2O2 systems, have shown promise in degrading recalcitrant organics such as DCF due to their ability to generate highly reactive hydroxyl radicals. The catalytic behavior of the UV/H2O2 system demonstrates significant advantages when compared to other advanced oxidation processes, such as ozonation, TiO2/UV photocatalysis, and the photo-Fenton reaction [11,12,13,14,15,16,17,18,19]. Unlike ozonation, which often requires the fine-tuning of pH and suffers from limited selectivity toward specific pollutants, the UV/H2O2 process rapidly generates non-selective hydroxyl radicals under UV light without introducing gas-phase reactants. Compared to TiO2-based photocatalysis, UV/H2O2 avoids issues related to catalyst recovery and surface fouling, offering a homogeneous reaction environment with faster degradation kinetics in many cases. Furthermore, in contrast to the photo-Fenton process, which depends on the presence of iron and is pH-sensitive (optimal around pH 3), the UV/H2O2 system operates effectively over a broader pH range and does not generate metal sludge, making it more environmentally benign and operationally simpler. These characteristics position UV/H2O2 as a highly efficient and practical method for the degradation of recalcitrant organic pollutants such as diclofenac, particularly in applications where low sludge production, rapid treatment times, and minimal system complexity are desired.
In this study, the UV/H2O2 advanced oxidation process was selected over catalyzed alternatives due to its high efficiency, simplicity, and suitability for modeling the degradation kinetics of micropollutants in aqueous environments. The photolysis of hydrogen peroxide under UV irradiation rapidly generates hydroxyl radicals (HO●), which are highly reactive and non-selective oxidants capable of effectively degrading aromatic pharmaceuticals such as DCF. Unlike heterogeneous photocatalytic systems (e.g., TiO2) or Fenton-based processes, the UV/H2O2 system does not introduce secondary contaminants or catalyst residues into the treated water, thereby minimizing the risk of post-treatment pollution. Moreover, the homogeneous nature of the reaction medium facilitates accurate kinetic modeling, particularly in relation to changes in aromaticity and color—key indicators of water quality deterioration during pharmaceutical oxidation. The operational simplicity and scalability of the UV/H2O2 process further support its relevance for practical water treatment applications, making it a suitable choice for investigating the mechanistic and environmental implications of diclofenac oxidation.
DCF exhibits complex behaviors in aquatic environments, particularly when subjected to AOPs such as UV/H2O2 treatment. In such systems, DCF can interact with both inorganic and organic co-contaminants through various mechanisms that influence its degradation kinetics and the nature of its transformation products. In the presence of inorganic constituents, DCF primarily undergoes indirect photodegradation via reactions with hydroxyl radicals (HO●). However, common anions such as bicarbonate (HCO3−), chloride (Cl−), nitrate (NO3−), and nitrite (NO2−) can act as radical scavengers, reducing the availability of HO● for DCF oxidation and consequently diminishing its degradation efficiency. Additionally, interactions with reactive nitrogen species derived from nitrite or nitrate can lead to nitration or nitrosation of the DCF molecule, potentially generating transformation products with enhanced toxicity [4,20].
Simultaneously, DCF can interact with co-existing organic pollutants through competitive and reactive pathways. Other organic compounds may compete for HO●, altering the degradation pathway and efficiency of DCF. Furthermore, intermediate species generated during DCF oxidation, such as phenoxy radicals or quinone-type structures, may engage in secondary reactions with the surrounding organic matter. These interactions can result in the formation of complex oligomeric products or other by-products with potentially greater environmental persistence or toxicity than the parent compound [21,22]. Overall, the presence of inorganic and organic contaminants in water matrices significantly affects the transformation pathways of DCF under oxidative conditions. These interactions highlight the importance of evaluating water quality parameters when assessing the efficiency and safety of DCF degradation in environmental and engineered treatment systems.
Several investigations have explored the DCF degradation kinetics and pathways under AOPs [22,23,24,25,26,27,28]. For instance, Vogna et al. (2004) [29] analyzed the oxidative degradation of DCF and identified hydroxylated intermediates and C–N cleavage products. However, despite advances in the identification of transformation products, many aspects of the process remain unresolved, including the fate of aromatic intermediates and their environmental relevance. Recent studies, such as those by Sohn et al. (2024) [30], have demonstrated the efficacy of UV/H2O2 in eliminating micropollutants in drinking water treatment facilities.
Building upon these studies, the present work introduces a novel and comprehensive kinetic modeling approach for the UV/H2O2 degradation of DCF, addressing not only the removal of the parent compound, but also the temporal evolution of aromaticity and visible coloration as indirect indicators of intermediate formation and potential toxicity. Unlike previous works [26,30], this study incorporates differentiated kinetic models for three key aspects of the oxidation process: the degradation of DCF, the evolution of aromacticity, and the formation and degradation of a chromofore, modeling the characteristic yellow coloration observed during DCF oxidation. This multi-kinetic approach enables a deeper understanding of the behavior of aromatic intermediates, whose persistence may have ecotoxicological implications.
Furthermore, the study explores the correlations between structural transformations, water quality indicators (aromaticity, color, and turbidity), and environmental compliance metrics, such as the turbidity limit established by Directive 2020/2184. The model is validated using experimental data and proposes a predictive framework that is applicable to other pharmaceuticals and aromatic pollutants, expanding the practical applicability of AOPs in water treatment. This integrated approach—combining environmental chemistry, reaction kinetics, and water quality assessments—provides original contributions in the field of sustainable water treatment. It delivers practical insights for the design and optimization of UV/H2O2 systems, and highlights the importance of monitoring not only the parent compound but also key intermediate indicators. These features make this work particularly relevant for environmental engineering, wastewater treatment, and applied chemistry.
The study presents a significant advancement in the field of pharmaceutical degradation by introducing a comprehensive multi-kinetic model for DCF degradation using the UV/H2O2 process. While previous research has often relied on simplified kinetic models to describe the degradation of DCF, this study expands upon that foundation by integrating distinct kinetic regimes for different aspects of the degradation process. Specifically, it models the parent compound degradation, the formation and breakdown of aromatic intermediates, and the evolution of color-causing species separately. This layered modeling approach allows for a more accurate and realistic depiction of the multistep oxidative degradation pathway, capturing the dynamic changes in reaction rates as intermediates form and degrade over time.
A key innovation of this work is its mechanistic resolution. The kinetic modeling is not limited to empirical rate constants but is instead directly linked to the specific chemical transformations occurring during the degradation process—such as hydroxylation, ring cleavage, and the formation of quinones or phenolic intermediates. By correlating the kinetic behavior with changes in aromaticity, turbidity, and color, the model provides valuable insight into how reaction conditions influence not only the removal of the parent compound but also the transformation and eventual mineralization of potentially toxic intermediates. This represents a considerable step forward in understanding the environmental fate of DCF and similar aromatic pollutants.
2. Materials and Methods
The experimental assays were conducted in a photocatalytic reactor equipped with a 150 W UV lamp (medium-pressure mercury lamp, TQ 150-Heraeus, 230 V/50 Hz, 2.5 A), and homogenized via a magnetic stirrer operating at 300 rpm. The emission spectrum of the lamp was suitable for the intended reactions, with approximately 95% transmission between 300 and 570 nm. A total of 1.0 L of a synthetic aqueous solution of sodium diclofenac (DCF; Fragon, purity > 99.5%) was prepared at an initial concentration of [DCF]0 = 50.0 mg/L and introduced into the reactor. The reaction was initiated by adding hydrogen peroxide (H2O2; PRS Panreac, 30% w/v) at varying initial concentrations ([H2O2]0 = 0–50.0 mM) specific to each experimental set. All experiments were conducted at the natural pH of the DCF solutions, approximately pH 5, and pH was monitored throughout using a calibrated pH meter (Kent EIL9142, ABB Kent-Taylor Ltd., Cambridge, UK). The temperature was maintained at a constant value of T = 25 °C using a 1150 W refrigerated thermostatic bath (Frigiterm-10, Selecta, Madrid, Spain), which circulated water through the reactor jacket housing the UV lamp [31].
An initial DCF concentration of 50.0 mg/L was selected to facilitate the detection and quantification of the parent compound and its transformation products using standard analytical techniques. Although environmental DCF concentrations are typically in the ng/L to low µg/L range, the elevated concentration enables a detailed analysis of the degradation kinetics, intermediate identification, and treatment optimization in a controlled laboratory setting. These findings can subsequently be extrapolated to environmentally relevant scenarios [32,33].
DCF concentration was quantified via High-Performance Liquid Chromatography (HPLC) using a Waters 2695 system coupled with a Dual Wavelength Absorbance Detector (Model 2487, Waters Cromatografía S.A., Cerdanyola del Vallès, Spain). Chromatographic separation was achieved with a Zorbax Eclipse PAH analytical column (150 mm × 4.6 mm; 5 μm particle size) and a matching guard column (4.6 mm × 12.5 mm), both supplied by Agilent Technologies (Santa Clara, CA, USA). The mobile phase consisted of water and acetonitrile (ACN), with a flow rate of 0.8 mL/min. The gradient elution began at 20% ACN, increased to 50% over 3 min, was maintained for 6 min, then returned to 20% at 10 min. The injection volume was 50 μL, and all separations were performed at ambient temperature. DCF was identified through comparison with the analytical standards and monitored at a detection wavelength of 275 nm.
Water aromaticity ([Arom], absorbance units, AU) at λ = 254 nm and color intensity ([Color], AU) at λ = 455 nm were assessed using a UV/Vis spectrophotometer (Jasco V-630) [32,33]. Turbidity ([Turbidity], NTU) was measured with a nephelometric turbidimeter (Hanna Instruments HI88703, Hanna Instruments Ltd., Leighton Buzzard, UK). Dissolved oxygen ([DO], mg/L) and temperature were monitored using a dissolved oxygen meter (Hanna Instruments HI9142).
