Abstract
Peptide pools are important research tools in different biomedical fields. They consist of a complex mixture of defined peptides, which places high demands on the production and quality control of these products. Previously it has been shown that the combination of UHPLC with high-resolution mass-spectrometry (HRMS) is a fast and powerful method to confirm the relative concentration and the structural identity of all peptides expected to be in the pool. In this work, the additional information contained in the UV chromatograms and mass spectra is used to search for impurities due to synthesis by-products and degradation during storage and transportation and to identify possible analytical artifacts. It was shown that most impurities are only present in trace amounts and can be considered uncritical for most applications. The most frequent and perhaps unexpected impurities were homo- and heterodimers caused by the free cysteines contained in these peptide pools. Furthermore, pyroglutamate and aspartimide formation, deamidation, methionine oxidation, and amino acid deletions could be found. This list is not intended to be comprehensive, but rather a brief guide to quickly identify impurities and, in the long term, to suggest possible changes in the composition of the peptide pools to avoid such impurities by design or by special precautions.
1. Introduction
Impurities in synthetic peptides are rarely discussed in detail. Scientists with less experience in analytical chemistry and organic synthesis often take the purity of a commercial product as granted and may not consider impurities at all. Others assume that a low percentage of a byproduct should be irrelevant per se, which may be not the case in biomedicine, where minute amounts of unwanted constituents may cause strong side effects, such as allergic reactions. Peptide pools consist of a defined combination of different peptides [1], which makes quality control particularly demanding. The number of potential contaminants is likely to be much higher than for a single compound. In the case of pre-clinical or clinical studies and toxicity tests, even trace impurities can lead to the failure of highly valuable drugs or other active substances. For instance, Beck et al. found that the activation of T cells was absent due to the pyroglutamate derivatization of an otherwise active peptide [2]. In addition to false negative results, there is also the risk of false positive outcomes. De Beukelaar et al. observed that non-reproducible T cell activation was caused by deletion peptides [3]. Verbeken et al. demonstrated in experiments with isolated tissue that impurities, specifically adducts of peptides and protecting groups, triggered a functional response in the tissue [4]. Currier et al. reported false positive results in an HIV vaccine study, which were attributed to cross-contamination by a structurally unrelated peptide [5]. Finder et al. noted stereochemical peptide impurities that led to altered aggregation kinetics and increased cytotoxicity [6]. The economic damage resulting from such misleading results can be enormous. Hence, in any biochemical or biomedical experiment, the purity of the used reagents and test materials need to be characterized and unexpected compounds should be examined as detailed as possible. Obviously, such examinations are often disregarded due to time or financial constraints. However, many scientists are not aware of the risks associated with such a hasty procedure. Additionally, some users may be unaware of the typical limitations associated with the analytical methods used for purity determination. This can lead to the misinterpretation of purity specifications presented in a product data sheet, resulting in a significant information gap [7]. On the other hand, additional purity examinations can be very laborious, and hence they are rarely performed. Therefore, we proposed a fast and easy approach for the analysis of peptide pools and other products containing synthetic peptides [8]. This involves the execution of a state-of-the-art UHPLC separation combined with UV detection and subsequent or parallel high-resolution mass-spectrometry, for example with an orbitrap or a TOF-MS system. This approach leads to a robust information about the relative quantity of a peptide and the confirmation of the chemical identity. This can be a sufficient characterization of a commercial product in the context of a quality control process. Nevertheless, such powerful analytical systems as those mentioned above can deliver much more information. In this paper, we reexamined the chromatograms and mass-spectra of a study conducted for the quality control of a typical peptide pool, known as CEF [9,10], which nominally consists of 32 peptides with a length of 8 to 12 amino acids (Table 1). By utilizing extracted ion chromatograms (XICs) with a maximum mass tolerance of 5 ppm, a targeted search was conducted for frequently occurring peptide-related impurities. This search included oxidized peptides, peptides containing cyclized amino acid residues and their derivatives, truncation sequences resulting from pyroglutamate formation during synthesis, and known deletion peptides identified from individual peptide measurements. An automated approach including algorithms known from non-target analysis seems to be lacking today, which obviously would facilitate this type of impurity analysis in the future, even for the less experienced scientist.

Table 1.
List of all expected peptides in the CEF pool.
2. Materials and Methods
2.1. Chemicals
The CEF peptide pool consists of 32 peptides derived from specific HLA class I-restricted T cell epitopes associated with cytomegalovirus (CMV), Epstein–Barr virus (EBV), and influenza virus. Due to its relevance, it is available from several manufacturers. Our samples have been described in more detail in our previous paper [8]. Peptide 5 (CLGGLLTMV, purity 95.2%, EP08605), peptide 20 (ELRSRYWAI, purity 95.1%, EP08620), and peptide 23 (QAKWRLQTL, purity 90.9%, EP08623) were acquired from Peptides & Elephants GmbH, Hennigsdorf, Germany. Trifluoroacetic acid (TFA, purity > 99.5%, 85183) was sourced from Thermo Fisher Scientific, Life Technologies GmbH. Acetonitrile (ACN, LC-MS grade, 2697.2500) was obtained from Th. Geyer GmbH & Co. KG, 71,272 Renningen, Germany. Helium 6.0 (10100530) was purchased from Linde AG. The LTQ ESI positive ion calibration solution (purity > 99.5%, 88322) was acquired from Thermo Fisher Scientific GmbH, 16,761 Hennigsdorf, Germany. Ammonium bicarbonate (ABC, purity > 99.0%, 09832) was obtained from Fluka. Tris(2-carboxyethyl)phosphine hydrochloride (TCEP, purity = 98%, AB121644) was purchased from ABCR GmbH, 76,189 Karlsruhe, Germany. Iodoacetic acid (IAC, purity > 97%, 211794) was sourced from J&K Scientific, San Jose, CA 95110, USA. Iodoacetamide (purity ≥ 99%, I1149) was obtained from Sigma-Aldrich, St. Louis, Missouri, USA. Sodium hydroxide (purity ≥ 98%, 1372.1000) was acquired from Th. Geyer GmbH & Co. KG, 71,272 Renningen, Germany. Water was purified using an Ultra-Pure Water System from Merck Millipore, Darmstadt, Germany, with a resistivity of 18.2 MΩ·cm.
2.2. Sample Preparation
2.2.1. Impurity Identification
CEF Peptide Pool
The lyophilized CEF peptide pool sample (0.8 mg) was stored at −20 °C and allowed to equilibrate at room temperature (RT) for 30 min prior to use. It was dissolved in 50 µL of ACN/TFA 0.05% (v/v) and then in 50 µL of water/TFA 0.05% (v/v) to create a stock solution of 8 µg/µL (approximately 0.25 µg/µL per peptide). Both solvents were degassed by sonication (45 kHz, 15 min, RT). Following a 15-min equilibration period, 20 µL of the stock solution were diluted with 180 µL of water/TFA 0.05% (v/v), achieving a final concentration of 0.8 µg/µL, which corresponds to approximately 0.025 µg/µL per peptide. After centrifugation (2000× g, 5 min, RT), 180 µL of the resulting supernatant was transferred into an amber HPLC glass vial containing a 200 µL glass insert. The solvents used for the peptide pool were also employed in the preparation of blank samples.
A 5% ACN blank solution was prepared by diluting 100 µL of a 1:1 mixture of water/TFA 0.05% (v/v) and ACN/TFA 0.05% (v/v) with 900 µL of water/TFA 0.05% (v/v). Following centrifugation at 2000× g for 5 min at room temperature, 800 µL of the supernatant was transferred to an amber HPLC glass vial.
Peptide 20 and 23
Lyophilized peptides 20 (ELRSRYWAI, 1 mg) and 23 (QAKWRLQTL, 1 mg) were stored at −20 °C and allowed to equilibrate at room temperature for 30 min before use. They were then dissolved in 500 µL of ACN/TFA 0.05% (v/v), followed by the addition of 500 µL of water/TFA 0.05% (v/v) to create a stock solution with a concentration of 1 µg/µL. Prior to use, both solvents were degassed by sonication at 45 kHz for 15 min at room temperature. A volume of 20 µL of the peptide stock solution was diluted with 750 µL of water/TFA 0.05% (v/v) and 30 µL of ACN/TFA 0.05% (v/v), resulting in a final concentration of 0.025 µg/µL. After centrifugation (2000× g, 5 min, RT), 700 µL of the supernatant was transferred to an amber HPLC glass vial.
A blank solution was prepared by diluting 100 µL of water/TFA 0.05% (v/v) with 100 µL of ACN/TFA 0.05% (v/v). Subsequently, 100 µL of this mixture was further diluted by adding 900 µL of water/TFA 0.05% (v/v), resulting in a 5% ACN blank solution. After centrifugation (2000× g, 5 min, RT), of the supernatant was transferred to an amber HPLC glass vial.
2.2.2. Alkylation of Cysteine-Residues
Reduced Peptide 5
Lyophilized peptide 5 (CLGGLLTMV, 1 mg) was stored at −20 °C and allowed to equilibrate at room temperature for 30 min before use. It was then dissolved in 500 µL of ACN/TFA 0.05% (v/v), followed by an additional 500 µL of water/TFA 0.05% (v/v) to create a stock solution with a concentration of 1 µg/µL. Prior to use, both solvents were degassed by sonication at 45 kHz for 15 min at room temperature. The stock solution was aliquoted into 100 µL portions and stored at −20 °C. Before LC-MS analysis, the aliquots were thawed for 30 min. Subsequently, 80 µL of the stock solution was diluted with 320 µL of water (1:5 ratio), resulting in a final concentration of 0.2 µg/µL. This solution was then combined with 100 µL of a 100 mM aqueous TCEP solution and shaken for 10 min at room temperature (950 rpm). After centrifugation (2000× g, 5 min, RT), 170 µL of the supernatant was transferred to an amber HPLC glass vial containing a 200 µL glass insert.
A blank solution was prepared by diluting 100 µL of a 0.05% (v/v) water/TFA solution with 100 µL of a 0.05% (v/v) ACN/TFA solution. Subsequently, 100 µL of this mixture was further diluted by adding 900 µL of a 0.05% (v/v) water/TFA solution, resulting in a 5% ACN blank solution. After centrifugation (2000× g, 5 min, RT), 800 µL of the supernatant was transferred to an amber HPLC glass vial. Additional blank aliquots were stored at −20 °C.
IAA-Alkylation of Peptide 5
Lyophilized peptide 5 (CLGGLLTMV, 1 mg) was stored at −20 °C and equilibrated at room temperature for 30 min prior to use. It was dissolved in 62.5 µL of ACN/TFA 0.05% (v/v) and then in an additional 62.5 µL of water/TFA 0.05% (v/v) to create an 8 µg/µL stock solution. Before use, both solvents were degassed by sonication at 45 kHz for 15 min. After a 15-min equilibration period, the stock solution was aliquoted into 10 µL portions and stored at −20 °C. Before LC-MS analysis, aliquots were thawed for 30 min and diluted with 355 µL of 1 M ammonium bicarbonate buffer (pH 8.0) and 35 µL of ACN to achieve a final concentration of 0.2 µg/µL. A total of 100 µL of this solution was combined with 100 µL of a 10 mM TCEP solution (dissolved in 1 M ABC buffer, pH 8.0) and shaken for 30 min at room temperature. Subsequently, 100 µL of the reduced peptide 5 solution was mixed with 100 µL of a 10 mM IAA solution (dissolved in 1 M ABC buffer, pH 8.0) and shaken for 30 min at room temperature in the dark. To prevent overalkylation, 100 µL of the alkylated peptide 5 was quenched with 100 µL of the previously used 10 mM TCEP solution (dissolved in 1 M ABC buffer, pH 8.0) and shaken for 15 min at room temperature in the dark. After centrifugation (2000× g, 5 min, RT), 180 µL of the supernatant was transferred to an amber HPLC glass vial with a 200 µL glass insert.
