Abstract
To determine the paclitaxel-related substances in a paclitaxel injection (albumin-bound), it is necessary to remove human blood albumin from the sample via solid-phase extraction and subsequently perform high-performance liquid chromatography analysis. The pre-processing operation is complicated, the duration of analysis is long, and much human power and material resources are needed in the sample testing process. Thus, the purpose of this study was to develop a quick two-dimensional liquid chromatography method that requires less consumables and possesses high sensitivity. Samples were directly injected into one-dimensional solid-phase extraction columns to remove human blood albumin. Paclitaxel and its related substances in the solid-phase extraction columns were then eluted into two-dimensional analytical columns through valve switching technology. The sample pretreatment step can be omitted in this method, which allows for the analysis of paclitaxel and its related substances to be completed more quickly, within only 15 min, under the developed two-dimensional elution conditions. The established method had good linearity (R2 = 0.9994), limit of detection (0.25 μg·mL−1), limit of quantification (0.5 μg·mL−1), precision (RSD = 0.5%), and accuracy (102.53–107.97%).
1. Introduction
Paclitaxel is a tricyclic diterpenoid and a natural secondary metabolite isolated and purified from the bark of taxus chinensis. It is considered an important microtubule stabilizer in chemotherapy. Paclitaxel can induce and promote tubulin polymerization and microtubule assembly, in addition to preventing depolymerization, blocking the cycle process of cancer cells, and inhibiting cancer cell mitosis, effectively preventing cancer cell proliferation and playing an anticancer role [1,2,3,4,5].
The application of paclitaxel to injections (albumin-bound) can not only retain its lethality to tumor cells, but also avoid the clinically severe allergic side reactions caused by the large amount of surfactant contained in traditional paclitaxel injections: polyoxyethylene hydrogenated castor oil. It can increase the concentration of paclitaxel and shorten the injection time, and induce a stronger anticancer effect. In recent years, various methods for the determination of paclitaxel in different matrices have been reported in the literature, for example, immunoassays, micellar electrokinetic chromatography, tubulin-based biochemical assays, and chromatography-based assays. Among them, high-performance liquid chromatography (HPLC) coupled with a UV or MS detector is widely used because of its high sensitivity and good separation capability [6]. However, when HPLC is used to investigate the related substances in paclitaxel for injection (albumin-bound), artificial solid-phase extraction is usually conducted to remove human blood albumin from the sample to reduce interference with the paclitaxel detection signal and prevent albumin from entering the column, thereby resulting in an increase in column pressure. This processing method is not only time-consuming, but also expensive with regard to human power and material resources, and is unable to meet the needs of large quantities of samples. Therefore, an online deproteinization method is urgently needed for the determination of paclitaxel-related substances in a human paclitaxel injection (albumin-bound).
In recent years, the application of two-dimensional high-performance liquid chromatography (2D-HPLC) has become increasingly extensive with its development regarding the theory, instrument, and methodology aspects [7,8,9,10,11,12,13]. Two-dimensional systems with different selectivities were constructed via two-dimensional liquid chromatography. This method combines two different methods to resolve complex mixtures on a two-dimensional plane, which enables the rapid, efficient, and comprehensive analysis of compounds [14]. Human albumin and other interfering matrices were removed from samples using a one-dimensional column and a two-dimensional column through the interface mode of a six-way valve, introducing the separated fractions of the one-dimensional column into the two-dimensional column for separation [15,16,17,18]. Solid-phase extraction (SPE) columns are widely used in 2D-HPLC technology to remove interfering substrates from samples. The target compound in the sample is separated from the interference matrix via a solid adsorbent when the sample solution passes through the SPE column, thus successfully separating and enriching the target compound. The use of SPE columns can achieve online automatic pretreatment and prevent manual errors, unlike when using the offline manual solid-phase extraction method [19].
Therefore, a new method for the online removal of human albumin from a sample and the determination of the related substances in paclitaxel preparations for injection (albumin bound) was developed via the 2D-HPLC technique combined with an SPE column. In contrast to the traditional sample pretreatment method, the proposed technique can directly analyze the sample, preserve the sample pretreatment time, and provide new methods and ideas for the quality control of paclitaxel for injection (albumin-bound).