3. Results
3.1. Oxidative Degradation of Diclofenac: Evolution of Water Quality Parameters Under UV/H2O2 Treatment
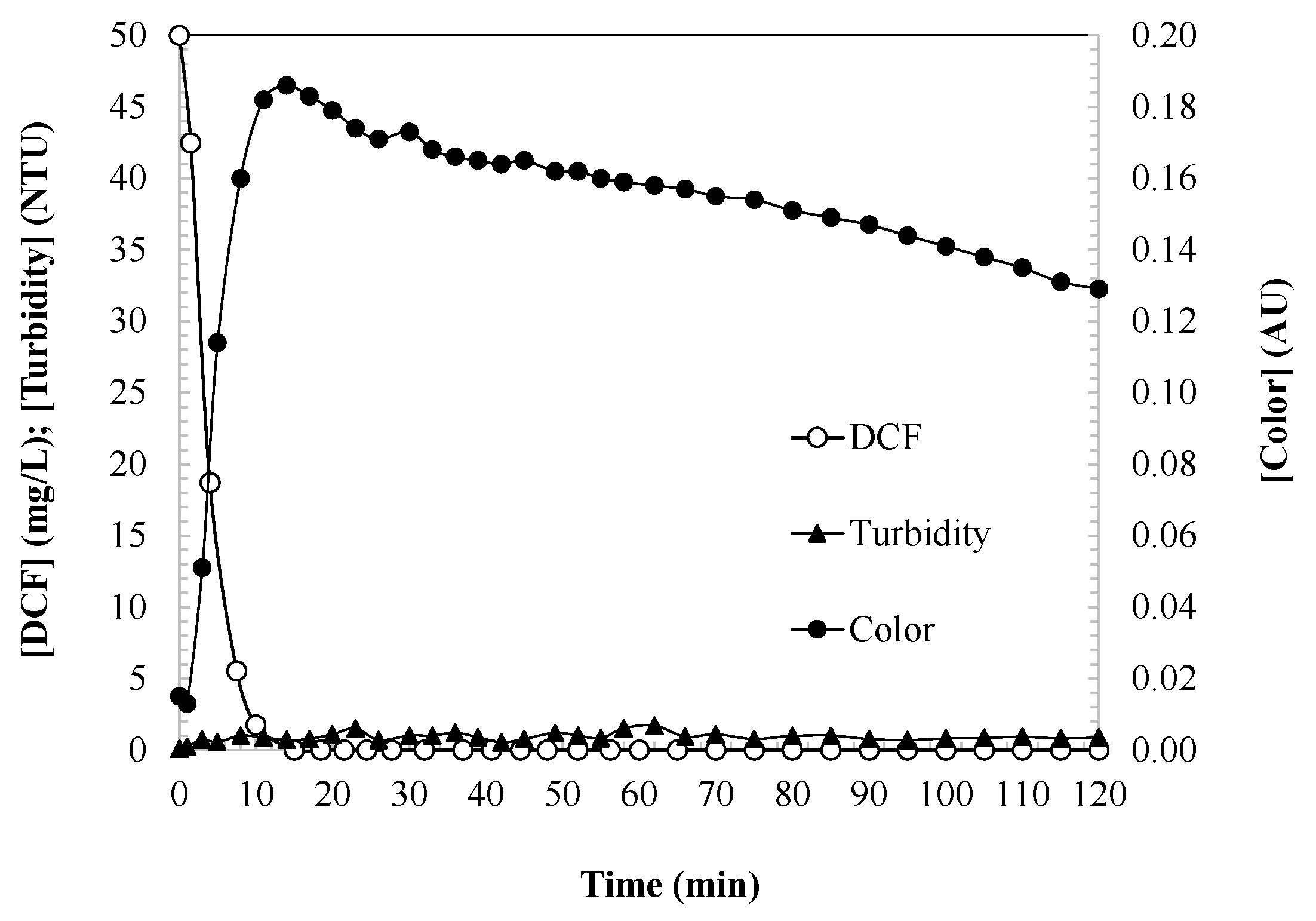
Figure 1 illustrates the concentration of DCF alongside the corresponding water color and turbidity. The UV/H2O2 system works through the generation of hydroxyl radicals via the photolysis of hydrogen peroxide under UV light. These radicals are responsible for the oxidative degradation of DCF, making the system effective for water purification, wastewater treatment, and environmental cleanup. The mechanism is continuous as long as the UV irradiation and H2O2 supply are maintained. The cycle of the UV/H2O2 photocatalytic activity mechanism can be described as having four general stages.
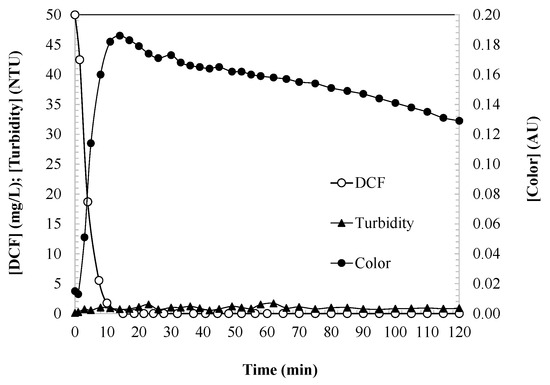
Figure 1.
DCF concentration, water color, and turbidity as indicators of DCF oxidation degree during the UV/H2O2 process. Experimental conditions: [DCF]0 = 50.0 mg/L; [H2O2]0 = 2.0 mM; [UV] = 150 W; and [T] = 25 °C.
UV Irradiation of H2O2: When hydrogen peroxide is exposed to UV light, it absorbs the UV photons, leading to its photosynthesis (breaking down of the molecule). The key reaction here is the decomposition of H2O2 into hydroxyl radicals and molecular oxygen. This reaction is typically initiated by UV light with wavelengths in the range of 200–300 nm, where H2O2 absorbs the photons and undergoes homolytic cleavage, producing two hydroxyl radicals (Equations (1)–(6)) [7,34].
Formation of Hydroxyl Radicals: The photolysis of H2O2 generates hydroxyl radicals, which are extremely reactive species that can initiate the oxidation of organic compounds. These radicals are among the strongest known oxidizers and are capable of breaking down many organic pollutants. The degradation of DCF in the presence of UV light and H2O2 occurs through the generation of hydroxyl radicals via the photolysis of H2O2 (Equation (1)). Hydroxyl radicals are highly reactive oxidizing agents that attack the chemical structure of DCF, breaking molecular bonds and forming oxidized degradation intermediates. The results demonstrate that UV light in combination with H2O2 is effective in oxidizing aqueous DCF molecules into degradation intermediates within 15 min. During the direct photolysis of hydrogen peroxide, a significant number of hydroxyl radicals are produced. These non-selective and highly reactive oxidants play a pivotal role in the rapid degradation of DCF, thereby influencing the overall efficiency of the treatment.
Oxidation of DCF: The generated hydroxyl radicals can attack DCF, leading to degradation (Equation (7)). The mechanism of this degradation depends on the nature of the pollutants, but in general, the hydroxyl radical can be added to various organic molecules, breaking them down into smaller, often less toxic or more easily degradable products.
The oxidative degradation of DCF initiates with hydroxyl radicals targeting the aromatic rings and amine groups of the molecule. These attacks typically lead to the hydroxylation of the aromatic structures and cleavage of the C–N bond, resulting in the formation of hydroxylated intermediates and various smaller nitrogen-containing fragments, such as anilines. These early transformations significantly alter the chemical structure and reactivity of DCF, thereby reducing its persistence in the aqueous phase. As oxidation progresses, stable aromatic intermediates are formed, including chlorinated phenols, quinone-type structures (oxidized catechols), and other conjugated compounds. These intermediates often contribute to the yellowish coloration observed in the treated solution, indicating that the complete degradation of aromaticity has not yet occurred. The presence of such chromophoric and potentially toxic compounds underscores the importance of controlling the reaction conditions to prevent the accumulation of harmful byproducts. With continued UV exposure and sufficient hydroxyl radical availability, the oxidation process proceeds through ring-opening reactions and the further transformation of intermediate products. Ultimately, the aromatic rings are degraded into aliphatic carboxylic acids, aldehydes, and other low-molecular-weight compounds. Under optimized conditions, this process can lead to the complete mineralization of DCF into carbon dioxide and water [23].
Regeneration of H2O2: This reaction is typically catalytic in nature, meaning H2O2 can be continuously regenerated and decomposed as long as UV light is supplied. In practice, the system does not require a constant supply of fresh H2O2 for each cycle; it just needs an ongoing UV irradiation to continue photolyzing H2O2 into hydroxyl radicals. As long as UV light is present, H2O2 is continuously decomposed to generate more hydroxyl radicals, maintaining the photocatalytic activity.
Color and turbidity are key parameters regulated by EU water quality standards (Directive 2000/60/EC). The aqueous DCF samples initially appear colorless, but during the first 15 min of the reaction, as DCF is degraded, a strong yellow tint develops in the water, intensifying until it reaches a maximum. DCF is susceptible to degradation due to its aromatic structure and amino group, which facilitate reactions with hydroxyl radicals. These radicals attack the double bonds of the aromatic rings, generating intermediate oxidized compounds. As degradation progresses, DCF is oxidized, leading to the formation of various intermediate products, such as quinones, hydroxylated aromatic rings, and compounds with conjugated double bonds. These products, particularly quinones and other oxidized aromatic derivatives, absorb visible light, imparting coloration to the water. If the reaction continues under appropriate conditions (sufficient reaction time, and adequate H2O2 and UV exposure), these oxidized intermediates further degrade to yield final products, including carbon dioxide, water, and inorganic ions such as chloride and nitrate. Subsequently, the color intensity decreases as the intermediate compounds evolve, stabilizing at a persistent steady-state value.
Turbidity is a critical parameter that is directly related to the efficacy of the disinfection processes, whether chemical (e.g., chlorine and ozone) or physical (e.g., UV radiation) [32]. The EU Directive 2020/2184 on the quality of water intended for human consumption stipulates that turbidity should remain below 1 NTU. Additionally, Regulation (EU) 2020/74 for water reuse specifies that regenerated water for agricultural irrigation must have a turbidity ≤ 5 NTU. In this study, the turbidity of the treated samples is consistently below 1 NTU, indicating that the DCF degradation intermediates do not contribute to turbidity.