A blank solution was prepared by diluting 100 µL of a 0.05% (v/v) water/TFA solution with 100 µL of a 0.05% (v/v) ACN/TFA solution. Subsequently, 100 µL of this mixture was further diluted by adding 900 µL of the previously used ABC buffer (1 M, pH 8.0). After centrifugation (2000× g, 5 min, RT), 800 µL of the supernatant was transferred to an amber HPLC glass vial.
IAC-Alkylation of Peptide 5
Lyophilized peptide 5 (CLGGLLTMV, 1 mg) was stored at −20 °C and equilibrated at room temperature for 30 min prior to use. It was dissolved in 62.5 µL of ACN/TFA 0.05% (v/v) and then in 62.5 µL of water/TFA 0.05% (v/v) to create a stock solution with a concentration of 8 µg/µL. Before use, both solvents were degassed by sonication at 45 kHz for 15 min at room temperature. After a 15-min equilibration period, the stock solution was divided into 10 µL aliquots and stored at −20 °C. Prior to LC-MS analysis, aliquots were thawed for 30 min. Subsequently, one aliquot was diluted with 355 µL of 0.1 M ammonium bicarbonate buffer (pH 8.0) and 35 µL of ACN, resulting in a final peptide 5 concentration of 0.2 µg/µL. A volume of 100 µL of this solution was combined with 100 µL of a 2.2 mM TCEP solution (dissolved in 0.1 M ABC buffer, pH 8.0) and shaken for 30 min. Following this, 100 µL of the reduced peptide 5 solution was mixed with 100 µL of a 4.4 mM IAC solution (in 0.1 M ABC buffer, pH 8.0) and shaken for 1 h at room temperature in the dark. Finally, 100 µL of the alkylated peptide 5 solution was diluted with 100 µL of the previously used ABC buffer (0.1 M, pH 8.0), and after centrifugation, 180 µL of the supernatant was transferred to an amber HPLC glass vial containing a 200 µL glass insert.
A blank solution was prepared by diluting 100 µL of a 0.05% (v/v) water/TFA solution with 100 µL of a 0.05% (v/v) ACN/TFA solution. Subsequently, 100 µL of this mixture was further diluted by adding 900 µL of the previously used ABC buffer (0.1 M, pH 8.0). After centrifugation (2000× g, 5 min, RT), 800 µL of the supernatant was transferred to an amber HPLC glass vial.
2.3. UHPLC-HRMS Analysis
Samples were analyzed using a Thermo Scientific UltiMate 3000 RSLC-nano UHPLC System coupled with a Thermo Fisher Scientific Exactive Orbitrap High-Resolution Mass Spectrometer. The autosampler temperature was maintained at 4 °C. Chromatographic separation was achieved with an Acclaim PepMap RSLC C18 column (100 Å, 2 µm, 0.3 mm × 150 mm) in conjunction with an Acclaim PepMap RSLC C18 trap precolumn (100 Å, 5 µm, 0.3 mm × 5 mm), both sourced from Thermo Scientific. The mobile phases for chromatography consisted of 0.05% (v/v) TFA in water (A) and 0.04% (v/v) TFA in acetonitrile (B). The mobile phases were degassed by purging with helium 6.0 for 5 min. Peptides were eluted using the following gradient: 4% B isocratic for 4 min (6 µL/min), followed by a linear increase to 44% B over 100 min (6 µL/min), then a linear increase to 95% B over 0.1 min (6 µL/min), held at 95% B for 6 min (10 µL/min), returned to initial conditions within 0.1 min (10 µL/min), and maintained at a flow rate of 10 µL/min for 11 min, followed by 6 µL/min for 1 min. For the stability studies of IAC-alkylated and IAA-alkylated peptide 5, a shorter HPLC method was employed. This method differed only in the gradient, which increased linearly from 4% B to 56% B over 26 min (6 µL/min) and then to 95% B in 1 min (6 µL/min). The column oven temperature was set to 55 °C. The injection volume was 1 µL, conducted in full loop mode, with a flush volume of 5 µL, a flush volume 2 of 3 µL, and a loop overfill of 2 µL. UV detection was performed at 214 nm. Prior to analyzing the peptides in the mass spectrometer, the eluate was split in a 1:10 ratio using a T-piece. Subsequent electrospray-ionization was conducted in positive mode (ESI+). The mass spectrometer settings were configured as follows: spray voltage at 4.5 kV, capillary voltage at 30 V, capillary temperature at 320 °C, tube lens voltage at 80 V, skimmer voltage at 24 V, and scan range from 200 to 1800 m/z. The MS scans were acquired with ultra-high resolution (100,000 at 200 m/z) at a scan rate of 1 Hz, a balanced automatic gain control (AGC) target (1 × 106), and a maximum injection time (IT) of 500 ms. External mass calibration using LTQ ESI positive ion calibration solution provided a mass accuracy of <5 ppm. Data acquisition was performed using Xcalibur 2.2 with Dionex Chromatography Mass Spectrometry Link (DCMSLink).
2.4. Data Processing
Analysis of mass spectrometry data was performed using Xcalibur 2.2. Theoretical exact masses and m/z values for the peptides and peptide-related impurities were calculated using the online tool Chem-Calc [11]. Experimental m/z values were validated by employing extracted ion chromatograms (XICs) with a maximum mass tolerance of 5 ppm.
UV spectrometry data were analyzed using Chromeleon 7.2.10. Baseline correction was achieved by subtracting the blank run injection. A baseline with specified start and end points was established. For peak detection and integration, a minimum area threshold was defined. Peak areas were measured using the perpendicular drop method.
3. Results
3.1. Impurity Identification
3.1.1. Deletion Peptides
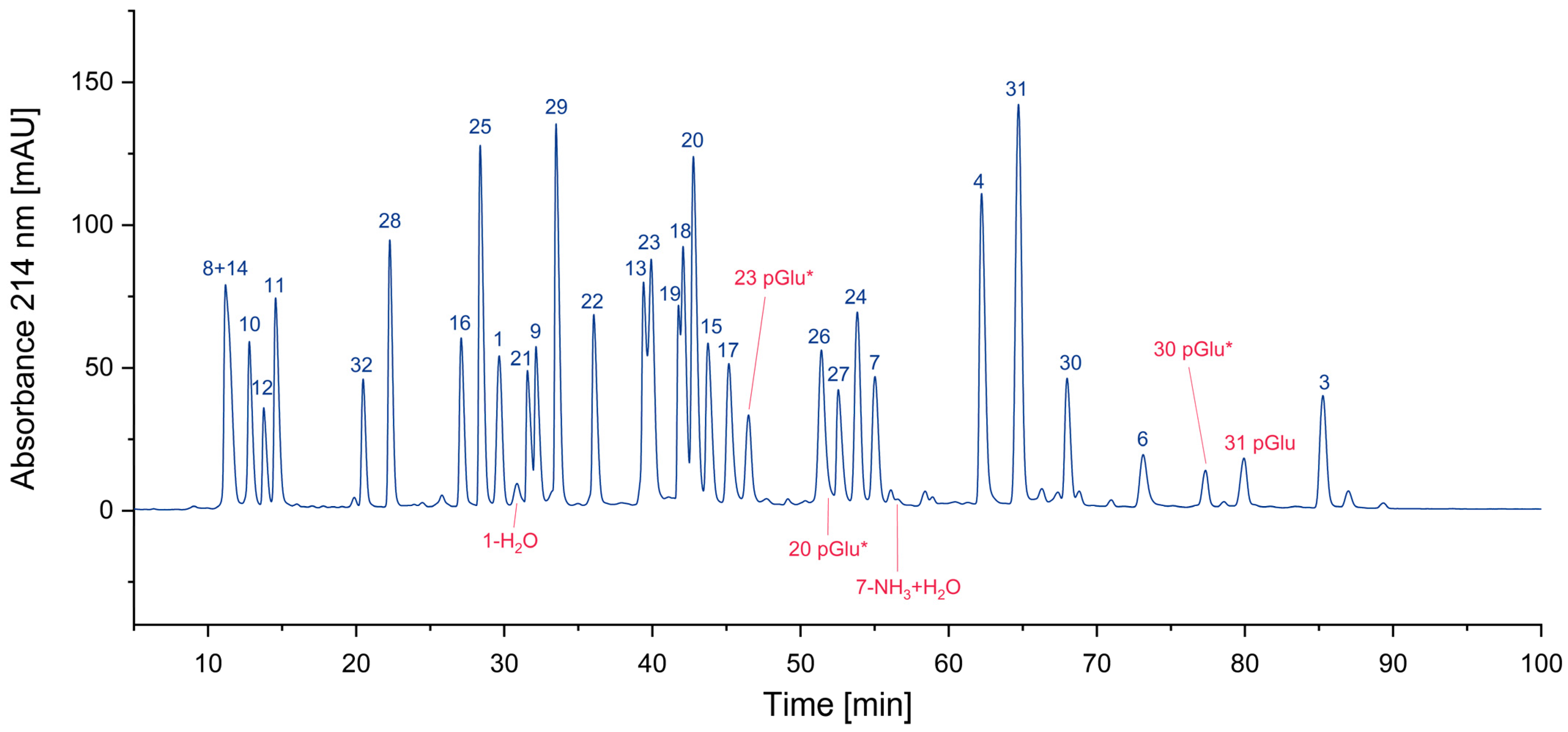
One of the most obvious impurities in synthetic peptides are deletion peptides. Since the synthesis of peptides is performed amino acid by amino acid, some coupling steps might be incomplete, due to factors such as steric hindrance or insufficient activation of the amino acid being coupled. Furthermore, incomplete deprotection of the N-α-amino group can also result in the formation of deletion peptides [13,14,15,16]. This is often overcome by repetition of the respective coupling step if this issue is known. Furthermore, uncoupled amino termini can be blocked by acetylation to avoid further growth, since truncated peptides can be removed more easily than peptides differing only by one amino acid. These impurities can nearly always be traced back to issues during synthesis and subsequent insufficient purification. In the case of this sample of the CEF pool, only very small traces of deletion peptides could be detected. This indicates a highly optimized synthesis process and/or an efficient purification of the individual peptides. In Figure 1, two examples of deletion peptides are indicated.
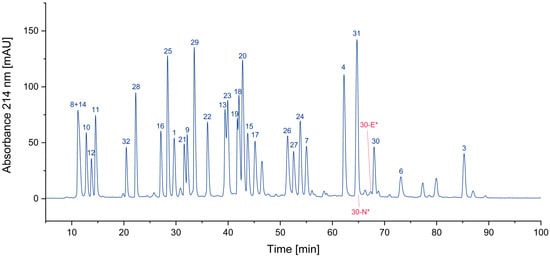
Figure 1.
Search for deletion peptides (* coelution with other peptides/impurities).
