1. Introduction
Emerging pollutants in aquatic ecosystems, including pharmaceuticals, insecticides, pesticides, dyes, and surfactants, have been identified as environmentally hazardous. These contaminants, which are common globally, are associated with serious environmental and health-related issues. Their widespread presence raises concerns about their potential impact on aquatic life [
1,
2,
3,
4,
5]. Addressing these challenges requires the development of novel catalysts capable of achieving efficient and complete pollutant mineralization [
6,
7,
8,
9]. The development of photocatalytic materials offers significant potential for efficient pollutant removal. In this context, TiO
2-based photocatalysts have been extensively studied due to their low toxicity, biological inertness, low operating temperatures, water insolubility, easy availability, and high chemical stability. However, TiO
2 has a wide band gap, limiting its activation to UV light, and exhibiting rapid charge carrier recombination, which reduces its efficiency [
10,
11,
12,
13]. To address these issues, research has focused on improving the structural design of TiO
2 nanomaterials. The shape and size of nanoparticles are crucial factors in catalytic reactions, as their dimensions, structure, and morphology significantly influence their catalytic activity. These efforts aim to mitigate the limitations of traditional nanomaterials while leveraging the benefits of shape-controlled TiO
2 nanomaterials.
Shape-controlled TiO
2 materials with unique morphologies and uniform size distributions are found to enhance light harvesting and facilitate unique heterojunction designs that improve charge separation. Nanostructures such as nanocuboids, bipyramids, and nanowires have shown significant photocatalytic activity in aqueous solutions [
14,
15,
16,
17,
18,
19,
20,
21]. TiO
2 exists in four crystalline polymorphs, namely anatase, rutile, brookite, and TiO
2 (B), each offering distinct structural and functional properties [
22]. Anatase is renowned for superior photocatalytic activity, and rutile for thermal stability, while brookite and TiO
2 (B) nanobelts possess unique electronic and optical characteristics. TiO
2 (B), with its orthorhombic crystal structure and high surface area, has gained attention for energy storage, lithium-ion batteries, and photocatalysis [
23,
24]. However, the limited active surface sites and charge carrier recombination hinder the photocatalytic performance of TiO
2 (B) nanobelts. Designing heterostructures with second-phase materials is a well-established strategy to overcome these challenges, improving photocatalytic efficiency while preserving the intrinsic properties of TiO
2. In this context, TiO
2 (B) has the potential to self-transform into anatase-based heterostructures through calcination, thereby enhancing its overall photocatalytic performance [
22]. Such a structure enhances photocatalytic processes by promoting efficient charge separation, reducing the recombination of electron–hole pairs that typically limit the efficiency of traditional TiO
2 photocatalysts. These heterojunctions leverage the differences in conduction band positions between TiO
2 (B) and anatase, promoting efficient charge transfer and enhancing the photocatalytic degradation of pollutants [
21,
25,
26]. This leads to the potential of TiO
2 (B)/anatase heterostructures for advance photocatalytic applications [
27,
28]. The calcination temperature plays a crucial role in determining the heterojunction structure, as it influences the ratio of TiO
2 (B) to anatase composites, thereby affecting the photodegradation rates of various contaminants [
29,
30,
31,
32,
33,
34,
35]. Previous studies have investigated the transformation of TiO
2 nanobelts (B) into TiO
2 nanobelt (B)/anatase heterojunctions at elevated temperatures [
22]. However, systematic studies on the photodegradation performance of TiO
2 (B)/anatase heterojunctions against emerging aqueous pollutants across different water matrices remain unexplored. This knowledge gap limits a thorough understanding of how varying TiO
2 (B)/anatase ratios influences their effectiveness and applicability in water treatment. Investigating their role in industrial wastewater treatment—an essential area for photocatalysis—requires an examination of the effects of emerging pollutants and different water matrices, such as rainwater and Milli-Q water, on their photocatalytic performance. Evaluating the heterojunction’s efficiency in degradation and mineralization processes is essential to optimizing TiO
2 (B) performance under diverse environmental conditions and establish their suitability for advanced wastewater treatment applications.