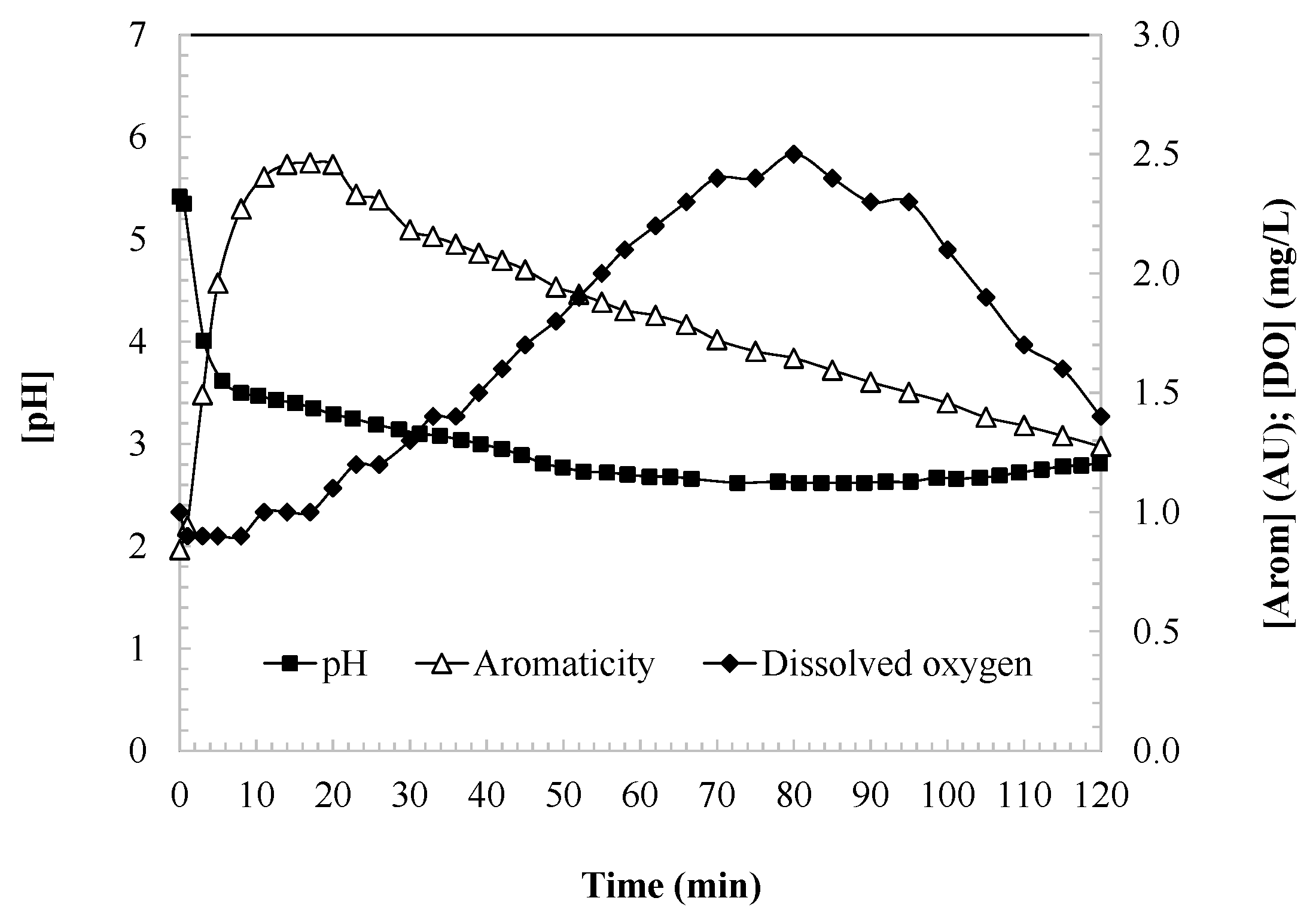
Figure 2 illustrates the evolution of aromaticity, the dissolved oxygen (DO), and the pH during the oxidation of DCF. In the first 15 min of the reaction, a significant increase in aromaticity is observed, reaching a peak that likely corresponds to the stoichiometric transformation of DCF into aromatic intermediates. DCF possesses an aromatic framework comprising a biphenyl nucleus (two fused benzene rings) and functional groups such as amino and carboxyl moieties. Hydroxyl radicals preferentially target the aromatic rings via hydroxylation, introducing hydroxyl groups into the structure, while the amino group is also susceptible to oxidation, facilitating ring opening or structural modification.
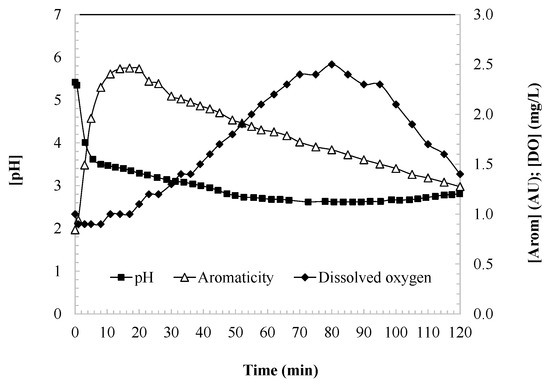
Figure 2.
Aromaticity, dissolved oxygen, and pH as indicators of the degree of DCF oxidation during the UV/H2O2 process. Experimental conditions: [DCF]0 = 50.0 mg/L; [H2O2]0 = 2.0 mM; [UV] = 150 W; ad [T] = 25 °C.
During the early stages of oxidation, hydroxylation of the aromatic rings occurs without immediate loss of aromaticity. Instead, these modifications lead to the formation of oxidized aromatic intermediates, such as catechols and quinones. These compounds are relatively stable and maintain a partial or complete aromatic character, thereby accounting for the observed increase in aromaticity within the first 20 min of treatment.
Following the peak in aromaticity, a gradual decline is observed as the oxidation process advances. This reduction corresponds to the progressive degradation of aromatic intermediates into non-aromatic compounds. Persistent aromatic species that resist further oxidation contribute to a residual, stable level of aromaticity. As hydroxyl radicals continue to act on these intermediates, the aromatic rings are ultimately cleaved, leading to partial mineralization and the formation of simpler compounds, such as carboxylic acids, carbon dioxide (CO2), and water (H2O).
Simultaneously, the concentration of dissolved oxygen (DO) increases due to the release of oxygen as a byproduct of the radical-based reaction mechanisms (Equations (4)–(6)) [35,36,37]. DO levels rise until a maximum is attained, at which point the consumption of hydrogen peroxide begins to outweigh the generation of oxygen, resulting in a gradual but sustained decrease in DO concentration.
Additionally, the pH of the solution decreases throughout the oxidation process until reaching a stable, long-term value. This decline in pH is attributed to reactions such as that described in Equation (3), in which hydroperoxy radical species decompose to generate superoxide anions and release protons into the aqueous medium [34].
While the observed pH drop—from an initial value of approximately 5.4 to below 3 (Figure 2)—has been attributed to the decomposition of hydroperoxy radicals into superoxide anions and protons (Equation (3)), this mechanism may not fully account for the magnitude of acidification observed. The formation of superoxide anions in aqueous systems typically occurs in much lower concentrations (nanomolar to micromolar range), which is unlikely to lead to proton accumulation corresponding to a [H+] of 10−3 mol/L (pH ≈ 3). A more plausible and significant contributor to the pH decrease is the formation of acidic organic by-products, particularly phenolic and carboxylic compounds. As the UV/H2O2 process progresses, aromatic rings in diclofenac and its intermediates undergo hydroxylation, ring opening, and further oxidation, ultimately yielding aliphatic carboxylic acids such as formic, acetic, and oxalic acids. These compounds are well known to significantly acidify aqueous media. This interpretation aligns with the chemical behavior commonly observed in advanced oxidation processes, where the oxidation of aromatic structures leads to the generation of acidic degradation products. Therefore, the decline in pH is likely the result of a combined effect, where radical-induced proton release is supplemented—and possibly dominated—by the accumulation of organic acids formed during the mineralization of the parent compound and its intermediates.
The gradual decrease in pH observed during the UV/H2O2 oxidation of DCF plays a critical role in modulating both reaction kinetics and the fate of oxidation intermediates. As the pH declines, the efficiency of hydroxyl radical generation improves due to the reduction in radical scavenging and enhanced H2O2 stability under mildly acidic conditions. This results in the accelerated degradation of DCF during the initial stages of the process. However, the acidification may also influence the stability of the intermediate byproducts; while some may degrade more readily under acidic conditions, others—particularly aromatic or halogenated compounds—could become more persistent. Therefore, a pH near its natural value (~5) offers a balance between effective hydroxyl radical production and favorable conditions for the further breakdown of intermediates, supporting efficient and complete pollutant removal.
3.2. Kinetic Evaluation of DCF Removal Based on a Pseudo-First-Order Model
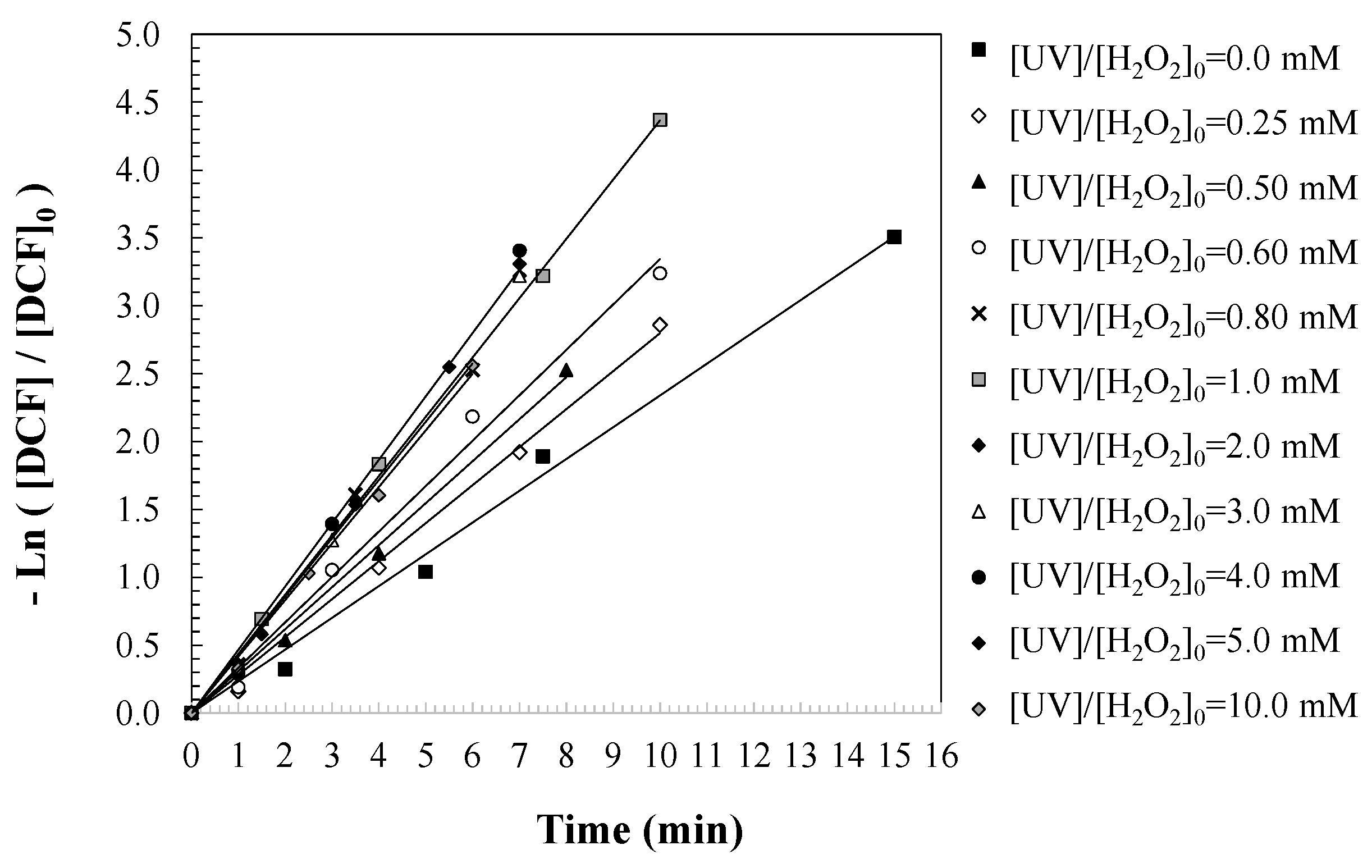
To evaluate the influence of initial hydrogen peroxide concentration on the degradation kinetics of DCF, a series of batch experiments were conducted at varying [H2O2]0 levels, while maintaining constant UV irradiation intensity and temperature. A pseudo-first-order kinetic model was applied to describe DCF degradation in the batch reactor, assuming that DCF is transformed into oxidized intermediates and by-products according to the reaction mechanism represented in Equation (8) [38,39]. By integrating the corresponding mass balance (Equation (9)), the first-order rate equation for DCF degradation was derived (Equation (10)). Temporal profiles of DCF concentration were monitored, and the apparent pseudo-first-order rate constants (kDCF, 1/min) were determined from the slopes of the linearized plots, as illustrated in Figure 3. These rate constants were then correlated with the respective initial [H2O2]0 values, as summarized in Table 1. It should be noted that a decrease in degradation rate was observed at H2O2 concentrations above 2.0 mM, with 50.0 mg/L of the pollutant being degraded. Similar results were observed at this concentration when degrading other contaminants, such as caffeine and sulfamethoxazole [36,40,41]. This phenomenon can be attributed to the scavenging effect of excess hydrogen peroxide, which reacts with hydroxyl radicals to form less reactive species such as HO2● and O2●−. These side reactions reduce the availability of hydroxyl radicals for DCF degradation and may also trigger non-selective oxidation pathways, potentially altering the formation and evolution of intermediate products. These findings highlight the importance of optimizing the oxidant dose to avoid radical-induced side reactions that impair treatment efficiency.