3.1.2. Cyclized Amino Acid Residues and Their Derivatives
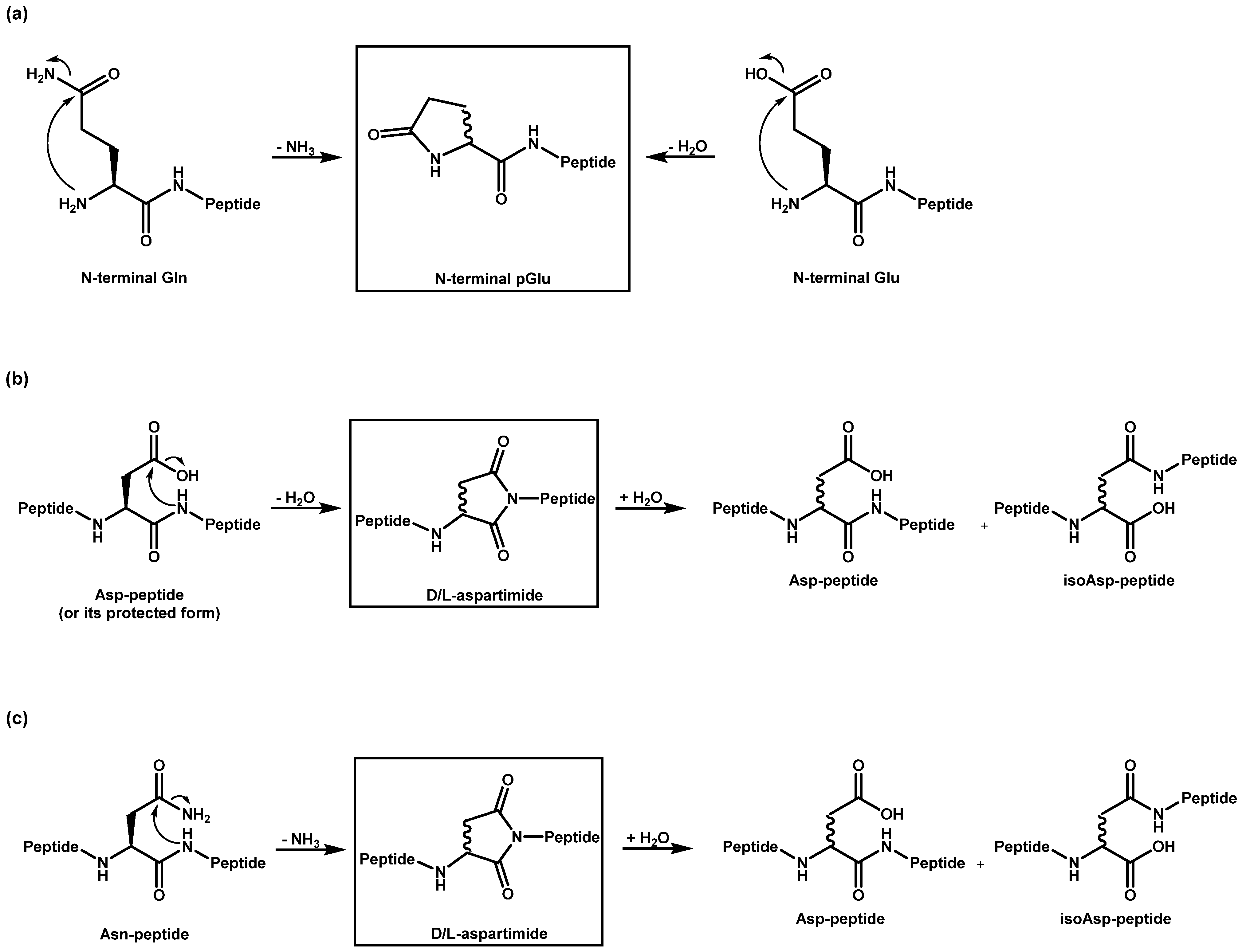
A known side reaction is also the formation of cyclized N-termini of glutamine (Q, Gln) and glutamic acid (E, Glu). In these cases, ammonia or water is released by the condensation reaction. Both reactions lead to the same product with a N-terminal pyroglutamate residue (Figure 2a).
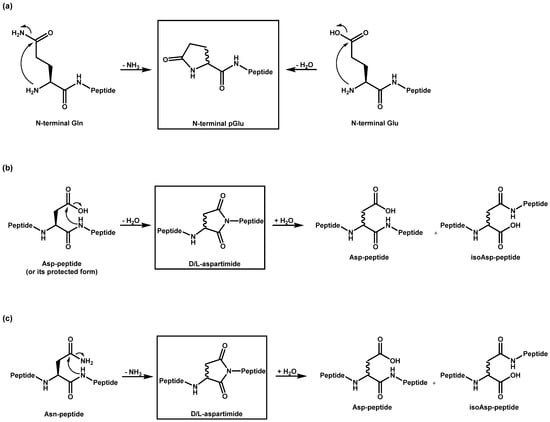
Figure 2.
Cyclization of amino acid residues and subsequent reactions: Reaction pathways for the formation of (a) pyroglutamate, and (b) aspartimide, as well as for (c) deamidation.
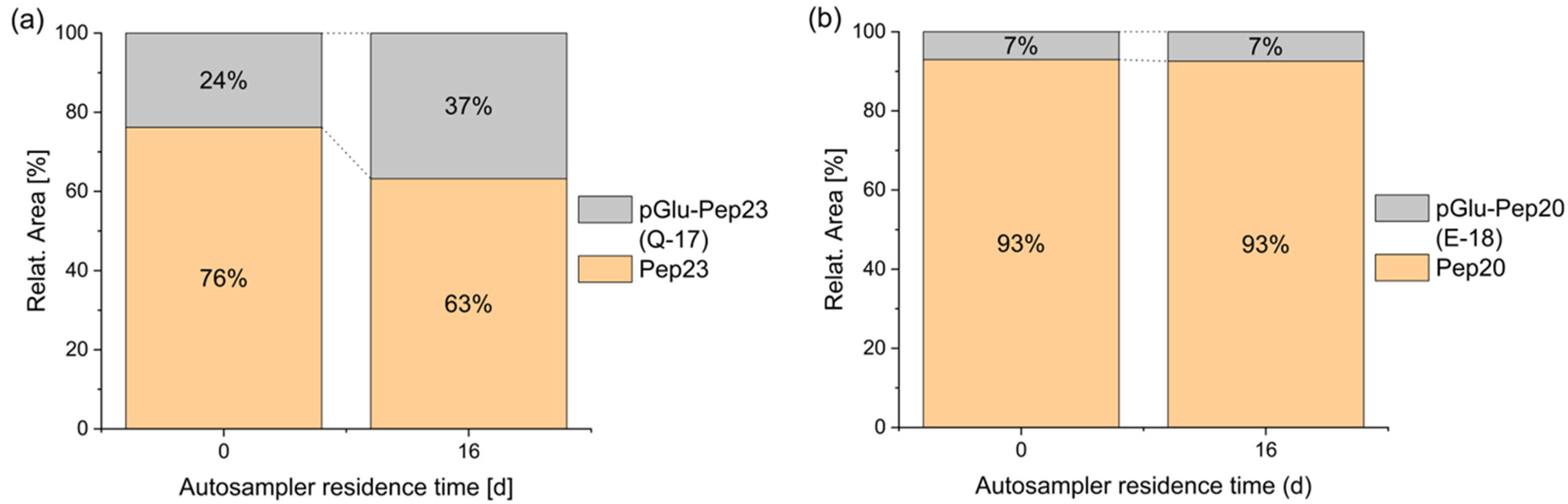
The formation of pyroglutamate usually occurs via acid catalysis [17]. Consequently, it can take place during solid-phase peptide synthesis (SPPS) when coupling an amino acid to the N-terminal glutamine or glutamic acid of the peptide, as well as during the final deprotection and cleavage of the completed peptide. Furthermore, this side reaction has been observed outside of peptide synthesis in acidic aqueous solutions at room temperature [18]. Beck et al. demonstrated that pyroglutamate formation can occur even in a freeze-dried state at elevated temperatures (37 °C), leading them to conclude that similar effects may be expected during long-term storage at lower temperatures [2]. Additionally, under neutral and basic conditions, the formation of pyroglutamate was observed in combination with elevated temperatures in N-terminal glutaminyl peptides [19,20]. The conversion of a peptide into a pyroglutamyl peptide can significantly influence its properties. For example, aminopeptidases are usually not able to cleave this terminal amino acid and the positive charge of the N-terminus is lost. Additionally, it has been shown that the cytotoxic T lymphocyte (CTL) activity of pyroglutamyl peptides may decrease compared to that of unmodified peptides [2]. In the current study, where a glutamic acid or a glutamine was expected at the N-terminus, the formation of some pyroglutamate peptide was detected in all cases. In a solvent consisting of water with 5% acetonitrile and 0.05% trifluoroacetic acid, terminal glutamic acid was fairly stable for several days, in contrast to glutamine, which significantly degraded into pyroglutamate during 16 days of storage (Figure 3). Complete datasets are presented in Figure S1 of the Supplementary Materials. A similar behavior was also reported in the literature before [21]. A comparison of the relative proportions of the pyroglutamate species (pGlu-Pep23: 24%; pGlu-Pep20: 7%) with the purity of the individual peptides, as indicated by the manufacturer (Peptide 23: 90.9%; Peptide 20: 95.1%), suggests that, in addition to the pyroglutamate present from the initial stages of synthesis and purification, storage and/or sample preparation are likely contributing factors to the observed pyroglutamate content.
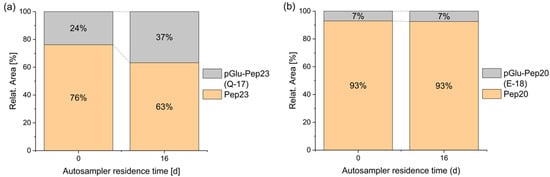
Figure 3.
Stability of N-terminal glutamine vs. glutamic acid during autosampler storage. Spontaneous cyclization of the N-terminal amino acid is faster for peptide 23 with glutamine (a) than of peptide 20 with glutamic acid (b), both in 5% ACN + 0.05% TFA at 4 °C.
Aspartimides (Figure 2b) are formed from aspartic acid (D, Asp) through a cyclization process that involves the loss of water [22]. They are typically considered a side reaction that occurs during synthesis. The formation of aspartimide can occur under both acidic [23] and basic [24] catalysis, with the reaction being more pronounced under basic conditions [25]. This process can take place during piperidine-mediated N-α deprotection, as well as during TFA-mediated global deprotection of peptide side chains in Fmoc/tBu (SPPS) or during TFA-mediated N-α deprotection and HF-mediated global deprotection of peptide side chains in Boc/Bzl SPPS. Additionally, the bases introduced during amino acid coupling can facilitate aspartimide formation [26]. Moreover, aspartimide formation is also possible during purification and storage, presenting a significant challenge. Particularly, the sequence -DG- has been shown to be sensitive to this type of reaction [27,28,29,30]. This could be confirmed in our study. All ten peptides with aspartic acids without a subsequent glycine were stable under the selected conditions and only one with the sequence -DG- showed the formation of the aspartimide. It should be noted that this mechanism also leads to epimerization and a further rearrangement of the amino acid into the iso form, both of which are not easily identified by MS. Furthermore, the aspartimide could be opened by the reagent piperidine during synthesis, leading to another epimerized byproduct. However, these products could not be found in our sample(s). Similar to the loss of water in aspartic acid (Asp), the deamidation of asparagine (Asn) also results in the formation of aspartimide [31], which can subsequently hydrolyze into aspartic acid and into isoaspartic acid (Figure 2c). Aspartimide can be formed under neutral or basic conditions, whereas under acidic conditions, direct hydrolysis into the Asp peptide takes place [32]. Due to this instability under varying conditions, the deamidation of Asn can occur not only during peptide synthesis but also during purification, storage, and application [33].