2. Materials and Methods
2.1. Apparatus and Reagents
The method of 2D-HPLC-UV was used to determine the paclitaxel-related substances in paclitaxel preparations. The experiment was carried out on a double-ternary-pump high-performance liquid chromatograph (Thermo Ultimate 3000, Thermo Fisher Scientific, Waltham, MA, USA) with the Chromeleon 7 software.
Paclitaxel for injection (albumin-bound) (lot number: 211225) was obtained from YiBai Pharmaceutical Co., Ltd. (Guizhou, China). The reference substance paclitaxel (99.8%) was purchased from the National Institutes for Food and Drug Control (Beijing, China). Acetonitrile was obtained from Oceanpak (Gothenburg, Sweden) and pure water was prepared using a field scientific instrument.
2.2. Experimental Conditions
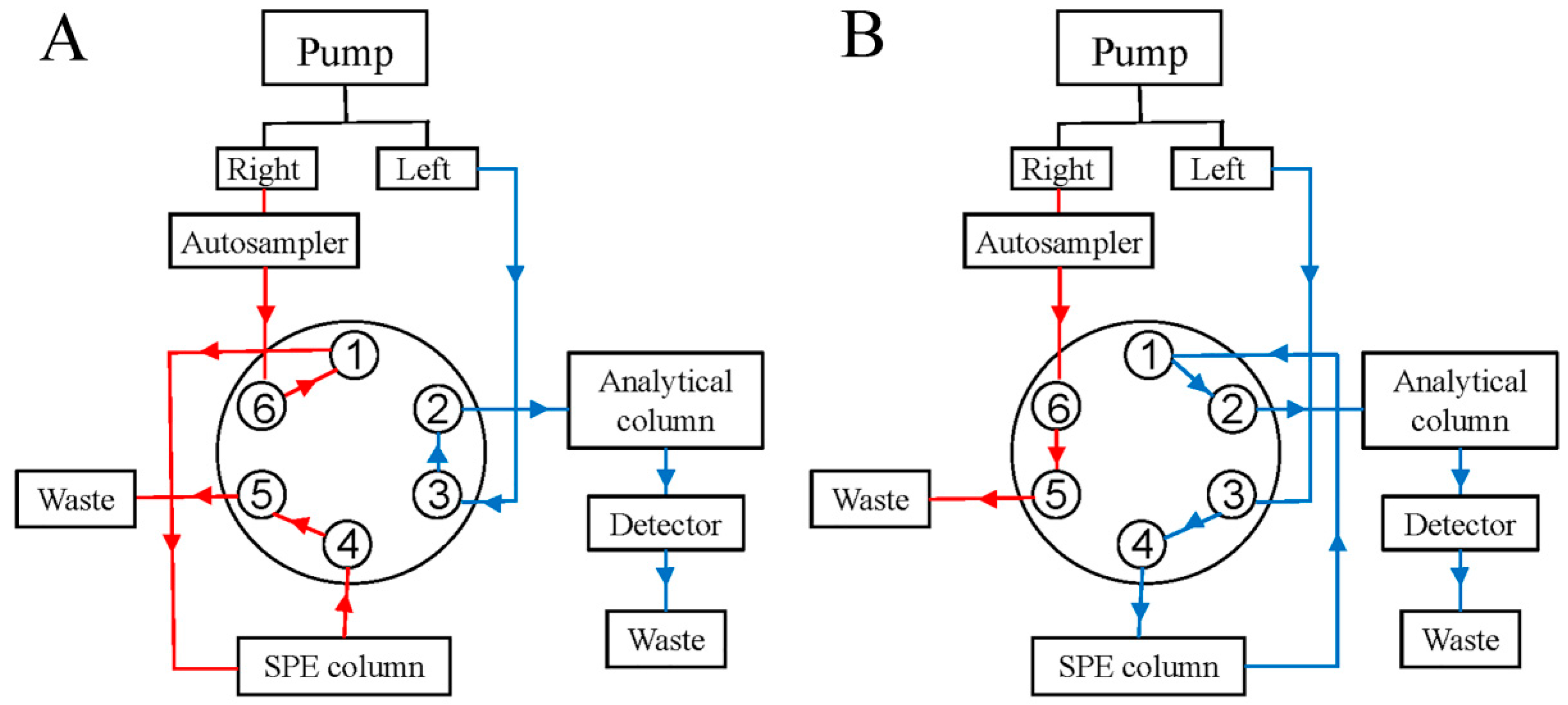
The method removed human albumin from the sample through a six-way valve. Figure 1 shows that the six-way valve was in the 1–6 connection state at 0–1.5 min, where the samples flew into the SPE column through the 1–6 connection. Within 1.5 min, human albumin was fully eluted out of the SPE column, while paclitaxel and its related substances were retained in the SPE column. At exactly 1.5 min, the six-way valve was converted to a 1–2 connection and the paclitaxel and its related substances retained in the SPE column were eluted with the mobile phase of the left pump into the C18 column (Hypersil GOLD, 100 × 2.1 mm, 1.9 μm) for separation analysis. By the third minute, all of the paclitaxel and related substances remaining on the SPE column were eluted to the analytical column connected to the left pump. The six-way valve returned to the 1–6 connection state, the mobile phase changed, and the SPE column was gradually returned to the initial state in preparation for the next injection. The mobile phase gradient and valve switching time are shown in Table 1; the detection wavelength was 227 nm, the column temperature was 40 °C, and the injection volume was 5 μL.