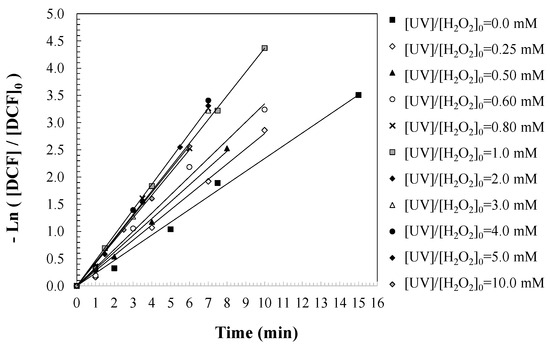
Figure 3.
Predictions of DCF degradation based on the first-order kinetic model under UV/H2O2 treatment. Experimental conditions: [DCF]0 = 50.0 mg/L; [UV] = 150 W; and [T] = 25 °C.
Mass balance for the degradation of DCF:
First-order kinetic equation for degradation of DCF:
To develop a predictive model for kDCF as a function of hydrogen peroxide concentration, a polynomial regression analysis was performed, treating [H2O2]0 as the independent variable. The resulting empirical equation (Equation (11)) incorporates terms up to the fifth order to accurately capture the observed non-linear dependency across the experimental range. This non-linearity reflects the complex dynamics of hydroxyl radical formation, competitive scavenging effects at elevated peroxide concentrations, and the influence of secondary reactions on the overall degradation process. The polynomial model was selected based on statistical performance metrics and its ability to maintain mechanistic consistency. The final expression of kDCF was subsequently integrated into a comprehensive kinetic modeling framework that also accounts for DCF removal and the temporal evolution of water aromaticity under UV/H2O2 treatment conditions.
These are as follows:
[DCF]0: initial concentration of DCF contained in the water (=50.0 mg/L).
[DCF]: concentration of DCF contained in the water (mg/L).
kDCF: first-order kinetic constant for the degradation of DCF (1/min).
t: time (min).
In a previous study, the efficacy of several advanced oxidation processes (UV, photo-Fenton) for DCF degradation was evaluated using a kinetic analysis comparable to the one performed in the present work [39]. This study concluded that the UV/H2O2 system showed superior performance in terms of degradation rate, especially under neutral pH conditions, where the photo-Fenton process typically presents operational challenges. Furthermore, the UV/H2O2 method offers several advantages over traditional technologies, such as hydrogel adsorption and degradation for DCF treatment [42]. It achieves a higher degradation efficiency, removing over 95% of DCF in just 30 min, which is faster and more complete than hydrogel adsorption. Additionally, it is capable of mineralization, breaking down DCF into harmless molecules like CO2 and H2O, unlike the adsorption methods that merely transfer pollutants to another phase. The UV/H2O2 system also reduces sludge production and produces fewer secondary pollutants, making it more sustainable and reducing contamination risks. Furthermore, it is characterized by its operational simplicity and speed, as it does not require adsorbent material regeneration and operates continuously with minimal complexity. These findings support the selection of the UV/H2O2 system as the focus of this research and justify the detailed kinetic modeling presented here, providing practical insights and academic relevance to the field.
3.3. Modeling the Aromaticity During the Degradation of DCF via Pseudo-First-Order Kinetics
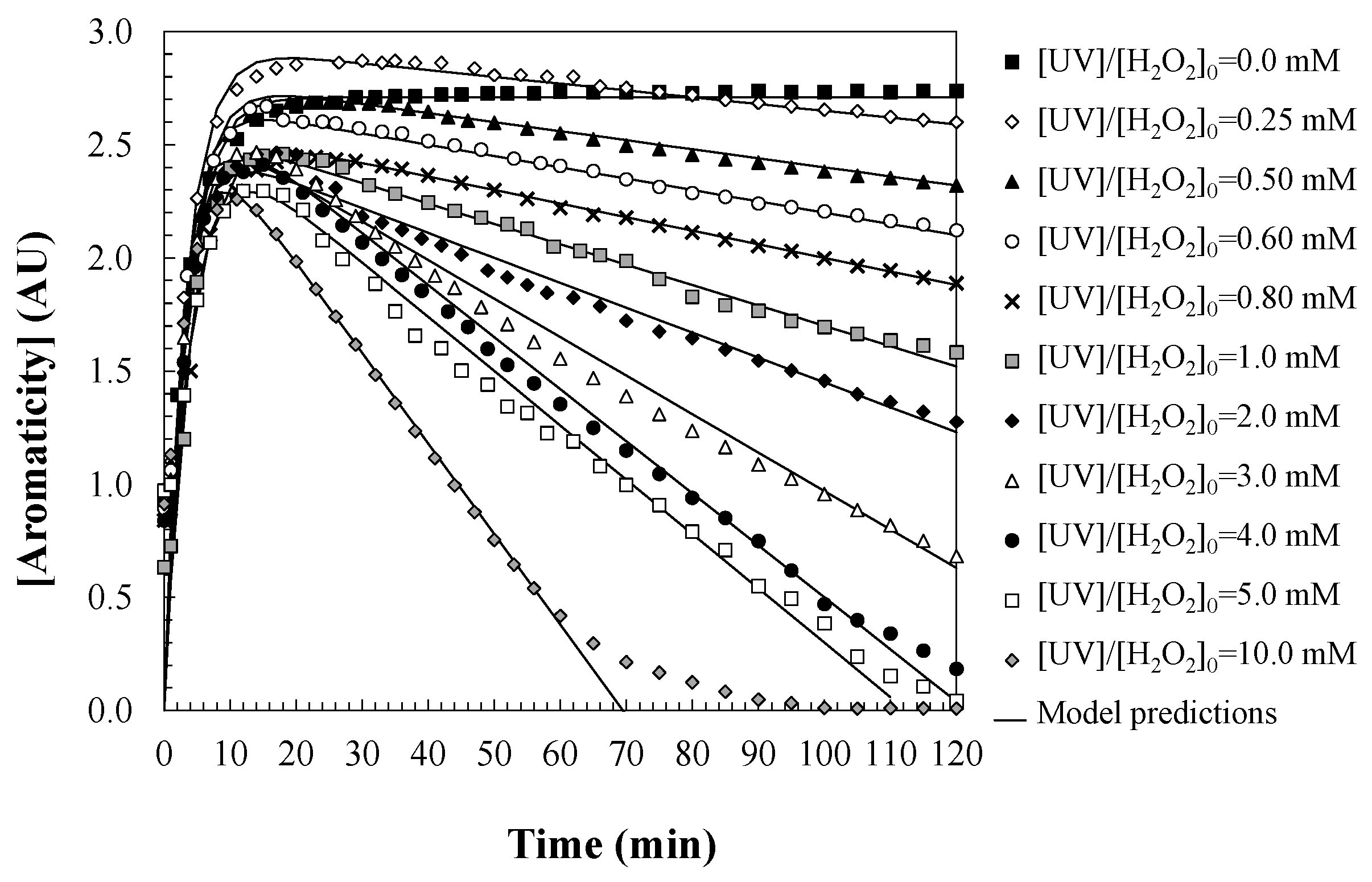
Figure 4 illustrates the temporal evolution of water aromaticity during the oxidation of DCF via the UV/H2O2 process, under varying initial concentrations of hydrogen peroxide. The experimental results reveal an initial increase in aromaticity within the first 10 min of the reaction, attributed to the formation of highly aromatic degradation intermediates derived from DCF. This phase is followed by a gradual decline in aromaticity, corresponding to the progressive oxidation of these intermediates into non-aromatic, low-molecular-weight compounds. The chemical nature of these aromatic intermediates was investigated, showing that DCF rapidly converts into two lactam derivatives, which subsequently lead to the formation of various aromatic species, including anilines and phenolic compounds [13]. Additionally, a persistent acidic byproduct, 2-hydroxyphenylacetic acid, is also formed. According to the literature, these intermediates exhibit higher toxicity than the parent DCF molecule [41]. Once the peak formation of aromatic intermediates is reached, water aromaticity begins to decrease, as these compounds are further degraded into polyhydroxylated species and small saturated and unsaturated molecules such as amines and carboxylic acids, indicating a significant degree of water detoxification [41].
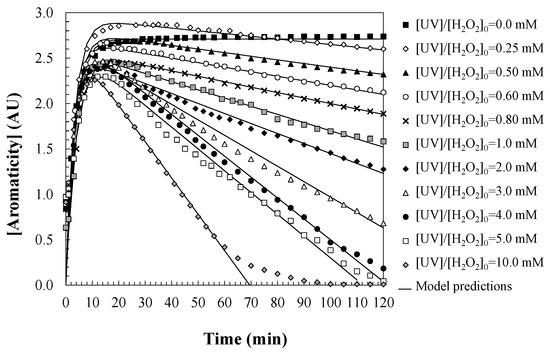
Figure 4.