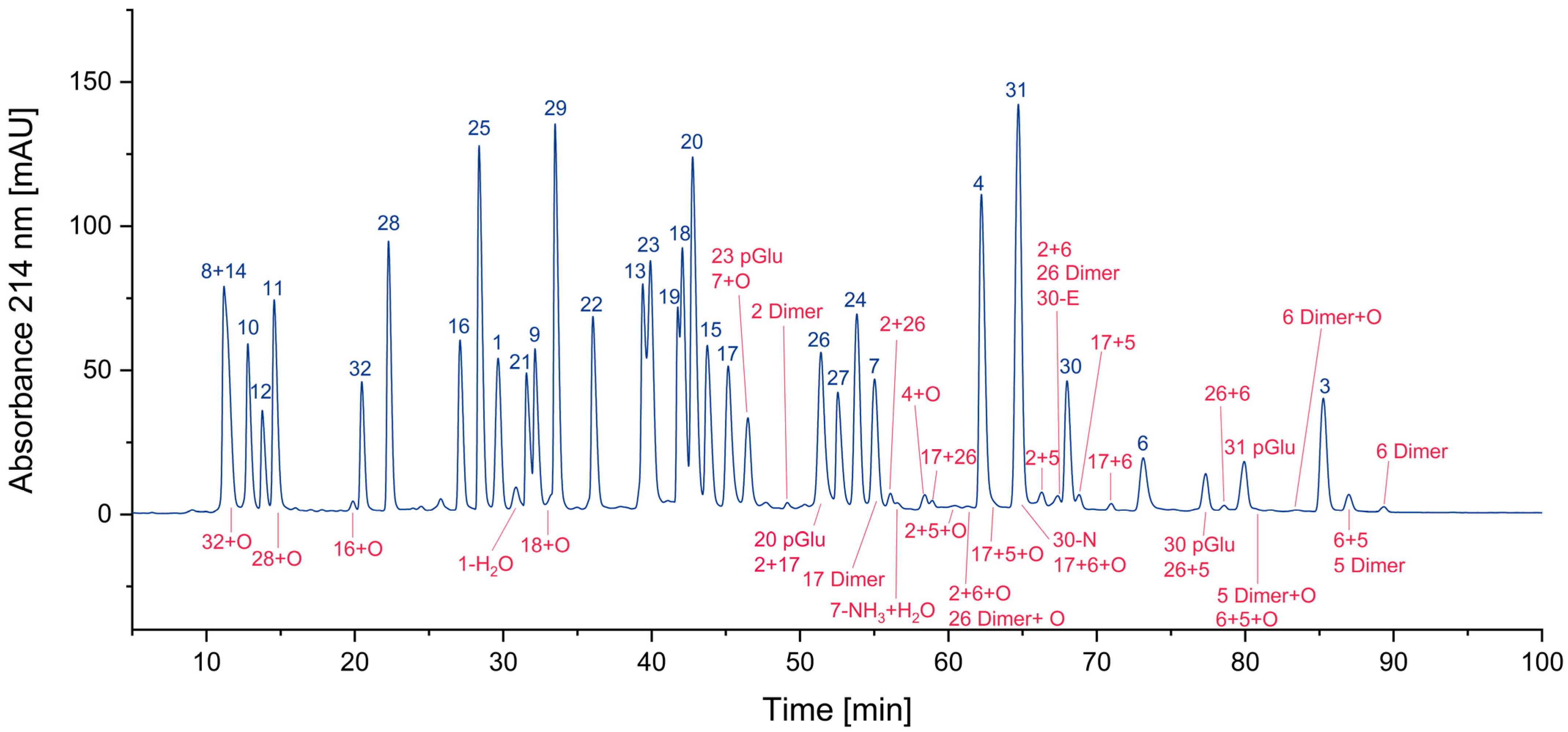
In Figure 4, all cyclized peptides, including those formed through a cyclic intermediate (deamidation), are indicated in red. Most of them are pyroglutamate-terminated peptides, only one is an aspartimide by-product, while another results from the deamidation of asparagine. They are separately listed in Table 2.
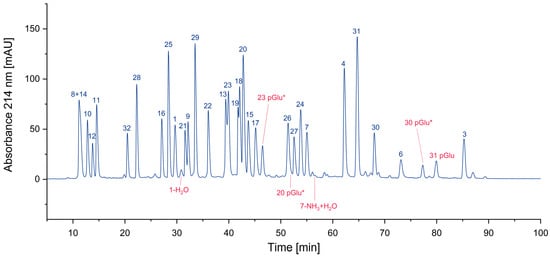
Figure 4.
Search for cyclized amino acid residues and their derivatives. (* coelution with other peptides).
Table 2.
Potential intra-residue cyclization of amino acids and subsequent reactions (-, not found).
3.1.3. Oxidized Peptides
In an oxygen-containing atmosphere, oxidation is a frequent reaction. Particularly, cysteine and methionine are sensitive to oxidative processes. A rarely examined reaction is the unwanted formation of cysteine dimers, since most protocols for peptide analysis include an alkylation step to avoid this side reaction. In proteins, cysteines are usually “protected” by the formation of defined disulfide bridges, which can be either analyzed in their native form or opened by reduction with tris(2-carboxyethyl)phosphine (TCEP) or dithiothreitol (DTT) and subsequent alkylation. In this way, the formation of cysteine dimers can be prevented. However, it should be borne in mind that each reduction step destroys the information of pre-existing disulfide bonds. In peptide pools, however, such a protection by alkylation or disulfides is not present, and hence, directly after the cleavage of the peptides from the solid phase and deprotection of the cysteine side chain, the formation of homodimers (particularly before mixing of the peptides) and heterodimers (likely after the combination of the peptides to the pool) can occur in significant amounts. For the five peptides containing cysteines, 23 dimers (including methionine-oxidized products) could be tentatively identified.
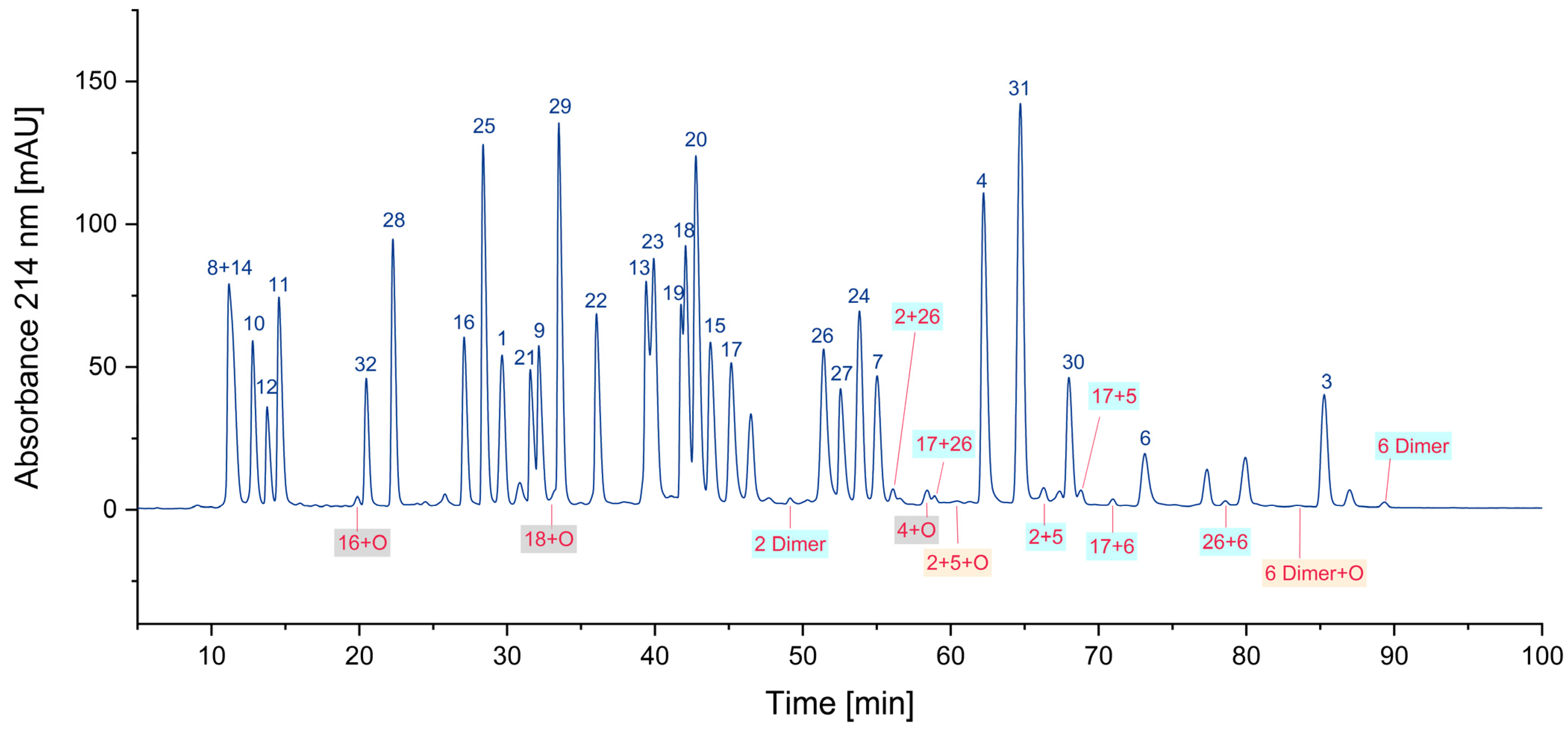
Oxidized peptides with a methionine sulfoxide can be found frequently. In our peptide pool sample, all methionine-containing peptides were also found in their oxidized form. For peptide 16, about 8% of the compound was found as the sulfoxide. Further oxidation was not significant for 77 h in the autosampler. In Figure 5, oxidized peptides are labeled: cysteine dimers in blue, methionine sulfoxides in grey, and dimers with an additional oxygen in orange. Only a selected number of oxidized peptides are highlighted in the figure.
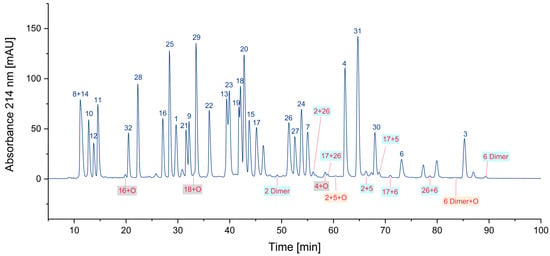
Figure 5.
Some oxidized peptides. Note: Only a few examples of the identified peptides are labeled in the chromatogram. Grey background: contains methionine sulfoxide; blue background: contains cysteine dimers (cystine); and orange background: shows peptides with both types of oxidations.