Pharmaceuticals and pesticides are two significant contaminants that have prompted concern; the EU has classified methomyl and diclofenac as emerging contaminants [
36] because of their high pollution levels, low environmental concentrations, propensity for bioaccumulation, and detrimental effects on humans and biota [
37]. This study explores the hydrothermal synthesis of TiO
2 nanobelt/anatase heterojunctions for the degradation of diclofenac, methomyl, and phenol contaminants. The photocatalytic efficacy of the developed nanomaterials was assessed in different water matrices, including rainwater from Garray, Spain. This study focuses on the assessment of the photocatalytic activity of self-transformed nanobelt (B)/anatase heterojunctions formed through TiO
2 (B) calcination at various temperatures (400–800 °C). The transition of TiO
2 (B) to TiO
2 (B)/anatase was systematically investigated, and the photodegradation behavior was reported. The systematic variation in the reaction conditions provides insights into the relationships between the processing variables and the resulting TiO
2 nanobelt structures, highlighting the potential influence of these variables on the photocatalytic activity.
2. Materials and Methods
Titania (P25, 80% anatase, 20% rutile) was procured from Evonik Industries, Germany, and sodium hydroxide (NaOH, reagent grade, 97%) was obtained from Acros (Baroda, India). Hydrochloric acid (HCl, 35.0–37.0%) and sulfuric acid (H
2SO
4, 95–98%) were obtained from Beijing Chemical Works (Beijing, China). For the chromatographic analyses, methanol (99.9%), acetonitrile (HPLC-grade), and orthophosphoric acid (85%) were supplied by Scharlab (Barcelona, Spain). Solutions were prepared using Milli-Q Type 1 water and Strom water. Rainwater samples were collected from a collection center located in Madrid, Spain. To remove solids and particles, glass fiber filters (0.7 μm) were employed, and the filtered samples were stored at 4 °C in the dark. The pesticide selected for treatment was methomyl, with a purity of 99.5%, obtained from Aragonesas Agro S.A. (Madrid, Spain) Diclofenac sodium (DCF, >99% purity) and phenol (99% purity) were selected as the model pharmaceutical and organic pollutants, respectively, both procured from Aldrich (Darmstadt, Germany). The characteristics of the pollutants are summarized in
Table S1. The physicochemical properties of the utilized water are listed in
Table S2.
2.1. Synthesis of Titanium Dioxide Nanobelts
TiO
2 nanobelts were developed utilizing a simple hydrothermal approach. A typical synthesis involved combining 0.75 g of TiO
2 P25 with 150 mL of 10 M NaOH aqueous solution. The mixture was magnetically agitated, transported to a Teflon-lined stainless-steel autoclave, heated at 180 °C for 48 h, and finally left to cool at an ambient temperature. The finished powder was thoroughly cleaned with deionized water and filtered. The wet powder was immersed (24 h) in a 0.1 M HCl aqueous solution and washed with distilled water to yield H
2Ti
3O
7 nanobelts (TB-100 °C). By annealing the H
2Ti
3O
7 nanobelts for 4 h at 400, 600, and 800 °C, TiO
2 nanobelts with different phases were obtained, referred to as TB-400 °C, TB-600 °C, and TB-800 °C, respectively. The H
2Ti
3O
7 nanobelts were also annealed at 900 °C under similar conditions, resulting in their transformation into pure anatase, referred to as TB-900 °C, to evaluate and compare their photodegradation behavior. The possible formation mechanism is presented in Equations (1)–(3) [
27].
2.2. Characterization
Morphological and microstructural features were examined via a scanning electron microscope (SEM) equipped with an energy-dispersive spectroscopy (EDS) detector and a JEOL-IT300 microscope (JEOL Ltd., Tokyo, Japan). X-ray diffraction (XRD) patterns were obtained via X’Pert high-score diffractometer (Rigaku, Tokyo, Japan) under ambient conditions, employing cobalt K-α radiation (λ = 1.54 Å) at 10 mA and 30 kV. Measurements were performed with a step size of 0.005° over a 2θ range of 20° to 80°. Surface functional groups and chemical bonding were analyzed using Fourier transform infrared spectroscopy (FTIR) (Agilent Technologies, Santa Clara, CA, USA) with an Excalibur series instrument in attenuated total reflectance (ATR) mode. Spectra were recorded over the range of 550–4000 cm−1 with a resolution of 4 cm−1 and 32 scans. A diamond crystal was utilized as the internal reflective element (IRE). X-ray photoelectron spectroscopy (XPS) was conducted via a Thermo Scientific ESCALAB 250Xi X-ray spectrometer (Waltam, MA, USA) with Al Kα radiation at 1486.6 eV. Additionally, UV-visible diffuse reflectance spectra (DRS) were recorded at room temperature using a Shimadzu UV-Vis-NIR spectrophotometer (Model UV-3600, (Kyoto, Japan)) over a wavelength range of 200–800 nm.