Predictions of the proposed kinetic model regarding changes in water aromaticity during DCF oxidation with UV/H2O2. Experimental conditions: [DCF]0 = 50.0 mg/L; [UV] = 150 W; and [T] = 25 °C.
To quantitatively describe this behavior, a kinetic model based on a series of first-order reactions is proposed (Equation (12)), encompassing two primary stages: (i) the formation of aromatic intermediates from DCF, and (ii) their subsequent transformation into non-aromatic species. The first stage follows first-order kinetics, as the degradation rate of DCF is directly proportional to its concentration. This assumption is supported by the stability of DCF and its exponential decay under radical-based oxidative mechanisms initiated by hydroxyl radicals generated Via H2O2 photolysis. Since the reaction occurs spontaneously at the substrate and the likelihood of degradation is concentration-dependent, first-order kinetics are applicable. The proposed kinetic model assumes that the formation of aromatic intermediates proceeds through pseudo-first-order kinetics with respect to DCF, under the condition of an excess oxidant (H2O2) [43,44]. This assumption is consistent with previous studies on the degradation of pharmaceuticals under AOPs, where the concentration of hydroxyl radicals is considered to remain quasi-steady due to the continuous production from hydrogen peroxide photolysis [42].
The corresponding rate constant for aromatic intermediate formation, karom,form (1/min), exhibits a linear correlation with the initial hydrogen peroxide concentration, indicating that higher oxidant availability accelerates the degradation process (Equation (15)). As DCF breaks down, aromatic intermediates such as hydroquinone and chlorinated aromatic derivatives are produced, which retain their aromatic features, although they are structurally distinct from the parent compound.
As these intermediates accumulate, their further degradation becomes increasingly reliant on the availability of hydroxyl radicals, continuously generated by UV-induced H2O2 photolysis. At this stage, the concentration of H2O2 plays a pivotal role. The oxidation of aromatic intermediates into less aromatic or fully aliphatic products—such as short-chain carboxylic acids and amines—requires a sustained presence of hydroxyl radicals, due to the generally higher recalcitrance of these intermediates compared to the original DCF molecule.
Accordingly, the kinetic constant governing the degradation of aromatic intermediates is significantly influenced by the hydrogen peroxide concentration in the system. An increased dosage of H2O2 enhances the generation of hydroxyl radicals, thereby accelerating the cleavage of aromatic rings and promoting a more rapid decline in water aromaticity. In contrast, insufficient H2O2 levels may lead to the accumulation of persistent aromatic intermediates and partially oxidized by-products, ultimately limiting the overall detoxification efficiency of the process.
Additionally, the competitive consumption of H2O2 by both the parent compound (DCF) and its transformation products plays a crucial role in determining the net oxidative capacity of the system. In H2O2-limited scenarios, the reactive species may preferentially oxidize the parent compound, thereby delaying the breakdown of aromatic intermediates. This dynamic can result in a sustained period of elevated aromaticity in the treated effluent. Consequently, optimizing the hydrogen peroxide dosage is essential not only to ensure the complete degradation of DCF, but also to facilitate the effective transformation of toxic aromatic by-products into less harmful species, thereby minimizing the long-term environmental burden of the treated water. When sufficient H2O2 is present, these aromatic intermediates are further degraded through kinetic pathways involving the dehalogenation, hydroxylation, and cleavage of NH-aromatic bonds, leading to the progressive loss of aromaticity.
The second stage of the process, corresponding to the oxidation of aromatic intermediates, is modeled using a zero-order kinetic approximation. This was selected based on the observation that, during the initial stage of degradation, the hydroxyl radical concentration is sufficiently high to maintain a constant degradation rate for these compounds. This behavior has been reported in similar photo-Fenton systems and is especially valid when intermediates are present at low concentrations relative to the oxidant. Moreover, fitting the experimental data to zero-order kinetics yielded the best correlation, supporting the validity of this approximation. Although empirical observations suggest that this stage follows fractional-order kinetics (approximately 0.3), a zero-order approach was adopted for mathematical simplicity and to facilitate model solvability. This approximation is justifiable in systems where the reaction rate becomes independent of the intermediate concentration and is instead limited by the availability of reactive species—in this case, hydroxyl radicals. Such kinetic behavior is commonly observed in complex environmental and chemical degradation processes.
The non-linear relationship between the intermediate concentration and degradation rate reflects the multi-step nature of aromatic breakdown, wherein aromatic moieties are incrementally removed through less efficient oxidative reactions. To simplify model development, zero-order kinetics were assumed for the degradation of aromatic intermediates, as expressed in Equation (13). This stage leads to the formation of non-aromatic end-products, including low-molecular-weight carboxylic acids (e.g., oxalic, acetic, and formic acids) and inorganic ions such as CO2, NO3−, Cl−, and NH4+ [42,44].
The mass balance for the degradation of aromatic intermediates is as follows:
Integrating Equation (12), the kinetic equation is obtained for the loss of aromaticity of the aqueous DCF solutions oxidized via treatment (Equation (14)). Due to the necessity of homogenizing the concentration units, the initial concentration of DCF causing aromaticity in water is expressed in aromaticity units through the parameter αArom (AU L/mg).
Being:
[Arom]: aromaticity of water (AU).
αArom: ratio of initial DCF concentration causing aromaticity in water (AU L/mg).
kArom,form: first-order kinetic constant for the degradation of DCF to aromatic intermediates (1/min).
kArom,deg: zero-order kinetic constant for loss of aromaticity during DCF degradation (AU/min).
Through these adjustments, the values of the kinetic parameters are estimated as a function of the hydrogen peroxide concentration (Equations (15)–(17)) and shown in Table 1.
This model highlights the temporal evolution of water aromaticity and underscores the importance of H2O2 as a key oxidant in the second stage of the process. The balance between these two steps ultimately determines the effectiveness of UV/H2O2 treatment in achieving the complete mineralization and detoxification of diclofenac-contaminated waters. The general expression of the model allows for the prediction of the aromaticity evolution over time, accounting for both the formation and degradation of aromatic compounds. Additionally, the kinetic parameters employed (Equations (15)–(17)) are expressed as functions of the initial H2O2 concentration, enabling their applicability across a range of process conditions. The corresponding rate constant, karom,deg (AU/min), exhibits a non-linear dependence on the initial H2O2 concentration, indicating a complex interaction governed by the concurrent consumption of the oxidant by both DCF and its degradation intermediates (Equation (16)). Overall, the model provides a useful tool for understanding and optimizing the UV/H2O2 system performance, not only in terms of DCF removal but also through reducing the toxicity associated with the formation of aromatic degradation by-products.
The predictions obtained show agreement with the experimental data across all conditions tested, thus validating the robustness of the proposed kinetic model. The observed deviation between model predictions and experimental data at an H2O2 concentration of 10.0 mM, particularly after 70 min of reaction, can be attributed to the elevated peroxide concentrations, as the excessive generation of hydroxyl radicals may lead to non-selective side reactions, which reduce the effective availability of hydroxyl radicals for the oxidation of aromatic intermediates. This phenomenon has been reported in AOPs and is commonly referred to as the radical quenching effect. Moreover, the accumulation of low-molecular-weight acids and other transformation products over time may lead to a slight buffering or scavenging of reactive species, further inhibiting the oxidative degradation of persistent aromatic intermediates. Additionally, at extended reaction times, changes in pH and ionic strength caused by the progressive mineralization of organic matter may alter the photolysis efficiency of H2O2 and the generation rate of hydroxyl radicals.
3.4. Kinetic Modeling of Water Color Changes Based on First-Order Consecutive Reactions
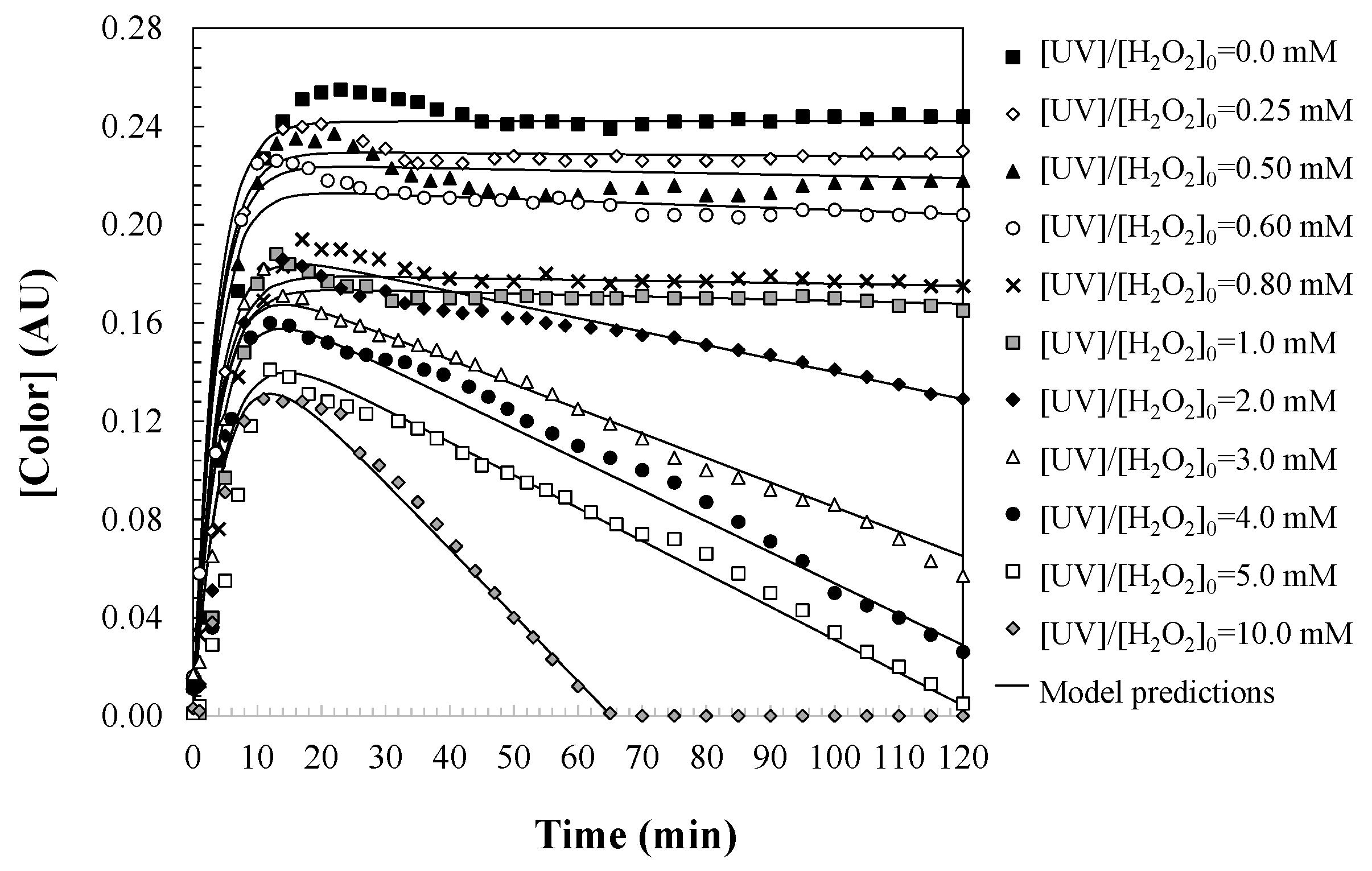
During the degradation of the initially colorless aqueous solutions of DCF, a pronounced yellow coloration develops over time (Figure 5), which corresponds to the kinetic formation of reaction intermediates (Figure 6). This color change arises due to the generation of oxidation by-products—specifically, aromatic compounds and quinones bearing chromophoric functional groups—that absorb visible light and impart a distinct hue to the solution.