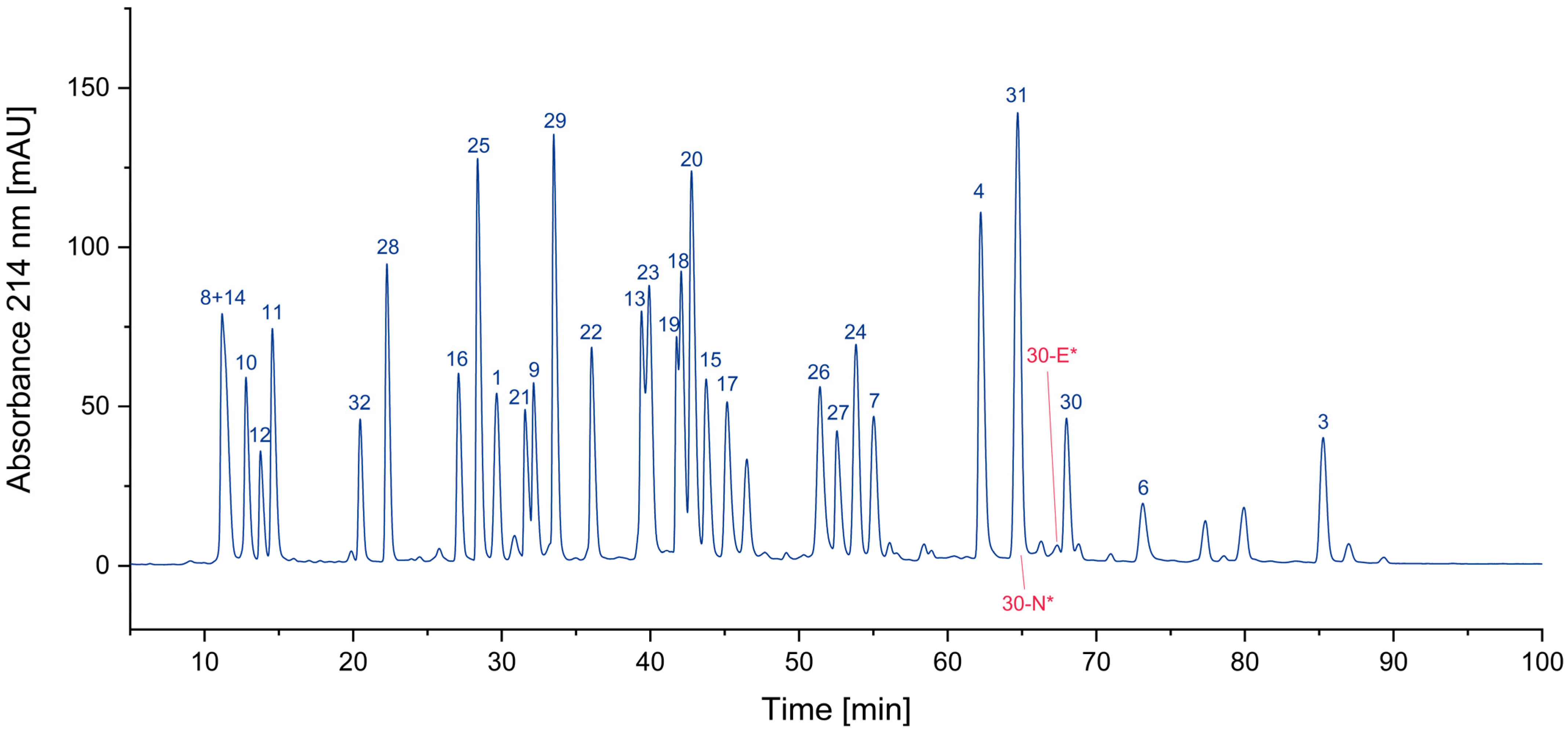
3.1.4. Overview
In Figure 6 all identified impurities are labeled in red. It is obvious that the 37 additional peptides, in addition to the 32 expected peptides, complicate the analysis of the peptide pool considerably by increasing the number of overlapping peaks. In addition, the presence of unwanted byproducts might lead to unexpected results when the peptide pool is used in a sensitive experimental setting. Hence, it might be concluded that the chemical composition of peptide pools should be optimized to reduce the number of additional products. Another option might be the improved purification of the synthesized peptides and the subsequent compliance of strict storage conditions, such as freezing at –80 °C or liquid nitrogen. Which approach may be considered more feasible depends on the user. The most trivial option could be to accept the impurities as “unavoidable” and to hope that the experiment will not be affected by the small percentage of additional components.
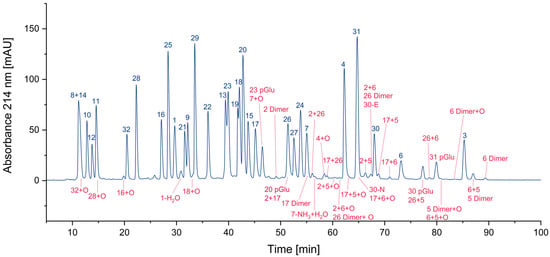
Figure 6.
Thirty-seven impurities were identified in the CEF peptide pool.
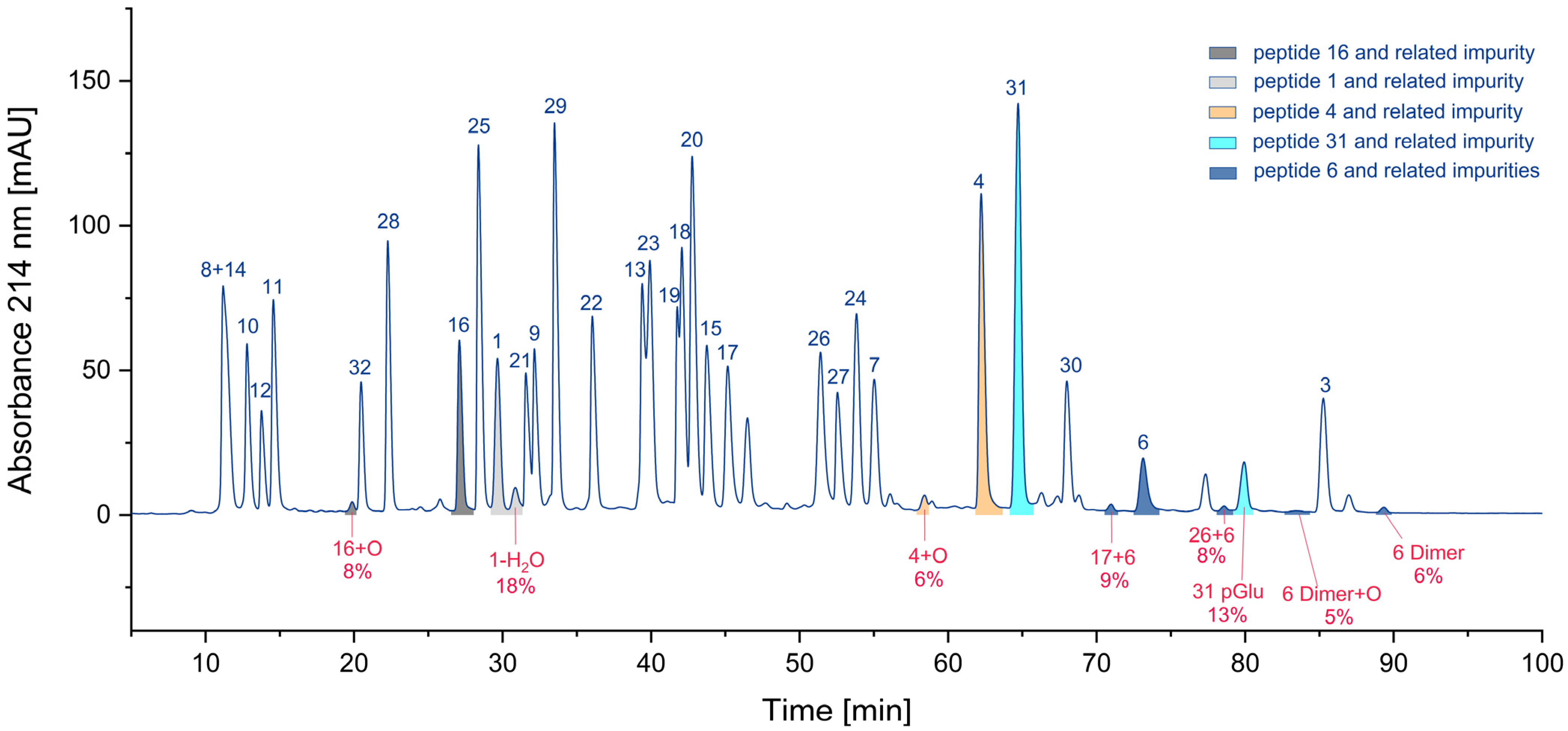
Considering the number of impurities of a specific class (Figure S2, Supplementary Materials), hetero- and homodimers of cysteine-containing peptides (23) are by far the most frequent. Also present are peptides with methionine sulfoxide (6) and N-terminal pyroglutamates (4). Deletion peptides (2), aspartimides (1), and deamidation products (1) are quite rare. Nevertheless, the number of impurities may be not the most relevant parameter. Assuming that the parent peptide is the only active one and the byproduct is inactive, the residual percentage of the parent peptide would be interesting. Hence, we calculated these percentages based on the UV area for some peptides. In Figure 7, the areas of various peptides and their corresponding impurities are presented in the chromatogram. The pairs of parent peptides and their corresponding impurities are shown in the same color.
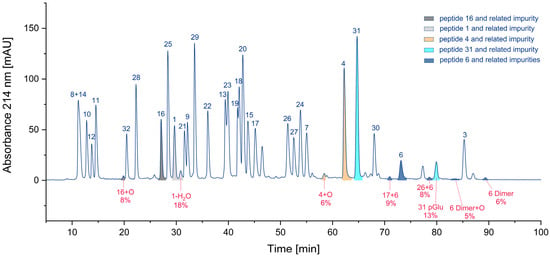
Figure 7.
Relative proportion of impurities and the corresponding parent peptides (colors indicate individual peptides and their related impurities).
For cysteine dimers, this calculation is relatively inaccurate, due to the formation of numerous products, some of which may be hidden in overlapping peaks. Consequently, the residual percentage of the parent peptide may be overestimated, if no reference measurement is available. In the worst case, only 71% of the parent peptide remained, while other calculations indicated that 82%, 87%, 92%, and 94% of the parent peptide were still present (Table 3). For peptide pools, such as the CEF peptide pool, where certain peptides are present in significant excess [9], a reduced concentration of the parent peptide may be irrelevant.
Table 3.
Relative proportion of impurities from the respective parent peptide. Note: For peptides that form dimers, the purity may be overestimated, as UV peak areas may be not accessible (n.a.) for some species (see peptide 6).