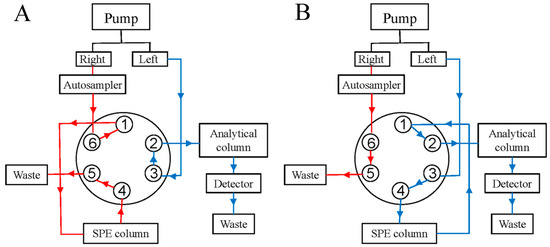
Figure 1.
Six-way valve conversion process: (A) is the six-way valve flow path when connecting 1–6, and (B) is the six-way valve flow path when connecting 1–2 (The total analysis time was 15 min. 0–1.5 min and 3–15 min, the six-way valve position is 1–6; 1.5–3 min, the six-way valve position is 1–2).

Table 1.
Pump flow phase gradient.
2.3. Preparation of the Sample and Reference Solutions
2.3.1. Sample Preparations
During the optimization and method validation processes, paclitaxel for injection (albumin-bound) was dissolved in 20 mL of normal saline to obtain the sample stock solution and was subsequently diluted tenfold to obtain a final paclitaxel concentration of approximately 0.5 mg·mL−1 as the sample.
The sample stock solution was exposed to a 1 M hydrochloric acid solution for 2 h, a 0.1 M sodium hydroxide solution for 0.5 h, 30% hydrogen peroxide for 2 h, a 60 °C water bath for 2 h, and a UV wavelength of 254 nm for 24 h in the experiment to investigate sample degradation. The prepared sample needed to be neutralized with regard to the acid–base conditions before being analyzed, and the corresponding blank solution was obtained via the same method.
2.3.2. Reference Solution Preparations
Paclitaxel was accurately weighed according to the reference standards to obtain 12.5 mg, which was dissolved in 10 mL of acetonitrile. The above solutions were subsequently diluted 100 times to produce a stock solution. The working standard solutions used for linearity were obtained by diluting the stock solution at concentrations of 1.25 μg·mL−1 (50%), 1.75 μg·mL−1 (70%), 2.5 μg·mL−1 (100%), 3.75 μg·mL−1 (150%), and 5 μg·mL−1 (200%), respectively. The solution with a concentration of 2.5 μg·mL−1 was used as a reference solution for the investigation of precision, repeatability, stability, accuracy, and durability. The stock solution was diluted 25 and 50 times to determine the limits of quantification and detection.
3. Results and Discussion
3.1. Establishment of the Analytical Methods
3.1.1. Confirmation of the Valve Switching Time
During online SPE sample processing, the accuracy of the sample determination depends on the complete elution of albumin from the SPE column for paclitaxel samples. There are two valve switching times in the sample determination process: one is the time, tA, when human albumin is completely drained from the SPE column, and the other is the time, tB, when paclitaxel and its related substances are completely drained from the SPE column, which is then re-balanced in preparation for the next sample injection. The valve switching time, tA, determines the degree to which human albumin is removed from the sample; residual human albumin will block the analysis column and make the pressure too high, resulting in consumption.