Figure 5.
DCF degradation scheme, highlighting the compounds that may contribute to aromaticity and color changes.
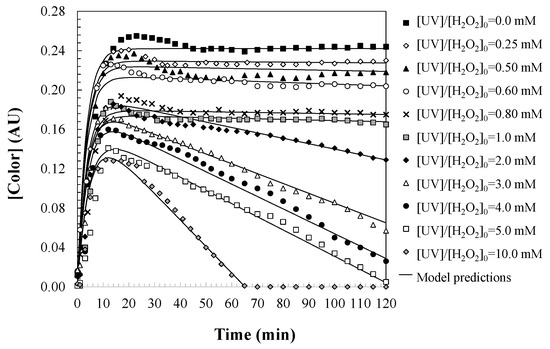
Figure 6.
Predictions of the proposed kinetic model for changes in water color during DCF oxidation with UV/H2O2. Experimental conditions: [DCF]0 = 50.0 mg/L; [UV] = 150 W; and [T] = 25 °C.
The transient increase in both aromaticity and visible coloration observed during the early stages of diclofenac oxidation can be explained through the formation of stable aromatic and chromophoric intermediates. Upon hydroxyl radical attack, diclofenac undergoes hydroxylation of its aromatic rings, yielding catechols, hydroquinones, and other phenolic derivatives. These compounds retain their aromatic structure and contribute to the enhanced UV absorbance at 254 nm. Further oxidation of these intermediates produces quinones, which are conjugated diketones with extended π-electron systems. Quinones exhibit strong absorbance in the visible range (around 455 nm), explaining the yellow coloration that is observed experimentally. In parallel, the cleavage of the amide bridge between the aromatic rings of diclofenac releases chlorinated anilines and amino-phenols. These compounds, along with their conjugated nitrogen derivatives, contribute both to aromaticity and coloration due to resonance effects involving amino and hydroxyl substituents. The combined presence of catechols, phenols, quinones, chlorinated anilines, amino-phenols, and conjugated nitrogen compounds leads to a temporary buildup of aromaticity and color in the solution. These intermediates represent a key transitional phase in the overall mineralization process, which ultimately degrades them into non-aromatic carboxylic acids and inorganic end-products.
Color formation is intrinsically linked to the oxidative degradation of DCF, which progresses as hydroxyl radicals successively attack both the parent compound and its transformation products. These phenomena have been previously reported by Chen et al. (2016) [44], who suggested the presence of a secondary oxidative pathway during radical-induced DCF degradation [13]. This route leads to the formation of phenolic intermediates, which are subsequently oxidized into quinone structures, thereby accounting for the yellowish tint observed in the treated water.
The oxidative degradation of DCF by hydroxyl radicals under UV/H2O2 conditions begins with the electrophilic attack of HO● on electron-rich sites of the molecule. The initial targets are typically the aromatic rings and the amino group linking the two phenyl rings. Specifically, hydroxyl radicals preferentially react via hydroxylation at ortho- and para-positions on the dichlorophenyl ring, leading to the formation of mono- and di-hydroxylated derivatives such as catechols. Simultaneously, HO● can induce C–N bond cleavage at the amine bridge, resulting in the formation of smaller aromatic fragments like anilines and phenols. As oxidation progresses, these intermediates undergo further transformation: catechols and hydroquinones can be oxidized to quinones through two-electron transfer mechanisms, while continued radical attack promotes ring-opening reactions and conversion into aliphatic carboxylic acids and low-molecular-weight organic acids. This sequential degradation process—starting from aromatic substitution and bond cleavage to ring fragmentation—explains the observed evolution of aromaticity and chromophoric compounds during treatment, as captured by the multi-kinetic model.
To quantitatively describe this process, a first-order consecutive reaction model is proposed (Equation (18)), in which DCF is converted into colored intermediates via a rate constant kColor,form (1/min). The adoption of first-order kinetics reflects the linear dependence of color formation on the concentration of DCF or its reactive intermediates. Since the generation of chromophoric species directly depends on the available DCF concentration, the rate of color formation naturally diminishes as DCF is progressively consumed during the reaction. To ensure dimensional consistency within the reaction rate expression (Equation (19)), the initial DCF concentration contributing to color formation is expressed in absorbance units at λ = 455 nm using the αColor parameter (AU·L/mg), which relates absorbance to mass concentration.
The mass balance for color changes is determined as follows:
Integrating Equation (19), the kinetic equation for color changes in aqueous DCF solutions oxidized by UV/H2O2 treatment is presented as Equation (20).
When sufficient hydrogen peroxide is present to ensure an adequate supply of hydroxyl radicals, the colored intermediates are further oxidized into non-chromophoric end-products, such as low-molecular-weight carboxylic acids and biodegradable inorganic species. This subsequent degradation step is approximated by a zero-order kinetic model, governed by the rate constant kColor,deg (AU/min), indicating that the rate is independent of intermediate concentration and primarily limited by the availability of reactive radicals. Zero-order kinetics indicate that the rate of color degradation is constant and independent of the concentration of the chromophoric compounds. This behavior is explained by the fact that once chromophore-containing intermediates are formed, their degradation rate depends primarily on the availability of hydroxyl radicals, and not on the concentration of the intermediates themselves. As long as an excess of H2O2 is present, generating a steady flux of radicals through photolysis, the degradation of color-causing compounds proceeds at a constant rate. On the other hand, hydroxyl radicals attack the intermediate products in a constant and non-selective manner, so the rate of color degradation is constant as long as there is a sufficient amount of hydroxyl radicals in the medium.
From the considered adjustments, the estimated kinetic parameters are obtained, as shown in Table 1. It is verified that the estimated kinetic parameters are functions of the concentration of hydrogen peroxide applied in the treatment (Equations (21)–(23)).
Being:
[Color]: water color (AU).
αColor: ratio of initial concentration of DCF containing in water that degrades to colored species (AU L/mg).
kColor,form: first-order kinetic constant for the degradation of DCF through UV/H2O2 treatment to colored intermediates (1/min).
kColor,deg: zero-order kinetic constant for the degradation of colored intermediates to colorless products (AU/min).
This highlights the critical role of H2O2: it not only initiates the oxidation of DCF, leading to chromophore formation, but also governs the extent and efficiency of the subsequent chromophore degradation. An insufficient amount of H2O2 may lead to the accumulation of quinonoid or phenolic intermediates, prolonging the presence of color in the water and compromising the visual and chemical quality of the effluent. However, a well-optimized H2O2 dosage ensures rapid consumption of these intermediates, shortening the duration of color expression and promoting the formation of non-colored end products.
In summary, H2O2 plays a dual and time-dependent role in the kinetics of color development during UV-induced oxidation: initially, it contributes to chromophore formation through partial oxidation pathways; subsequently, it becomes essential in breaking down those same chromophores, enabling complete decolorization. The balance between these competing effects is finely regulated by the concentration of H2O2, the intensity of UV light, and the residence time in the reactor. Therefore, careful control of these parameters is crucial to achieving an efficient and acceptable treatment process.
A clear correlation was observed between the kinetic parameters describing aromaticity loss and those governing color change during the UV/H2O2 treatment process. In both cases, the formation of intermediate species—either aromatic or colored—follows first-order kinetics, while their subsequent degradation into non-aromatic or colorless products is well-described by zero-order kinetics. Furthermore, the values of the kinetic constants for both formation and degradation exhibit a strong dependence on the initial hydrogen peroxide concentration, highlighting the dual and critical role of this oxidant in both stages of the process. The similarity in kinetic behavior suggests that the compounds responsible for the transient increase in aromaticity may largely overlap with those contributing to color formation, such as quinonoid and phenolic intermediates. Therefore, the concurrent evolution of aromaticity and color supports the hypothesis that both phenomena are closely linked to the formation and subsequent breakdown of partially oxidized intermediates, whose transformation is primarily governed by the availability of hydroxyl radicals in the system.

Table 1.
Estimated kinetic parameters for modeling DCF oxidation using UV/H2O2 technology. Experimental conditions: [DCF]0 = 50.0 mg/L; [UV] = 150 W; and [T] = 25 °C.
Table 1.