3.2. Alkylation of Cysteine Residues
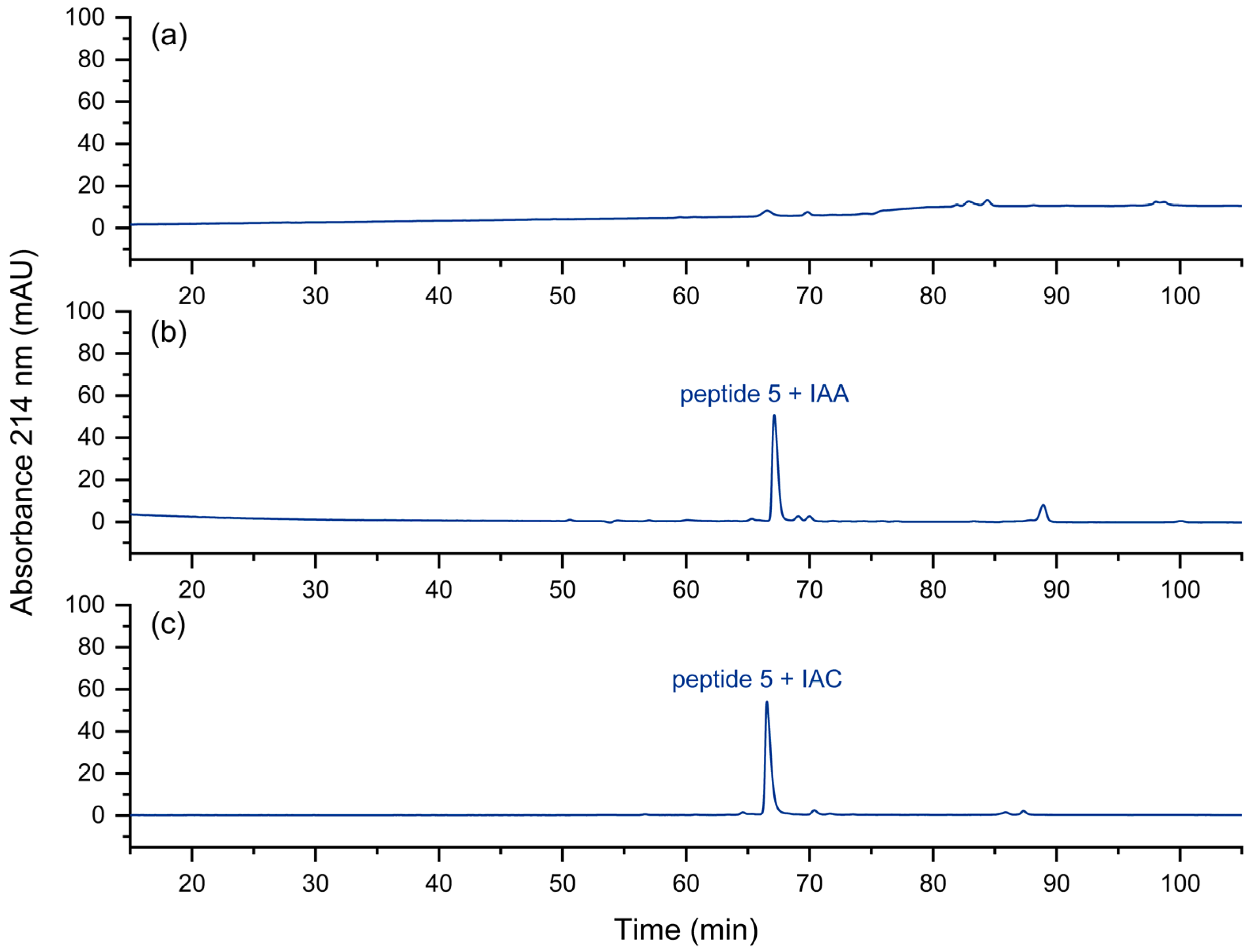
In the fast approach to the quality control of peptide pools [8], the reduction and the alkylation steps are omitted for the sake of simplicity and speed. However, it could be shown that peptides with free cysteine residues may display some unwanted effects during chromatography [34]. The corresponding peptide peak may be barely visible under certain separation conditions (Figure 8a). Peptides with non-terminal cysteines also showed a tendency towards low recovery. These issues could be removed by alkylation with iodoacetamide (IAA) or iodoacetic acid (IAC). In Figure 8b,c, this effect is shown. We assume strong interactions of the N-terminal thiol group with metal or other surfaces. The thioether formed after alkylation seems to be less prone to such interactions.
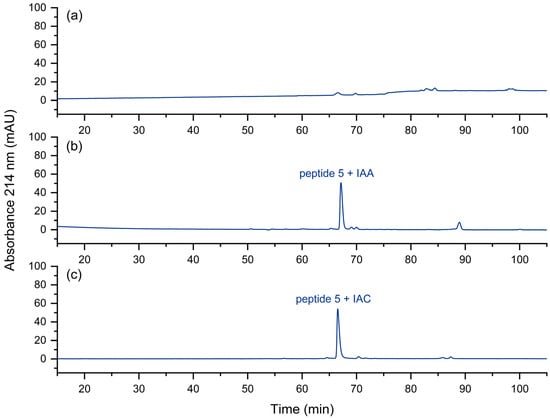
Figure 8.
UV chromatogram of (a) the cysteine-containing peptide 5, (b) iodoacetamide-alkylated peptide 5, (c) iodoacetic acid-alkylated peptide 5. No peak is obtained for peptide 5 with a free, N-terminal thiol group. Relative to the total peak areas (214 nm) of each chromatogram, with IAA a purity of 73% was obtained, in contrast to 84% with IAC.
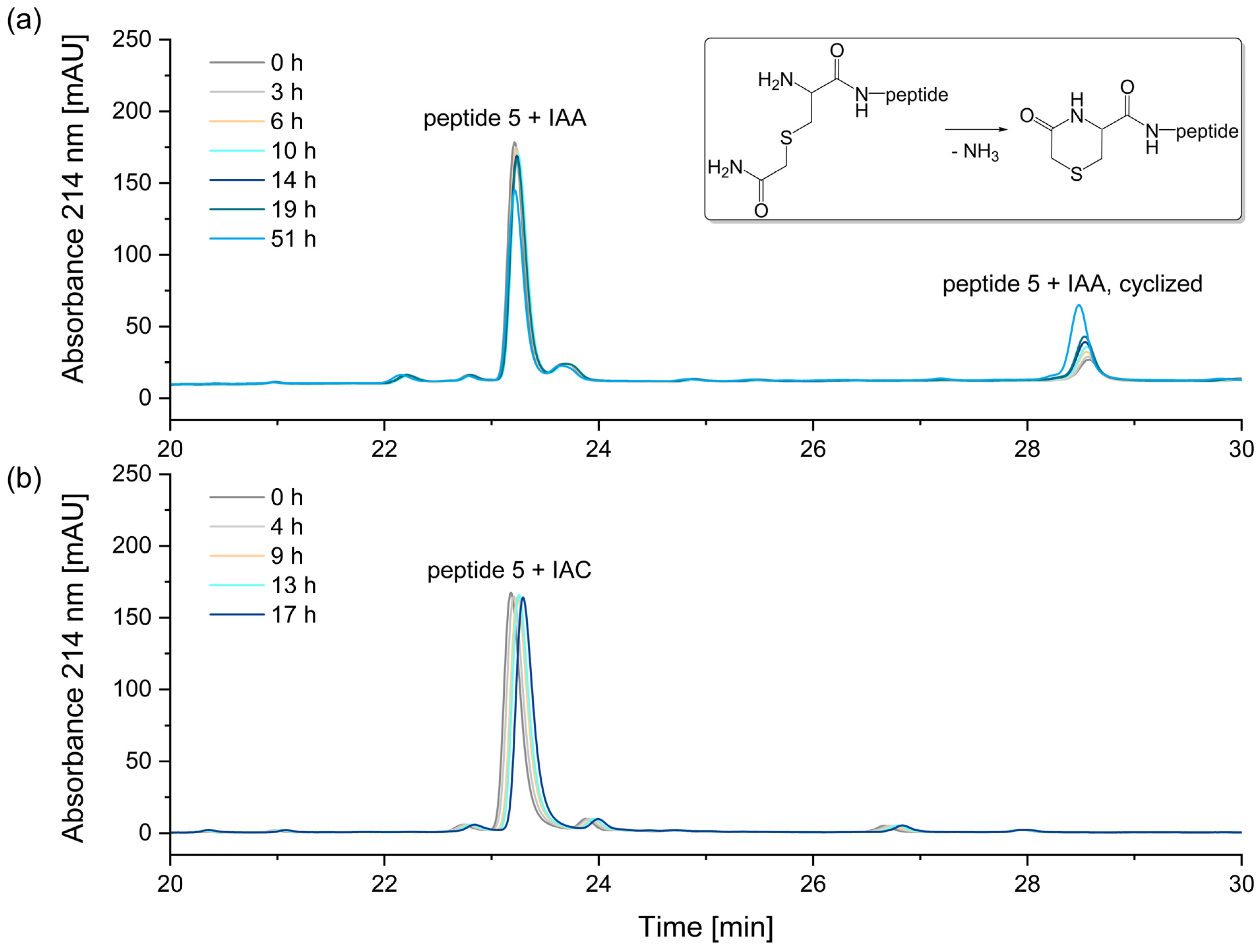
A comparison of the two alkylation reagents revealed that the alkylation reagent IAA, which is most used in proteomic studies [35], has a significant disadvantage compared to IAC: when alkylating an N-terminal cysteine, the resulting reaction product is unstable (Figure 9).
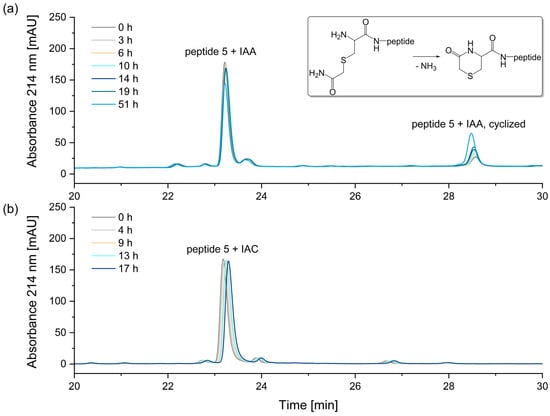
Figure 9.
Stability of alkylated peptide 5 containing an N-terminal cysteine. (a) Peptide 5 + iodoacetamide (IAA), (b) Peptide 5 + iodoacetic acid (IAC). The N-terminal cysteine of peptide 5 alkylated with iodoacetamide reacts to form N-terminal 5-oxohydro-1,4-thiazine-3-carboxylic acid, as indicated by a mass difference of −17 Da. After 51 h, the relative peak area of the peptide alkylated with iodoacetamide decreased by 18%. In contrast, no significant degradation was observed for peptide 5 alkylated with iodoacetic acid.
The instability of the IAA-alkylated peptide 5 is attributed to the cyclization of the alkylated cysteine at the N-terminus. In this process, the N-terminal carbamidomethylated cysteine reacts to form an N-terminal 5-oxohydro-1,4-thiazin-3-carboxylic acid, as indicated by a mass difference of −17 Da [36,37,38]. After 51 h in the autosampler, the relative peak area of the peptide alkylated with iodoacetamide decreased by 18%. In contrast, the alkylation of peptide 5 with iodoacetic acid (IAC) resulted in a stable peptide, with no significant degradation of the N-terminal carboxymethyl-cysteine observed. The corresponding peak areas are illustrated in Figure S3 in the Supplementary Materials. Therefore, in the alkylation of N-terminal cysteines, the reagent iodoacetic acid (IAC) is preferable.