2.3. Photocatalytic Activity
A series of controlled studies were conducted to assess the photocatalytic activity of the developed nano catalysts. Suspensions containing the catalyst at a concentration of 1 g/L were introduced into a photoreactor and subjected to radiation. The light source consisted of ten UVA LED lamps (Seoul Viosys, Ansan-si, Republic of Korea) emitting at a wavelength of 385 nm. These LEDs were strategically positioned to ensure uniform illumination across the reactor surface. Each LED was operated at a current intensity of 250 mA, consuming 8.38 W of electrical power. The power output of the irradiation lamp, which was positioned 4.5 cm above the solution surface, was quantified via the potassium ferrioxalate actinometry technique, which yielded an emission rate of 1682.8 ± 77.1 μmol m2 s−1 photons.
The photocatalytic activity was evaluated by monitoring the degradation of target pollutants, initially at a concentration of 0.1 mM, and fitting the degradation curves to an exponential decay model. All the experiments were performed under natural pH conditions. Before irradiation, the reaction mixture was mechanically stirred and incubated in the dark for 30 min to establish adsorption equilibrium. During the photocatalytic process, 0.8 mL aliquots of the irradiated solution were withdrawn via a syringe fitted with a 0.45 μm nylon filter to remove the suspended photocatalyst particles.
The concentrations of pollutants were determined via high-performance liquid chromatography (HPLC), Agilent Technologies (Santa Clara, CA, USA), with a diode array detector, binary gradient high-pressure pump, and automated sampler. After 120 min of irradiation, the total organic carbon (TOC) content was measured with a TOC analyzer (TOC-VCSH/CSN, Shimadzu, Kyoto, Japan).
The studies were conducted using two types of water: Milli-Q ultrapure water (UW) and rainwater (RW) collected in Madrid, Spain. The water sources used for the studies were Milli-Q ultrapure water (pH = 6.5) and stormwater (pH = 8.1–8.6), which was collected from the retention tank of a sustainable urban drainage system (SUDS) established in Madrid, Spain. Physicochemical analyses indicated that compared with UW, RW contained a greater concentration of ions, resulting in greater conductivity and total dissolved solids. Additionally, RW presented a more basic pH (8.1–8.6) than did UW.
Equation (4) presents the results of the Langmuir–Hinshelwood model analysis of the reaction kinetics for photodegradation.
where C
0 is the starting pollutant concentration, (t) is the duration, (k
r) is the intrinsic rate constant, and (k
ad) is the adsorption equilibrium constant. The initial rates that were collected were used to evaluate the efficiency of the photocatalytic process under different scenarios. All experiments were conducted twice, with the observed deviation being less than 3%, demonstrating the reproducibility and reliability of the results.
3. Results
The scanning electron microscopy (SEM) images elucidate the morphology of the TiO
2 nanobelts synthesized under different calcination conditions. The as-prepared nanobelts (
Figure 1a,b) have widths ranging from 40 to 80 nm and length extending up to several micrometers while maintaining a notably thin profile. The morphologies of the TiO
2 nanobelts calcined at temperatures of 400, 600, and 800 °C are depicted in
Figure 1c–h. Notably, there was no significant alteration in the morphology of the nanobelts with increasing calcination temperature, indicating the excellent thermal stability of these TiO
2 nanostructures. However, the nanobelt population density appears to increase with higher calcination temperatures, particularly for TB-600 °C and TB-800 °C, due to the thinning of the nanobelts upon calcination, which in turn leads to a higher population density per unit length. It is well aligned with previous studies, which report that TiO
2 nanobelts retain their structural characteristics at calcination temperatures between 400 and 800 °C, while further calcination to 900 °C merely results in smooth edges at their ends [
22].