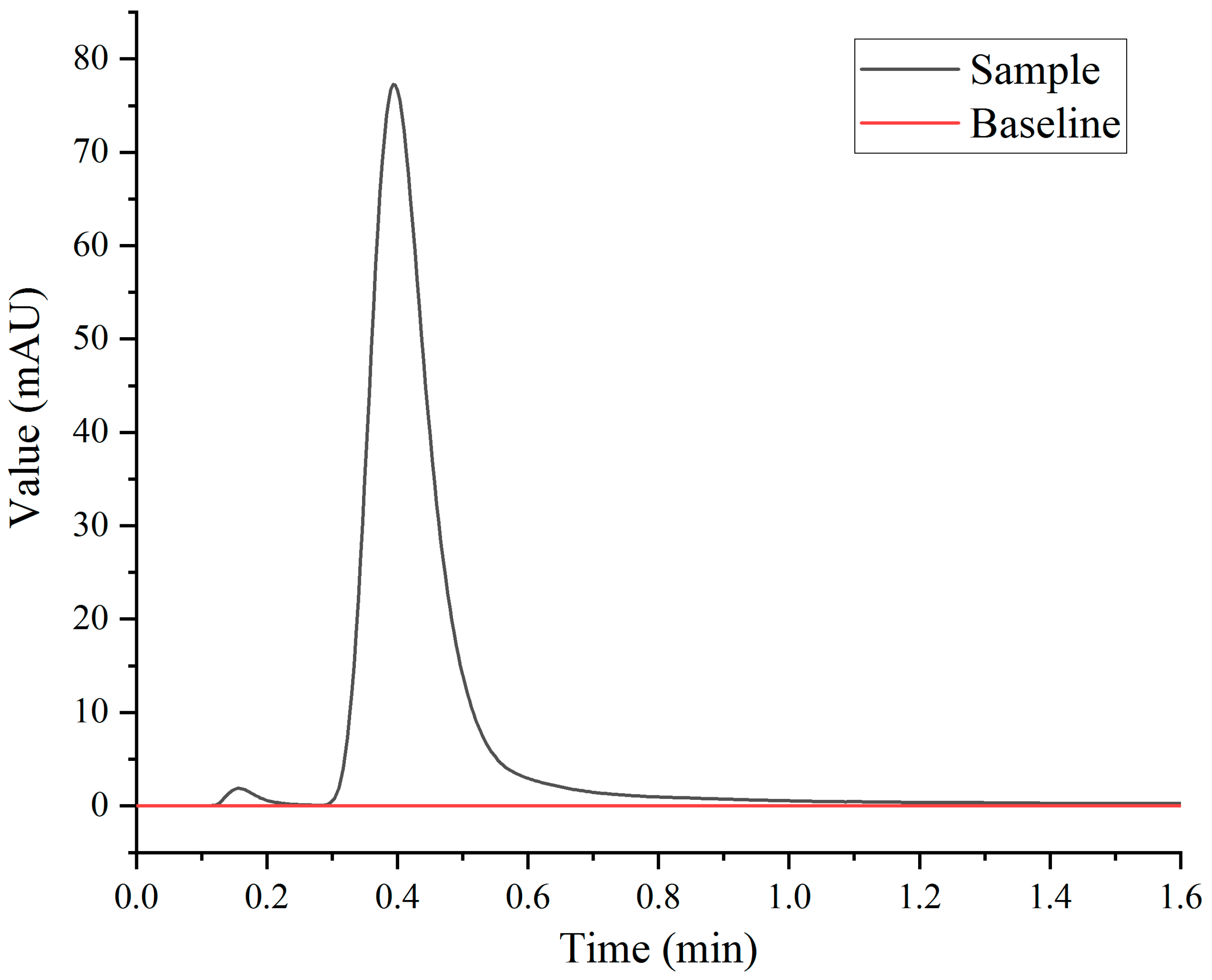
In this experiment, the initial state of the six-way valve was the 1–6 connection, and the valve was transformed into the 1–2 connection at tA (1.5 min). All the albumin in the sample flowed out of the SPE column, and the paclitaxel and related substances were then carried out of the SPE column through the mobile phase of the left pump. At tB (3 min), the six-way valve was converted to the 1–6 connection, and all the paclitaxel and related substances flowed out of the SPE column, which gradually returned to the initial state and waited for the next sample injection. Figure 2 shows that all of the human albumin was eluted within 1.5 min. The calculation of the SPE column volume shows that paclitaxel and its related substances can be eluted within 3 min.