4. Discussion
Research on peptide impurities in synthetic products is rarely carried out and therefore knowledge is limited, especially for complex products such as peptide pools. In this work, we used data obtained by high-resolution mass spectrometry to tentatively assign structural data to unknown peaks in a reversed-phase chromatogram. The results do not claim to be comprehensive, but are focused on impurities, which seem to be frequent and occurring in relevant concentrations. Perhaps the most interesting finding is the occurrence of numerous homo- or hetero-dimers resulting from unprotected cysteine residues. This result should not be ignored, as the formation of these impurities is to be expected and is difficult to avoid. Retrospectively, it is quite surprising that this issue is largely disregarded in most papers. This should be seen in the context of papers stressing the relevance of cysteine and cysteine dimers in MHC binding [39,40] or peptide pools. Furthermore, the loss of peptides with N-terminal cysteines was striking. This was observed in analytical methods without cysteine alkylation. Additionally, the formation of N-terminal pyroglutamyl residues was observed with relative high frequency. This aspect is also significant in the context of biological activity, as the positive charge of the terminus is lost and all processes that depend on a free N-terminus could be strongly influenced. For example, Beck et al. [2] demonstrated that CTL activity can be reduced by pyroglutamic peptides compared to their unmodified counterparts. A small proportion of certain deletion peptides was identified; however, their significance may be minimal due to their similar properties compared to the expected peptides and their low concentrations. Finally, only one instance of aspartimide formation and one instance of deamidation were detected. This is a positive outcome, as aspartimides typically leads to the formation of several isomers and other by-products that are difficult to distinguish.
5. Conclusions
First of all, awareness of impurities in peptide products should be raised. These products are used in various applications, and therefore it is difficult for the manufacturer to make a final judgment as to whether the product is suitable for the intended purpose. This means that a quick quality control of the product should be carried out by the user [8], regardless of whether the peptide pool has been freshly obtained from the manufacturer or the mixture has been stored in the laboratory for some time. Based on the protocol presented in this paper, the main impurities should be identified, and a brief risk analysis performed. Nevertheless, avoidance strategies are almost always preferable to reduce the number and concentration of undesired compounds in the peptide pool.
Starting with the occurrence of hetero- or homodimers of cysteine-containing peptides, for example, various means could be considered:
- Choice of different peptide sequences, avoiding any cysteines
- Replacement of cysteine by a more stable amino acid, such as alanine, valine [41], or methylcysteine
- Alkylation of the cysteines by iodoacetamide, iodoacetic acid or other known reagents. This seems to be particularly relevant in the case of analytical studies.
- Addition of reductants, such as DTT, TCEP, cysteine, glutathione, and others
- Strict avoidance of any oxidant, such as oxygen by use of protecting gases, such as helium, argon or nitrogen during synthesis, purification, storage, transportation, and use
- The use of dimethyl sulfoxide (DMSO), which is often recommended as a universal solvent for peptides and peptide pools [42,43,44], should be used with caution, as DMSO can act as an effective reagent for cysteine dimerization under certain conditions [45,46]. Similarly, methionine can be oxidized to methionine sulfoxide under acidic conditions with DMSO [47,48]. The occurrence of oxidized methionine could also be avoided by some of the previously mentioned approaches, particularly points 1, 2 and 5.
The formation of pyroglutamyl peptides can be mitigated through improved synthesis protocols. For example, the coupling reaction can be accelerated by employing highly efficient coupling reagents and/or utilizing a significant excess of the incoming amino acid. Additionally, preactivating the incoming amino acid to form an active ester may reduce acid-catalyzed pyroglutamate formation. Furthermore, using a (bulky) protecting group for glutamine or glutamic acid can be advantageous [49]. Formulating the compound as a hydrochloride salt is more effective in minimizing pyroglutamate formation compared to using acetate or trifluoroacetate salts [2]. N-Acetylation may also serve as a viable option to prevent pyroglutamate formation. Finally, the incorporation of glutamine or glutamic acid at the N-terminus of peptides could be avoided if possible. Similar approaches, such as the substitution or N-methylation of glycine, might be advisable in the (rare) case of an Asp-Gly sequence, which is highly prone to aspartimide formation and the generation of other by-products. Also, Fmoc-Asp(OtBu)-(Dmb)Gly-OH [28] or β-trialkyl-methyl ester protected aspartic acid [50] could be used. The FMOC cleavage reagent piperidine could be replaced with piperazine or 1-hydroxypiperidine [51]. The application of peptide backbone amide protecting groups has proven to be an effective strategy for mitigating aspartimide formation [52]. Furthermore, the use of innovative side chain protecting groups could provide a viable solution [53]. Deamidation can be minimized by controlling the pH and lowering the temperature to 4 °C or below [33]. Additionally, avoiding or substituting asparagine and glutamine in the peptide sequence could prevent deamidation.
Finally, the presence of deletion peptides could be prevented by improved synthesis protocols. These may include, for instance, extending the deprotection step, increasing the concentration of the deprotection reagent [13], and/or implementing double additions of activating reagents [16]. Additionally, more effective purification methods could also be employed. It should be kept in mind that smaller amounts of a peptide can be purified on analytical (U)HPLC columns, which have a much better separation performance than preparative columns. In addition, by injecting smaller amounts of sample and sharply cutting the peptide peak in the fraction collector, the purity of the final product can generally be improved, of course at the expense of lower yield and higher costs.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/separations12020036/s1, Figure S1: Stability of N-terminal glutamine vs. glutamic acid during autosampler storage. Spontaneous cyclization of the N-terminal amino acid is faster for (a) peptide 23, which contains glutamine, than of (b) peptide 20, which contains glutamic acid., both in 5% ACN + 0.05% TFA at 4 °C. The linear regression analysis yielded an R2 value of 0.999; Figure S2: Among the 37 impurities preliminarily assigned in this work, cysteine homodimers and heterodimers were the most common; Figure S3: Lack of stability of IAA-alkylated peptide 5 compared to that of IAC-alkylated peptide 5 during autosampler storage. Spontaneous cyclization to the N-terminal 5-oxohydro-1,4-thiazine-3-carboxylic acid occurs in (a) IAA-alkylated peptide 5; however, no significant degradation was observed in (b) IAC-alkylated peptide 5. The linear regression analysis conducted for IAA-alkylated peptide 5 over a time range of 0 to 51 h yielded an R2 value of 0.992.
Author Contributions
Conceptualization, M.G.W.; methodology, M.G.W.; validation, G.B.-B.; formal analysis, G.B.-B.; investigation, G.B.-B.; resources, M.G.W.; data curation, G.B.-B.; writing—original draft preparation, M.G.W.; writing—review and editing, G.B.-B. and M.G.W.; visualization, G.B.-B.; supervision, M.G.W.; project administration, M.G.W.; funding acquisition, M.G.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the funding line of the Bundesanstalt für Materialforschung und -prüfung (BAM) MI-Ideen Typ 3 under the grant number Ideen_2016_15.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tobery, T.W.; Wang, S.; Wang, X.M.; Neeper, M.P.; Jansen, K.U.; McClements, W.L.; Caulfield, M.J. A simple and efficient method for the monitoring of antigen-specific T cell responses using peptide pool arrays in a modified ELISpot assay. J. Immunol. Methods 2001, 254, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Bussat, M.C.; Klinguer-Hamour, C.; Goetsch, L.; Aubry, J.P.; Champion, T.; Julien, E.; Haeuw, J.F.; Bonnefoy, J.Y.; Corvaia, N. Stability and CTL activity of N-terminal glutamic acid containing peptides. J. Pept. Res. 2001, 57, 528–538. [Google Scholar] [CrossRef]
- de Beukelaar, J.W.; Gratama, J.W.; Smitt, P.A.S.; Verjans, G.M.; Kraan, J.; Luider, T.M.; Burgers, P.C. The impact of impurities in synthetic peptides on the outcome of T-cell stimulation assays. Rapid Commun. Mass Spectrom. 2007, 21, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Verbeken, M.; Wynendaele, E.; Lefebvre, R.A.; Goossens, E.; De Spiegeleer, B. The influence of peptide impurity profiles on functional tissue-organ bath response: The 11-mer peptide INSL6[151-161] case. Anal. Biochem. 2012, 421, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Currier, J.R.; Galley, L.M.; Wenschuh, H.; Morafo, V.; Ratto-Kim, S.; Gray, C.M.; Maboko, L.; Hoelscher, M.; Marovich, M.A.; Cox, J.H. Peptide impurities in commercial synthetic peptides and their implications for vaccine trial assessment. Clin. Vaccine Immunol. 2008, 15, 267–276. [Google Scholar] [CrossRef]
- Finder, V.H.; Vodopivec, I.; Nitsch, R.M.; Glockshuber, R. The Recombinant Amyloid-β Peptide Aβ1-42 Aggregates Faster and Is More Neurotoxic than Synthetic Aβ1-42. J. Mol. Biol. 2010, 396, 9–18. [Google Scholar] [CrossRef]
- Weller, M.G. The Protocol Gap. Methods Protoc. 2021, 4, 12. [Google Scholar] [CrossRef]
- Bosc-Bierne, G.; Ewald, S.; Kreuzer, O.J.; Weller, M.G. Efficient Quality Control of Peptide Pools by UHPLC and Simultaneous UV and HRMS Detection. Separations 2024, 11, 156. [Google Scholar] [CrossRef]
- Zhang, W.J.; Moldovan, I.; Targoni, O.S.; Subbramanian, R.A.; Lehmann, P.V. How much of Virus-Specific CD8 T Cell Reactivity is Detected with a Peptide Pool when Compared to Individual Peptides? Viruses 2012, 4, 2636–2649. [Google Scholar] [CrossRef]