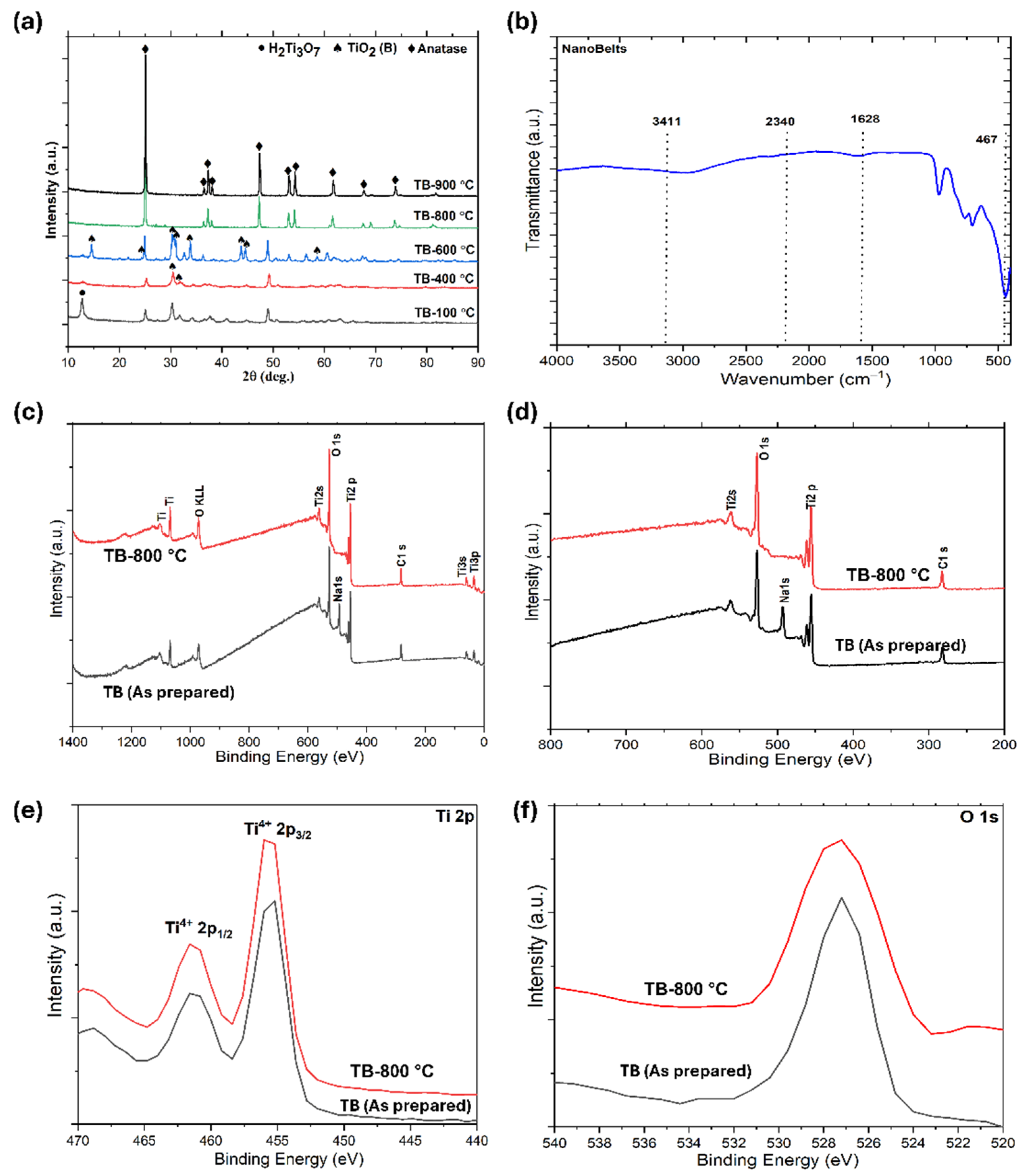
In
Figure 2a, the X-ray diffraction (XRD) analysis of TiO
2 nanobelts and those calcined at different temperatures highlight significant phase transitions. Initially, a peak at 11° corresponds to the H
2Ti
3O
7 phase (TB-100 °C), with weak peaks at 25° and 48° indicating a partial transition to TiO
2 due to dehydration. TiO
2 (B) is characterized by distinct peaks at 14.19°, 24.93°, 30.67°, 33.19°, 44.50°, 45.34°, 58.51°, and 60.86° (JCPDS file no. is 46-1237). After calcination at 400 °C (TB-400 °C), the H
2Ti
3O
7 phase nearly disappears, with TiO
2 (B) becoming the predominant phase, suggesting a mixture of TiO
2 (B) and H
2Ti
3O
7. At 600 °C (TB-600 °C), the TiO
2 (B) peak intensity remains dominant, while anatase peaks begin to emerge, resulting in a composition of approximately 60% anatase and 40% TiO
2 (B). By 800 °C (TB-800 °C), anatase becomes the predominant phase, constituting ~92% of the material, with only ~8% TiO
2 (B), confirming that TiO
2 (B) acts as a metastable phase. The characteristic peaks of anatase are observed at 25.28°, 36.94°, 37.89°, 38.57°, 48.04°, 53.89°, 55.06°, 55.18°, 61.11°, 70.30°, and 75.02°, corresponding to the standard diffraction data from JCPDS file no. 21-1272. Anatase is identified as a tetragonal crystal structure with lattice parameters of
a =
b = 0.38 nm and
c = 0.95 nm. These findings demonstrate the thermal stability and phase evolution of TiO
2 nanobelts, which are critical for optimizing their photocatalytic performance. Upon calcination at 900 °C, a pure anatase phase is formed (TB-900 °C), which is then investigated for photodegradation to be used as a reference for comparing the degradation performance of the TiO
2 (nanobelt)/anatase heterostructures. The combination of TiO
2 phases, such as anatase and nanobelts, can exhibit synergistic effects that enhance the interphase charge transfer.
Figure 2b shows the infrared spectra of the nanobelts (TB-800 °C), highlighting bands at 3400 and 1630 cm