Estimated kinetic parameters for modeling DCF oxidation using UV/H2O2 technology. Experimental conditions: [DCF]0 = 50.0 mg/L; [UV] = 150 W; and [T] = 25 °C.
| [H2O2]0 | kDCF | αArom | kArom,form | kArom,deg | αColor | kColor,form | kColor,deg |
|---|---|---|---|---|---|---|---|
| (mM) | (1/min) | (AU L/mg) | (1/min) | (AU/min) | (AU L/mg) | (1/min) | (AU/min) |
| 0.00 | 0.2341 | 0.0542 | 0.30 | 0.000 | 0.0048 | 0.30 | 0.0 |
| 0.25 | 0.2798 | 0.0590 | 0.30 | 0.003 | 0.0046 | 0.30 | 2.0 × 10−5 |
| 0.50 | 0.3093 | 0.0560 | 0.30 | 0.004 | 0.0045 | 0.30 | 5.0 × 10−5 |
| 0.60 | 0.3345 | 0.0540 | 0.35 | 0.005 | 0.0043 | 0.30 | 9.0 × 10−5 |
| 0.80 | 0.4293 | 0.0520 | 0.25 | 0.006 | 0.0036 | 0.29 | 4.0 × 10−5 |
| 1.00 | 0.4367 | 0.0520 | 0.35 | 0.009 | 0.0035 | 0.29 | 6.0 × 10−5 |
| 2.00 | 0.4540 | 0.0510 | 0.35 | 0.011 | 0.0039 | 0.28 | 5.5 × 10−5 |
| 3.00 | 0.4518 | 0.0534 | 0.38 | 0.017 | 0.0037 | 0.28 | 1.0 × 10−5 |
| 4.00 | 0.4799 | 0.0560 | 0.29 | 0.023 | 0.0036 | 0.26 | 1.3 × 10−5 |
| 5.00 | 0.4666 | 0.0540 | 0.26 | 0.024 | 0.0033 | 0.23 | 1.3 × 10−5 |
| 10.00 | 0.4163 | 0.0556 | 0.35 | 0.040 | 0.0035 | 0.22 | 2.7 × 10−5 |
The kinetic model developed in this study offers novel contributions that go beyond previous kinetic analyses of diclofenac (DCF) degradation. Unlike earlier models focused solely on the parent compound’s decay, this work presents a multi-kinetic framework that captures three interconnected processes: DCF degradation, the evolution of aromatic intermediates, and the formation and removal of chromophoric compounds. This provides a more complete and accurate representation of the oxidative treatment pathway, including intermediate behavior, with potential ecotoxicological relevance.
A key innovation is the linkage of reaction kinetics to water quality indicators such as aromaticity and color, which are modeled as functions of hydrogen peroxide concentration. This allows for insight into how oxidant levels affect not only degradation rates but also the transformation of toxic intermediates and water appearance. The compatibility of the model with spectrophotometric monitoring also enables practical, real-time assessment of treatment performance.
4. Conclusions
This study confirms the effectiveness of the UV/H2O2 process for the degradation of diclofenac (DCF) in aqueous media, emphasizing the central role of hydroxyl radicals in the oxidative mechanism. DCF degradation proceeds primarily through radical attack on its aromatic structure, leading to the formation of oxidized intermediates. Notably, an intense yellow coloration develops within the first 15 min, attributable to chromophoric intermediates such as quinones and conjugated compounds. These intermediates subsequently undergo further oxidation, resulting in the formation of final products including CO2, water, and inorganic ions, and thereby reducing the color of the solutions.
Throughout treatment, water turbidity remains below 1 NTU, meeting European water quality standards. The oxidation process initially increases aromaticity due to the generation of stable aromatic intermediates (e.g., catechols and quinones), followed by a decline associated with ring cleavage and partial mineralization. Concurrently, dissolved oxygen (DO) levels increase early in the process due to radical reactions and later decrease as H2O2 is consumed. The pH of the solution progressively decreases and stabilizes due to proton release during oxidation.
A kinetic model based on the first-order degradation of DCF was developed, demonstrating good agreement with experimental results. The model incorporates the formation and transformation of aromatic intermediates, with the initial degradation of DCF following first-order kinetics and the subsequent degradation of intermediates approximated by fractional-order kinetics (~0.3). For modeling purposes, the overall process was simplified to zero-order kinetics. Additionally, the formation and removal of colored species were modeled separately, with formation governed by first-order kinetics (dependent on DCF and intermediate concentrations) and decolorization described by zero-order kinetics (dependent on hydroxyl radical availability). The results demonstrate that the kinetic parameters governing DCF degradation, aromaticity changes, and color evolution are influenced by the concentration of hydrogen peroxide and can be modulated by adjusting the operational parameters. The proposed model effectively captures the multistep dynamics of the oxidation process and provides a valuable predictive tool for process optimization.
Although the UV/H2O2 process has some limitations, such as the possible presence of residual hydrogen peroxide in the effluent or the energy consumption associated with UV irradiation, these aspects can be controlled through proper process optimization and correct sizing in real-life applications. Furthermore, its high efficiency in removing diclofenac and transforming potentially toxic aromatic compounds into less harmful species, as demonstrated in this study, makes it a very useful technology as a tertiary treatment in advanced wastewater treatment systems. Therefore, the benefits in terms of improved water quality and reduced environmental risks outweigh the potential operational drawbacks.
Based on these findings, several avenues for future research are recommended. First, a comprehensive toxicological assessment of both the intermediate and final oxidation products (e.g., anilines, catechols, quinones) is essential to ensure the environmental safety of treated water. Second, validation of the kinetic model in complex real-world matrices—such as municipal wastewater, surface waters, or hospital effluents—is necessary to assess the influence of natural organic matter and co-contaminants. Third, the integration of UV/H2O2 with complementary advanced oxidation processes (e.g., photo-Fenton, ozonation, or TiO2 photocatalysis) could enhance removal efficiency and reduce reagent consumption. The modeling approach presented here can be adapted to describe such hybrid systems.
It is important to acknowledge that actual water bodies often contain a complex mixture of organic and inorganic pollutants, which can compete with DCF for reactive species such as hydroxyl radicals. This competition may alter the degradation pathway and affect the overall kinetics of DCF oxidation. Consequently, the first-order kinetic model employed in this study to describe the changes in aromaticity that occur during DCF degradation has inherent limitations, as it does not fully account for the influence of co-contaminants or matrix effects. Future work should aim to refine the kinetic model by incorporating competitive reactions and validating its applicability in more realistic environmental conditions.
Lastly, future research should also explore the application of artificial intelligence (AI) and machine learning (ML) to further enhance the modeling, prediction, and control of UV/H2O2-based treatment systems. Data-driven approaches can improve the quality of kinetic models, identify non-obvious patterns in complex reaction networks, and optimize operational parameters in real time. AI and ML can also assist in the classification of oxidation intermediates, toxicity prediction, and decision-making under variable influent conditions. Recent studies have demonstrated the potential of such strategies to advance water treatment technologies. Incorporating these tools represents a promising step toward the development of smart, adaptive, and sustainable treatment systems for emerging contaminants [45,46].
Author Contributions
Conceptualization, N.V.; methodology, U.D. and B.E.; software, U.D., B.E. and N.V.; validation, N.V.; formal analysis, U.D. and N.V.; investigation, N.V., B.E. and U.D.; resources, U.D., B.E., N.V. and A.M.d.L.; data curation, U.D. and N.V.; writing—original draft preparation, N.V., A.M.d.L. and U.D; writing—review and editing, N.V.; visualization, N.V., A.M.d.L. and U.D.; supervision, A.M.d.L. and N.V.; project administration, N.V. and A.M.d.L.; funding acquisition, N.V. and A.M.d.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by PIBA_2023_01_0032—Basic and/or Applied Research Project. Financing entity: Basque Government 2023.
Data Availability Statement
The data supporting the findings of this study can be found within the article.
Acknowledgments
The authors thank the Department of Chemical and Environmental Engineering and the Department of Chemical Engineering of the University of the Basque Country UPV/EHU for their support.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Antonopoulou, M.; Kosma, C.; Albanis, T.; Konstantinou, I. An overview of homogeneous and heterogeneous photocatalysis applications for the removal of pharmaceutical compounds from real or synthetic hospital wastewaters under lab or pilot scale. Sci. Total Environ. 2021, 765, 144163. [Google Scholar] [CrossRef] [PubMed]
- Landsdorp, D.; Vree, T.B.; Janssen, T.J.; Guelen, P.J. Pharmacokinetics of rectal diclofenac and its hydroxyl metabolites in man. Int. J. Clin. Pharmacol. Ther. Toxicol. 1990, 28, 298–302. [Google Scholar]
- Huber, C.; Preis, M.; Harvey, P.J.; Grosse, S.; Letzel, T.; Schröder, P. Emerging pollutants and plants—Metabolic activation of diclofenac by peroxidases. Chemosphere 2016, 146, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Lonappan, L.; Brar, S.K.; Das, R.K.; Verma, M.; Surampalli, R.Y. Diclofenac and its transformation products: Environmental occurrence and toxicity—A review. Environ. Int. 2016, 96, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Perisic, D.J.; Gilja, V.; Stankov, M.N.; Katancic, Z.; Kusic, H.; Stangar, U.L.; Dionysiou, D.D.; Bozic, A.L. Removal of diclofenac from water by zeolite-assisted advanced oxidation processes. J. Photochem. Photobiol. A Chem. 2016, 321, 238–247. [Google Scholar] [CrossRef]
- European Geosciences Union (EGU). New Global Models Predict Increasing Pollution of Rivers. In Proceedings of the Press Conference, Vienna, Austria, 8–13 April 2018. [Google Scholar]