- Castro, A.; Holenya, P.; Eckey, M.; Schulz, M.; Wenschuh, H.; Reimer, U.; Tech, T.; Chan, K.; Janani, R.; Broaten, B.; et al. A novel peptide pool with broad infectious antigen and MHC coverage for use as a positive stimulation control or as a means to elicit general T-cell responsiveness. J. Immunol. 2018, 200, 120.15. [Google Scholar] [CrossRef]
- Patiny, L.; Borel, A. ChemCalc: A Building Block for Tomorrow’s Chemical Infrastructure. J. Chem. Inf. Model. 2013, 53, 1223–1228. [Google Scholar] [CrossRef] [PubMed]
- Currier, J.R.; Kuta, E.G.; Turk, E.; Earhart, L.B.; Loomis-Price, L.; Janetzki, S.; Ferrari, G.; Birx, D.L.; Cox, J.H. A panel of MHC class I restricted viral peptides for use as a quality control for vaccine trial ELISPOT assays. J. Immunol. Methods 2002, 260, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa-Kumagaye, K.; Nishiuchi, Y.; Nishio, H.; Kimura, T. Amino acid deletion products resulting from incomplete deprotection of the Boc group from benzyloxy-methylhistidine residues during solid-phase peptide synthesis. J. Pept. Sci. 2005, 11, 512–515. [Google Scholar] [CrossRef]
- D’Hondt, M.; Bracke, N.; Taevernier, L.; Gevaert, B.; Verbeke, F.; Wynendaele, E.; De Spiegeleer, B. Related impurities in peptide medicines. J. Pharm. Biomed. 2014, 101, 2–30. [Google Scholar] [CrossRef]
- Jad, Y.E.; Govender, T.; Kruger, H.G.; El-Faham, A.; de la Torre, B.G.; Albericio, F. Green Solid-Phase Peptide Synthesis (GSPPS) 3. Green Solvents for Fmoc Removal in Peptide Chemistry. Org. Process Res. Dev. 2017, 21, 365–369. [Google Scholar] [CrossRef]
- Mthethwa, N.; Nandhini, K.P.; Kumar, A.; Sharma, A.; de la Torre, B.G.; Albericio, F. Toward sustainable solid-phase peptide synthesis strategy—Fmoc removal. Green Chem. Lett. Rev. 2024, 17, 2325993. [Google Scholar] [CrossRef]
- Dimarchi, R.D.; Tam, J.P.; Kent, S.B.H.; Merrifield, R.B. Weak Acid-Catalyzed Pyrrolidone Carboxylic-Acid Formation from Glutamine during Solid-Phase Peptide-Synthesis—Minimization by Rapid Coupling. Int. J. Pept. Prot. Res. 1982, 19, 88–93. [Google Scholar] [CrossRef]
- Viau, M.; Létourneau, M.; Sirois-Deslongchamps, A.; Boulanger, Y.; Fournier, A. Study of solid-phase synthesis and purification strategies for the preparation of polyglutamine peptides. Biopolymers 2007, 88, 754–763. [Google Scholar] [CrossRef]
- Khandke, K.M.; Fairwell, T.; Chait, B.T.; Manjula, B.N. Influence of Ions on Cyclization of the Amino Terminal Glutamine Residues of Tryptic Peptides of Streptococcal Pepm49 Protein—Resolution of Cyclized Peptides by Hplc and Characterization by Mass-Spectrometry. Int. J. Pept. Prot. Res. 1989, 34, 118–123. [Google Scholar] [CrossRef]
- Fernandez Garcia, A.; Butz, P.; Trierweiler, B.; Zöller, H.; Stärke, J.; Pfaff, E.; Tauscher, B. Pressure/temperature combined treatments of precursors yield hormone-like peptides with pyroglutamate at the N terminus. J. Agr. Food Chem. 2003, 51, 8093–8097. [Google Scholar] [CrossRef]
- Gazme, B.; Boachie, R.T.; Tsopmo, A.; Udenigwe, C.C. Occurrence, properties and biological significance of pyroglutamyl peptides derived from different food sources. Food Sci. Hum. Well. 2019, 8, 268–274. [Google Scholar] [CrossRef]
- Behrendt, R.; White, P.; Offer, J. Advances in Fmoc solid-phase peptide synthesis. J. Pept. Sci. 2016, 22, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Bodanszky, M.; Tolle, J.C.; Deshmane, S.S.; Bodanszky, A. Side reactions in peptide synthesis. VI. A reexamination of the benzyl group in the protection of the side chains of tyrosine and aspartic acid. Int. J. Pept. Prot. Res. 1978, 12, 57–68. [Google Scholar] [CrossRef]
- Yang, Y.; Sweeney, W.V.; Schneider, K.; Thornqvist, S.; Chait, B.T.; Tam, J.P. Aspartimide Formation in Base-Driven 9-Fluorenylmethoxycarbonyl Chemistry. Tetrahedron Lett. 1994, 35, 9689–9692. [Google Scholar] [CrossRef]
- Nicolas, E.; Pedroso, E.; Giralt, E. Formation of Aspartimide Peptides in Asp-Gly Sequences. Tetrahedron Lett. 1989, 30, 497–500. [Google Scholar] [CrossRef]
- Tam, J.P.; Wong, T.W.; Riemen, M.W.; Tjoeng, F.S.; Merrifield, R.B. Cyclohexyl Ester as a New Protecting Group for Aspartyl Peptides to Minimize Aspartimide Formation in Acidic and Basic Treatments. Tetrahedron Lett. 1979, 20, 4033–4036. [Google Scholar] [CrossRef]
- Lauer, J.L.; Fields, C.G.; Fields, G.B. Sequence dependence of aspartimide formation during 9-fluorenylmethoxycarbonyl solid-phase peptide synthesis. Lett. Pept. Sci. 1995, 1, 197–205. [Google Scholar] [CrossRef]
- Mergler, M.; Dick, F.; Sax, B.; Weiler, P.; Vorherr, T. The aspartimide problem in Fmoc-based SPPS. Part I. J. Pept. Sci. 2003, 9, 36–46. [Google Scholar] [CrossRef]
- Mergler, M.; Dick, F.; Sax, B.; Stähelin, C.; Vorherr, T. The aspartimide problem in Fmoc-based SPPS.: Part II. J. Pept. Sci. 2003, 9, 518–526. [Google Scholar] [CrossRef]
- Mergler, M.; Dick, F. The aspartimide problem in Fmoc-based SPPS. Part III. J. Pept. Sci. 2005, 11, 650–657. [Google Scholar] [CrossRef]
- Liu, S.; Moulton, K.R.; Auclair, J.R.; Zhou, Z.S. Mildly acidic conditions eliminate deamidation artifact during proteolysis: Digestion with endoprotease Glu-C at pH 4.5. Amino Acids 2016, 48, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Borchardt, R.T. Chemical Pathways of Peptide Degradation. III. Effect of Primary Sequence on the Pathways of Deamidation of Asparaginyl Residues in Hexapeptides. Pharm. Res. 1990, 7, 787–793. [Google Scholar] [CrossRef] [PubMed]
- McKerrow, J.H.; Robinson, A.B. Deamidation of Asparaginyl Residues as a Hazard in Experimental Protein and Peptide Procedures. Anal. Biochem. 1971, 42, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Kreuzer, O.J.; Weller, M.G.; Bosc-Bierne, G. Stabilization of N-terminal cysteines in HPLC-HRMS quality control of peptide pools. J. Immunol. 2023, 210, 159.09. [Google Scholar] [CrossRef]
- Müller, T.; Winter, D. Systematic Evaluation of Protein Reduction and Alkylation Reveals Massive Unspecific Side Effects by Iodine-containing Reagents. Mol. Cell Proteom. 2017, 16, 1173–1187. [Google Scholar] [CrossRef]
- Geoghegan, K.F.; Hoth, L.R.; Tan, D.H.; Borzillerl, K.A.; Withka, J.M.; Boyd, J.G. Cyclization of N-terminal S-carbamoylmethylcysteine causing loss of 17 Da from peptides and extra peaks in peptide maps. J. Proteome Res. 2002, 1, 181–187. [Google Scholar] [CrossRef]
- Krokhin, O.V.; Ens, W.; Standing, K.G. Characterizing degradation products of peptides containing N-terminal Cys residues by (off-line high-performance liquid chromatography)/matrix-assisted laser desorption/ionization quadrupole time-of-flight measurements. Rapid Commun. Mass Spectrom. 2003, 17, 2528–2534. [Google Scholar] [CrossRef]
- Reimer, J.; Shamshurin, D.; Harder, M.; Yamchuk, A.; Spicer, V.; Krokhin, O.V. Effect of cyclization of N-terminal glutamine and carbamidomethyl-cysteine (residues) on the chromatographic behavior of peptides in reversed-phase chromatography. J. Chromatogr. A 2011, 1218, 5101–5107. [Google Scholar] [CrossRef]
- Sachs, A.; Moore, E.; Kosaloglu-Yalcin, Z.; Peters, B.; Sidney, J.; Rosenberg, S.A.; Robbins, P.F.; Sette, A. Impact of Cysteine Residues on MHC Binding Predictions and Recognition by Tumor-Reactive T Cells. J. Immunol. 2020, 205, 539–549. [Google Scholar] [CrossRef]
- Bruno, P.M.; Timms, R.T.; Abdelfattah, N.S.; Leng, Y.M.; Lelis, F.J.N.; Wesemann, D.R.; Yu, X.G.; Elledge, S.J. High-throughput, targeted MHC class I immunopeptidomics using a functional genetics screening platform. Nat. Biotechnol. 2023, 41, 980–992. [Google Scholar] [CrossRef]
- Karapetyan, A.R.; Chaipan, C.; Winkelbach, K.; Wimberger, S.; Jeong, J.S.; Joshi, B.; Stein, R.B.; Underwood, D.; Castle, J.C.; Van Dijk, M.A.; et al. TCR Fingerprinting and Off-Target Peptide Identification. Front. Immunol. 2019, 10, 2501. [Google Scholar] [CrossRef] [PubMed]
- Suneetha, P.V.; Schlaphoff, V.; Wang, C.; Stegmann, K.A.; Fytili, P.; Sarin, S.K.; Manns, M.P.; Cornberg, M.; Wedemeyer, H. Effect of peptide pools on effector functions of antigen-specific CD8+T cells. J. Immunol. Methods 2009, 342, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Ay, B.; Streitz, M.; Boisguerin, P.; Schlosser, A.; Mahrenholz, C.C.; Schuck, S.D.; Kern, F.; Volkmer, R. Sorting and pooling strategy: A novel tool to map a virus proteome for CD8 T-cell epitopes. Biopolymers 2007, 88, 64–75. [Google Scholar] [CrossRef]
- Al-kolla, R.; Grifoni, A.; Crotty, S.; Sette, A.; Gianella, S.; Dan, J. Design and validation of HIV peptide pools for detection of HIV-specific CD4 and CD8 T cells. PLoS ONE 2022, 17, e0268370. [Google Scholar] [CrossRef]
- Tam, J.P.; Wu, C.R.; Liu, W.; Zhang, J.W. Disulfide Bond Formation in Peptides by Dimethyl-Sulfoxide—Scope and Applications. J. Am. Chem. Soc. 1991, 113, 6657–6662. [Google Scholar] [CrossRef]
- Otaka, A.; Koide, T.; Shide, A.; Fujii, N. Application of Dimethylsulphoxide (DMSO)/Trifluoroacetic Acid (TFA) Oxidation to the Synthesis of Cystine-Containing Peptide. Tetrahedron Lett. 1991, 32, 1223–1226. [Google Scholar] [CrossRef]
- Shechter, Y. Selective Oxidation and Reduction of Methionine Residues in Peptides and Proteins by Oxygen-Exchange between Sulfoxide and Sulfide. J. Biol. Chem. 1986, 261, 66–70. [Google Scholar] [CrossRef]
- Lipton, S.H.; Bodwell, C.E. Specific Oxidation of Methionine to Methionine Sulfoxide by Dimethyl-Sulfoxide. J. Agr. Food Chem. 1976, 24, 26–31. [Google Scholar] [CrossRef]
- Yang, Y. Side Reactions in Peptide Synthesis; Academic Press: Cambridge, MA, USA, 2016; p. 146. [Google Scholar]
- Behrendt, R.; Huber, S.; White, P. Preventing aspartimide formation in Fmoc SPPS of Asp-Gly containing peptides practical aspects of new trialkylcarbinol based protecting groups. J. Pept. Sci. 2016, 22, 92–97. [Google Scholar] [CrossRef]
- Wade, J.D.; Mathieu, M.N.; Macris, M.; Tregear, G.W. Base-induced side reactions in Fmoc-solid phase peptide synthesis: Minimization by use of piperazine as N-deprotection reagent. Lett. Pept. Sci. 2000, 7, 107–112. [Google Scholar] [CrossRef]
- Quibell, M.; Owen, D.; Packman, L.C.; Johnson, T. Suppression of Piperidine-Mediated Side Product Formation for Asp(Obu(T))-Containing Peptides by the Use of N-(2-Hydroxy-4-methoxybenzyl) (Hmb) Backbone Amide Protection. J. Chem. Soc. Chem. Comm. 1994, 7, 2343–2344. [Google Scholar] [CrossRef]
- Neumann, K.; Farnung, J.; Baldauf, S.; Bode, J.W. Prevention of aspartimide formation during peptide synthesis using cyanosulfurylides as carboxylic acid-protecting groups. Nat. Commun. 2020, 11, 982. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).