−1 attributed to the weak signals from –OH groups on the material surface and the bending of water molecules linked by weak hydrogen bonds. The bands between 400 and 1000 cm
−1 represent the Ti–O stretching and Ti–O–Ti bridging stretching modes. The spectrum corresponds to a nanobelt in the anatase phase, identified by its characteristic vibration bands at 492 and 791 cm
−1, which are associated with the Ti-O-Ti bond vibrations. Additionally, the band at 960 cm
−1 corresponds to the characteristic band of the titanium tetrahedral framework. The broadband between 3000 and 3600 cm
−1 reflects the O–H stretching mode of the hydroxyl groups, whereas the band at 1659 cm
−1 is due to the O–C–O–Ti link within the carbonate structure, suggesting the incorporation of carbon at the surface Ti atoms.
X-ray photoelectron spectroscopy (XPS) was used to analyze the chemical states of Ti and O in the TiO
2 nanobelts, where trace amounts of sodium in the as-prepared nanobelts (without washing) are also observed (
Figure 2c), The analysis observed the corresponding photoelectron peaks at 281.6 eV (C 1s), 492.8 eV (Na), 456.8 eV (Ti 2p), and 528.8 eV (O 1s) [
38,
39]. The Ti 2p
3/
2 and Ti 2p
1/
2 peaks were observed at binding energies of 456.6 eV and 461.6 eV [
39], respectively, aligning with standard TiO
2 values (
Figure 2d,e). The two main doublet Ti 2p peaks are characteristic of Ti
4+ in TiO
2. The O 1s peak at 528.8 eV corresponds to lattice oxygen (Ti–O bonds) and surface Ti–OH groups (
Figure 2f) [
38]. These surface Ti–OH groups contribute to the hydrophilicity of the catalyst, potentially enhancing its photocatalytic activity. The TB-800 °C spectra showed the absence of a Na peak, indicating that washing Na
2Ti
3O
7 with HCl resulted in the replacement of Na
+ ions with H
+ ions (
Figure 2d). The use of HCl effectively removes alkaline ions, facilitating the synthesis of TiO
2 nanobelts (TiO
2 (B)). These findings underscore the significance of Na
+ ions in forming the unique TiO
2 nanobelt morphology [
40]. The Ti 2p doublet remained almost unchanged before and after calcination, indicating that Ti maintained its tetravalent state throughout the process.