- Acuña, V.; Ginebreda, A.; Mor, J.R.; Petrovic, M.; Sabater, S.; Sumpter, J.; Barceló, D. Balancing the health benefits and environmental risks of pharmaceuticals: Diclofenac as an example. Environ. Int. 2015, 85, 327–333. [Google Scholar] [CrossRef]
- Pan, D.; Zhang, C.; Wang, C.S.; Zhang, P.; Jiao, X.Y.; Ma, Q.R.; Wang, L.T.; Li, D.J.; Li, L.P. Unravelling hidden threats of water disinfection: Toxicity evaluation and toxic products identification during diclofenac degradation. Environ. Pollut. 2024, 345, 123424. [Google Scholar] [CrossRef]
- Castiglioni, S.; Bagnati, R.; Fanelli, R.; Pomati, F.; Calamari, D.; Zuccato, E. Removal of Pharmaceuticals on Sewage Treatment Plants in Italy. Environ. Sci. Technol. 2006, 40, 357–363. [Google Scholar] [CrossRef]
- Yusuf, A.O.; Mohamed, G.H.; Al-Sakkaf, R.; Rodríguez, J.; Zerjav, G.; Pintar, A.; Palmisano, G. Photocatalytic degradation of diclofenac amide in a fixed-bed reactor using TiO2/β-Bi2O3: Process optimization and stability analysis. J. Photochem. Photobiol. A Chem. 2024, 450, 115470. [Google Scholar] [CrossRef]
- Capodaglio, A.G.; Bojanowska-Czajka, A.; Trojanowicz, M. Comparison of different advanced degradation processes for the removal of the pharmaceutical compounds diclofenac and carbamazepine from liquid solutions. Environ. Sci. Pollut. Res. 2018, 25, 27704–27723. [Google Scholar] [CrossRef]
- De Luis, A.; Lombraña, J.I. pH-Based Strategies for an Efficient Addition of H2O2 During Ozonation to Improve the Mineralisation of Two Contaminants with Different Degradation Resistances. Water Air Soil Pollut. 2018, 229, 372. [Google Scholar] [CrossRef]
- Leyva, E.; Moctezuma, E.; Baines, K.M.; Noriega, S.; Zarazúa, E. A review on chemical advanced oxidation processes for pharmaceuticals with paracetamol as a model compound. Reaction conditions, intermediates and total mechanism. Curr. Org. Chem. 2018, 22, 2–17. [Google Scholar] [CrossRef]
- Wang, Y.; Zhong, M.; Ma, F.; Wang, C.; Lu, X. Shell-induced enhancement of Fenton-like catalytic performance towards advanced oxidation processes: Concept, mechanism, and properties. Water Res. 2024, 268, 122655. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.D.; Pierens, X.; Benhabib, K. Experimental and numerical study of methylparaben decomposition in aqueous solution using the UV/H2O2 process. J. Environ. Sci. Health Part B 2019, 54, 357–365. [Google Scholar] [CrossRef]
- Dhawle, R.; Mantzavinos, D.; Lianos, P. UV/H2O2 degradation of diclofenac in a photocatalytic fuel cell. Appl. Catal. B Environ. 2021, 299, 120706. [Google Scholar] [CrossRef]
- Graumans, M.H.F.; Hoeben, W.F.L.M.; van Dael, M.F.P.; Anzion, R.B.M.; Russel, F.G.M.; Scheepers, P.T.J. Thermal plasma activation and UV/H2O2 oxidative degradation of pharmaceutical residues. Environ. Res. 2021, 195, 110884. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Lei, Y.; Zhang, X.; Yang, X. Treating disinfection byproducts with UV or solar irradiation and in UV advanced oxidation processes: A review. J. Hazard. Mater. 2021, 408, 124435. [Google Scholar] [CrossRef]
- Pan, S.; Cao, B.; Yuan, D.; Jiao, T.; Zhang, Q.; Tang, S. Complexes of Cupric Ion and Tartaric Acid Enhanced Calcium Peroxide Fenton-Like Reaction for Metronidazole Degradation. Chin. Chem. Lett. 2024, 35, 109185. [Google Scholar] [CrossRef]
- Gao, L.; Zhou, B.; Wang, F.; Wang, Y.; Yuan, R.; Chen, H.; Han, X. Effect of dissolved organic matters and inorganic ions on TiO₂ photocatalysis of diclofenac: Mechanistic study and degradation pathways. Environ. Sci. Pollut. Res. 2020, 27, 2044–2053. [Google Scholar] [CrossRef]
- Li, J.; Ma, L.Y.; Li, L.S.; Li, X. Photodegradation Kinetics, Transformation, and Toxicity Prediction of Ketoprofen, Carprofen, and Diclofenac Acid in Aqueous Solutions. Environ. Toxicol. Chem. 2017, 36, 3232–3239. [Google Scholar] [CrossRef]
- Lu, X.; Shao, Y.; Gao, N.; Chen, J.; Zhang, Y.; Xiang, H.; Guo, Y. Degradation of Diclofenac by UV-Activated Persulfate Process: Kinetic Studies, Degradation Pathways and Toxicity Assessments. Ecotoxicol. Environ. Saf. 2017, 141, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.Y.; Kim, M.K.; Yoon, Y.; Im, J.K.; Zoh, K.D. Kinetics and degradation mechanism of clofibric acid and diclofenac in UV photolysis and UV/H2O2 reaction. Desalination Water Treat. 2014, 52, 6211–6218. [Google Scholar] [CrossRef]
- Alharbi, S.K.; Kang, J.; Nghiem, L.D.; van de Merwe, J.P.; Leusch, F.D.L.; Price, W.E. Photolysis and UV/H2O2 of diclofenac, sulfamethoxazole, carbamazepine, and trimethoprim: Identification of their major degradation products by ESI–LC–MS and assessment of the toxicity of reaction mixtures. Process. Saf. Environ. Prot. 2017, 112, 222–234. [Google Scholar] [CrossRef]
- Perisic, D.J.; Belet, A.; Kusic, H.; Stangar, U.L.; Bozic, A.L. Comparative study on photocatalytic treatment of diclofenac: Slurry vs. immobilized processes. Desalination Water Treat. 2017, 81, 170–185. [Google Scholar] [CrossRef]
- Ledakowicz, S.; Drozdek, E.; Boruta, T.; Foszpańczyk, M.; Olak-Kucharczyk, M.; Żylla, R.; Gmurek, M. Impact of Hydrogen Peroxide on the UVC Photolysis of Diclofenac and Toxicity of the Phototransformation Products. Int. J. Photoenergy 2019, 2019, 1086704. [Google Scholar] [CrossRef]
- Pizzichetti, R.; Reynolds, K.; Pablos, C.; Casado, C.; Moore, E.; Stanley, S.; Marugán, J. Removal of diclofenac by UV-B and UV-C light-emitting diodes (LEDs) driven advanced oxidation processes (AOPs): Wavelength dependence, kinetic modelling and energy consumption. Chem. Eng. J. 2023, 471, 144520. [Google Scholar] [CrossRef]
- Conte, F.; Tommasi, M.; Degerli, S.N.; Forame, E.; Parolini, M.; De Felice, B.; Ramis, G.; Rossetti, I. Comparison of Different Advanced Oxidation Processes (AOPs) and Photocatalysts for the Degradation of Diclofenac. Chem. Photo. Chem. 2024, 8, e202300177. [Google Scholar] [CrossRef]
- Vogna, D.; Marotta, R.; Napolitano, A.; Andreozzi, R.; d’Ischia, M. Advanced oxidation of the pharmaceutical drug diclofenac with UV/H2O2 and ozone. Water Res. 2004, 38, 414–422. [Google Scholar] [CrossRef]
- Sohn, S.; Kim, M.K.; Lee, Y.M.; Sohn, E.J.; Choi, G.Y.; Chae, S.H.; Zoh, K.D. Removal characteristics of 53 micropollutants during ozonation, chlorination, and UV/H2O2 processes used in drinking water treatment plant. Chemosphere 2024, 352, 141360. [Google Scholar] [CrossRef]
- Villota, N.; Ferreiro, C.; Qulatein, H.A.; Lomas, J.M.; Camarero, L.M.; Lombraña, J.I. Color Changes during the Carbamazepine Oxidation by Photo-Fenton. Catalysts 2021, 11, 386. [Google Scholar] [CrossRef]
- Lekkerkerker-Teunissen, K.; Benotti, M.J.; Snyder, S.A.; van Dijk, H.C. Transformation of Atrazine, Carbamazepine, Diclofenac and Sulfamethoxazole by Low and Medium Pressure UV and UV/H2O2 Treatment. Sep. Purif. Technol. 2012, 96, 33–43. [Google Scholar] [CrossRef]
- Villota, N.; Duoandicoechea, U.; Lombraña, J.I.; De Luis, A.M. Kinetic Modelling of Aromaticity and Color Changes during the Degradation of Sulfamethoxazole Using Photo-Fenton Technology. Catalysts 2024, 14, 718. [Google Scholar] [CrossRef]
- Stefan, M.I.; Hoy, A.R.; Bolton, J.R. Kinetics and mechanism of the degradation and mineralization of acetone in dilute aqueous solution sensitized by the UV photolysis of hydrogen peroxide. Environ. Sci. Technol. 1996, 30, 2382–2390. [Google Scholar] [CrossRef]
- Villota, N.; Echevarria, B.; De Luis, A. Kinetic Modelling of Aromaticity and Water Colour Changes during Diclofenac Oxidation by UV/H2O2. arXiv 2024. [Google Scholar] [CrossRef]
- Villota, N.; Coralli, I.; Lomas, J.M. Changes of Dissolved Oxygen in Aqueous Solutions of Caffeine Oxidized by Photo-Fenton Reagent. Environ. Technol. 2021, 42, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Bilbao-García, E.; Duoandicoechea, U.; Villota, N. Dissolved Oxygen Changes in Wastewater During Sulfamethoxazole Degradation by Photo-Fenton Treatment. Sustainability 2025, 17, 3333. [Google Scholar] [CrossRef]
- Boutiti, A.; Zouaghi, R.; Bendjabeur, S.E.; Guittonneau, S.; Sehili, T. Photodegradation of 1-hexyl-3-methylimidazolium by UV/H2O2 and UV/TiO2: Influence of pH and chloride. J. Photochem. Photobiol. A Chem. 2017, 336, 164–169. [Google Scholar] [CrossRef]
- Khan, S.A.; Siddiqui, M.F.; Alam Khan, T. Synthesis of Poly(methacrylic acid)/Montmorillonite Hydrogel Nanocomposite for Efficient Adsorption of Amoxicillin and Diclofenac from Aqueous Environment: Kinetic, Isotherm, Reusability, and Thermodynamic Investigations. ACS Omega 2020, 5, 2843–2855. [Google Scholar] [CrossRef] [PubMed]
- Villota, N.; Echevarria, B.; Duoandicoechea, U.; Lombraña, J.I.; De Luis, A.M. Kinetic Study of the Water Quality Parameters during the Oxidation of Diclofenac by UV Photocatalytic Variants. Catalysts 2024, 14, 580. [Google Scholar] [CrossRef]
- Villota, N.; Cruz-Alcalde, A.; Ferreiro, C.; Lombraña, J.I.; Esplugas, S. Changes in solution turbidity and color during paracetamol removal in laboratory and pilot-scale semicontinuous ozonation reactors. Sci. Total Environ. 2023, 854, 158682. [Google Scholar] [CrossRef]
- Liang, J.; Zhang, W.; Zhao, Z.; Liu, W.; Ye, J.; Tong, M.; Li, Y. Different degradation mechanisms of carbamazepine and diclofenac by single-atom Barium embedded g-C3N4: The role of photosensitation-like mechanism. J. Hazard. Mater. 2021, 416, 125936. [Google Scholar] [CrossRef] [PubMed]
- Villota, N.; Mijangos, F.; Varona, F.; Andrés, J. Kinetic Modelling of Toxic Compounds Generated during Phenol Elimination in Wastewaters. Int. J. Chem. React. Eng. 2007, 5, A63. [Google Scholar] [CrossRef]
- Chen, W.; Li, X.; Pan, Z.; Ma, S.; Li, L. Effective mineralization of diclofenac by catalytic ozonation using Fe-MCM-41 catalyst. Chem. Eng. J. 2016, 304, 594–601. [Google Scholar] [CrossRef]
- Wang, H.; Cao, H.; Yang, L. Machine Learning-Driven Multidomain Nanomaterial Design: From Bibliometric Analysis to Applications. ACS Appl. Nano Mater. 2024, 7, 26579–26600. [Google Scholar] [CrossRef]
- Yang, L.; Wang, H.; Leng, D.; Fang, S.; Yang, Y.; Du, Y. Machine Learning Applications in Nanomaterials: Recent Advances and Future Perspectives. Chem. Eng. J. 2024, 500, 156687. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).