The UV–visible diffuse reflectance spectroscopy (DRS) results and energy band structures of TiO
2 nanobelts synthesized at various temperatures reveal distinct absorption edges and band gap values (
Figure 3). A phase transition from TiO
2 (B) to TiO
2 (B)/anatase led to an increase in band gap values, with TiO
2 (B) exhibiting a band gap of 2.8 eV and the TiO
2 (B)/anatase heterostructure showing a band gap of 3.18 eV (
Table 1). This anatase–TiO
2 (B) heterostructure promotes the separation of photogenerated charges, enhancing photocatalytic activity. The increase in anatase content during calcination, particularly for TB-800 °C, resulted in a higher band gap. Due to their heterostructure, TiO
2 nanobelts (TiO
2 (B)) effectively reduce charge carrier recombination, facilitating their movement toward the photocatalyst surface. Tauc plots indicated band gap values of 3.08, 3.10, 3.18, and 3.20 eV for TiO
2 nanocrystals calcined at 400 °C, 600 °C, 800 °C, and 900 °C, respectively. The TiO
2 heterostructure (TB-800 °C) exhibits a unified band gap (closer to the band gap value of anatase ~3.20–3.23 ((TB-900 °C)) [
41]) due to the charge transfer and electronic interactions at the anatase–nanobelt interface. This hybridization aligns the conduction and valence bands, producing a dominant optical response reflective of the heterostructure, rather than individual phase band gaps. Strong coupling typically merges optical transitions into a single band gap, but weak interfaces may retain distinct absorption edges [
22]. The transition from TiO
2 (B) to TiO
2 (B)/anatase is well-aligned with previous studies [
41], which allows photogenerated charges to migrate from anatase to TiO
2 (B), boosting the photocatalytic activity of the TiO
2 nanobelts.
The photocatalytic performance of TiO
2 (B) was assessed by examining the degradation of phenol, diclofenac, and methomyl (
Figure 4). Total organic carbon (TOC) measurements were taken before and after the degradation process, with each catalyst being tested for 120 min under UV illumination. Preliminary adsorption experiments conducted in the dark for 30 min demonstrated that the catalysts exhibited negligible adsorption, with less than 1% of the pollutants adsorbed. This study evaluated the effectiveness of TiO
2 catalysts in ultrapure and highly alkaline water matrices, with a phenol concentration of 0.1 mM. As the photodegradation of these pollutants follows first-order photochemical lifetimes, their degradation rates may vary significantly under the investigated irradiance conditions. Monitoring the extent of mineralization is essential, as the degradation of contaminants like diclofenac and methomyl can lead to the formation of intermediate compounds. These byproducts may exhibit toxicity levels comparable to or even greater than the original pollutants, posing significant environmental and health risks. Mineralization refers to the percentage of carbon from organic matter that is converted into CO
2. The photocatalytic process at TB-400 °C (
Figure 4) resulted in the complete degradation of phenol (60 min) and diclofenac (90 min), while a 78% degradation of methomyl (120 min) was achieved. Interesting findings are obtained for TB-800 °C (
Figure 4a–f), where phenol (30 min), diclofenac (40 min), and methomyl (90 min) were completely abated. This enhanced activity can be attributed to the improved charge transfer at the interface, reduced recombination rates of photogenerated charges, and better light absorption properties of the materials. For TiO
2 (B)/anatase heterostructures, the anatase phase facilitates internal diffusion along the nanobelt edges, developing a unique anatase–TiO
2 (B)/anatase structure that enhances pollutant degradation. Optimizing calcination conditions is thus critical for maintaining this heterostructure, ensuring the efficient photodegradation of diverse and persistent emerging pollutants in complex environmental matrices. The TB-900 °C specimen exhibited a significantly higher photodegradation performance for all three investigated pollutants (phenol, diclofenac, and methomyl) compared to TB-400 °C and TB-600 °C. However, its degradation performance is lower than that of TB-800 °C, which demonstrated the highest photodegradation rates among all the specimens investigated. These results highlight the potential of shape-controlled nanobelts/anatase heterostructures as effective photocatalysts.
Tables S3–S5 exhibits numerical data on reaction kinetics rate constants and degradation efficiencies for photocatalytic degradation of phenol, methomyl, and diclofenac in Milli-Q water.
The rates of photodegradation are substantially controlled by water chemistry, which is another crucial parameter affected by nature of water contents. Specifically, a substantial association is expected between water scarcity and photodegradation processes involving pollutants such as phenol, methomyl, and diclofenac; therefore, this exploration extends further to the photodegradation behavior of developed nanocatalysts in rainwater matrices [
3].
A similar pattern of photocatalytic performance was observed for rainwater (
Figure 5), where complete degradation for phenol and diclofenac was observed, albeit at a comparatively prolonged time. Catalytic performance studies showed that TB-800 °C exhibit higher photodegradation efficiency compared to TB-400 °C, TB-600 °C, and TB-900 °C. This improvement is attributed to the increased active sites and nanoparticle surface area, which facilitate a more effective charge transfer at the interface, slower recombination rates of photogenerated charges, and enhanced light absorption characteristics of the materials. Under similar conditions, methomyl degraded more slowly in both water matrices, primarily due to its more complex molecular structure. The TOC value demonstrates the robust photodegradation response of the examined nanobelts, whereas the heterostructure TiO
2 is shown to be equally efficient for the mineralization of contaminants in rainwater and ultrapure water.
Table 2 illustrates the photodegradation rates of TB-800 °C, highlighting the importance of achieving complete degradation under the tested conditions. A detailed analysis of reaction kinetics rate constants and degradation efficiencies for the photocatalytic decomposition of the investigated pollutants across all specimens is provided in the
supplementary data (Tables S3–S5).
Given this, ultrapure water (UW) has shown markedly superior performance, achieving total organic carbon (TOC) removal rates of approximately 89% and complete pollutant degradation. Notably, phenol and diclofenac exhibit faster photodegradation kinetics than methomyl, possibly because of the dependence on the reduction potential of the chemical species. Compared with ultrapure water, photodegradation was somewhat slower in rainwater, although the overall photodegradation behavior remained the same, in the following order: phenol > diclofenac > methomyl. The photocatalytic activity of TB-800 °C is higher than that of other TiO
2 nanobelts, and further, the sedimentation properties of TiO
2 (B) than commercial TiO
2, make it easier to separate from the aqueous solution [
25]. In conclusion, the TiO
2 nanobelt/anatase heterostructure improves the photocatalytic effectiveness by combining one-dimensional TiO
2 nanobelts with the strong photocatalytic activity of the self-transformed anatase phase. The nanobelts’ large surface area and shape improve light absorption, while the developed heterojunction between the nanobelts and self-transformed anatase promotes effective electron–hole separation, lowering recombination and improving the charge transfer.
4. Discussion
The transformation of TiO
2 (B) to the anatase base heterostructure occurs during calcination ranging from 400 to 800 °C, where TiO
2 (B) progressively transitions into nanobelt/anatase heterostructures. The self-transformation process developed a well-aligned and stable phase TiO
2 (B)/anatase, as confirmed by XRD, SEM, and UV-Vis analysis. This transformation results in a TiO
2 (B)/anatase heterostructure, which exhibits enhanced photocatalytic performance compared to pure TiO
2 (B) against three different ranges of pollutant categories. The self-transformed TiO
2 heterostructure nanobelts (TB-800 °C) facilitate internal diffusion along nanobelt edges, forming a unique anatase–TiO
2 (B)/anatase heterostructure that significantly enhanced the pollutant degradation (
Figure 6), positioning them as promising candidates for practical photocatalytic applications [
24,
25]. The TiO
2 (B) phase has higher positions for both the valence band and conduction band compared to pure anatase. Thus, in the case of TiO
2 (B)/anatase heterojunctions, photoexcited electrons migrate to the anatase phase, facilitating the photogenerated electrons in the conduction band to reduce molecular oxygen to form superoxide radicals (•O
2−), as described in Eq. (8). These reactive oxygen species are essential intermediates that drive the degradation of pollutants through oxidative pathways, contributing significantly to the observed photocatalytic performance of the material, while holes in the valence band move to the TiO
2 (B) phase, where they interact with pollutants [
28,
38,
39,
40]. This process improves the separation of charge carriers and enhances light absorption through reflection and refraction, resulting in increased photon absorption. The synergistic effect of these mechanisms promotes efficient charge separation, prolongs charge carrier lifetimes, increases the number of active sites, and enhances light absorption, boosting the performance of TiO
2 nanobelts synthesized at 800 °C. However, calcination at temperatures beyond 800 °C, such as 900 °C, leads to the formation of pure anatase. This results in the loss of the beneficial heterojunction interface, thereby causing comparatively lower photodegradation efficiency. The degradation mechanisms of phenol, diclofenac, and methomyl under TiO
2 photocatalysis demonstrate distinct variations in efficiency due to their reaction pathways [
42], despite their similar advanced oxidation processes. Phenol, owing to its simple structure and high reactivity, undergoes the fastest degradation (within 30 min) and achieves nearly complete mineralization under UV light using TB-800 °C. This results in highly efficient water purification in both Milli-Q and rainwater systems. Diclofenac, an anti-inflammatory drug, degrades at a moderate yet promising rate (45 min), achieving effective mineralization while potentially minimizing harmful by-products, such as hydroxy-diclofenac and dichlorodiclofenac [
43], as confirmed by a TOC analysis and rapid degradation rates. Methomyl, a pesticide, undergoes a complex degradation pathway [
44] and achieves complete mineralization within 90 min under similar conditions. TiO
2 (B)/anatase has proven to be a highly effective photocatalyst across a diverse range of pollutants, demonstrating its potential for photodegradation within 30–90 min in both Milli-Q and rainwater. This highlights its suitability as a material of choice for further studies aimed at comprehensively understanding reaction pathways. Advanced TiO
2 (B)/anatase materials hold the potential for sustainable pollutant removal, particularly in achieving better reaction rates and reduced toxic by-product formation [
45]. Although a comprehensive evaluation of TiO
2 (B)/anatase versus P25 (commercial) requires consideration of factors such as byproduct formation, irradiation intensity, regeneration, and reaction conditions, both materials effectively demonstrate phenol degradation. As a reference, P25 (C
0 = 0.53 mM, 0.5 g/L TiO
2) achieves complete degradation in 180 min under a 400 W lamp [
46], whereas TiO
2 (B)/anatase (TB-800 °C, C
0 = 0.1 mM, 1 g/L TiO
2) achieves complete degradation in 45 min while showing comparable total organic carbon (TOC) removal [
47], indicating complete mineralization. The unique shape-controlled morphology of TiO
2 (B)/anatase (size > 40 nm) enhances light harvesting, improves charge separation, and facilitates electron transfer, significantly reducing electron–hole recombination.
Figure 6 illustrates a proposed mechanism outlining the reactions involved in the photocatalytic process. When the photocatalyst is excited by light irradiation with energy exceeding its band gap, a hole–electron pair is generated. The subsequent reactions may proceed as follows: redox reactions occur, leading to the formation of superoxide ions and hydroxyl radicals (Equation (5)), which serve as oxidizing agents. The highly reactive holes (h
+) may react with water (H
2O) or hydroxyl groups adsorbed on the surface (Equations (6) and (7)), resulting in the generation of additional hydroxyl radicals that also exhibit strong oxidizing properties. Moreover, electrons (e
−) may interact with the oxygen (O
2) adsorbed on the semiconductor surface. Both holes (h
+) and electrons (e
−) can further react with contaminant molecules, facilitating their degradation and subsequent mineralization (Equations (8)–(10)). The photocatalytic degradation was modeled as a pseudo-first-order reaction, allowing its kinetics to be expressed by the following reactions (Equations (5)–(10)) [
48,
49]:


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}