Figure 2.
Elution time of human albumin on SPE column.
3.1.2. Establishment of the Separation Analysis Method
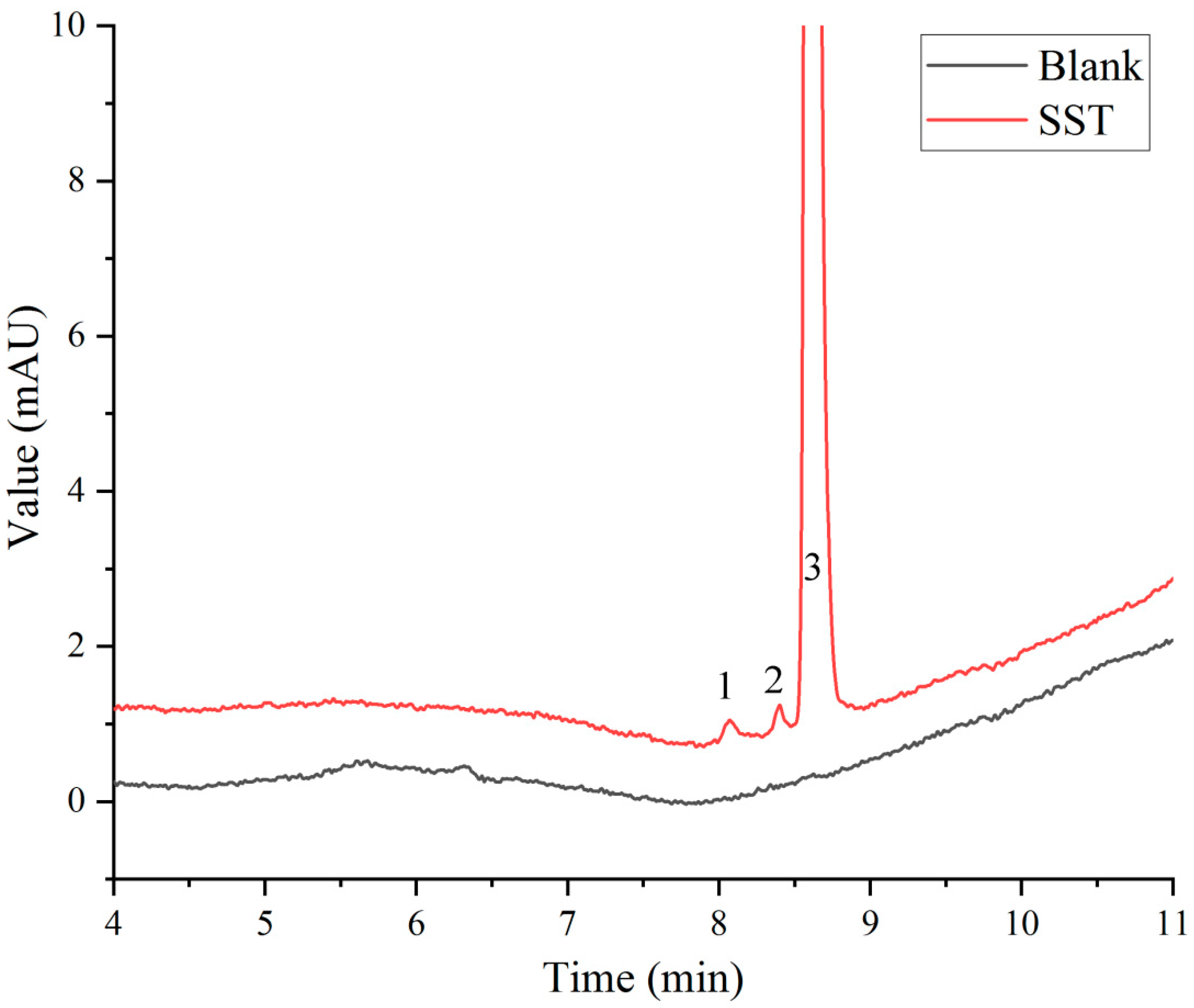
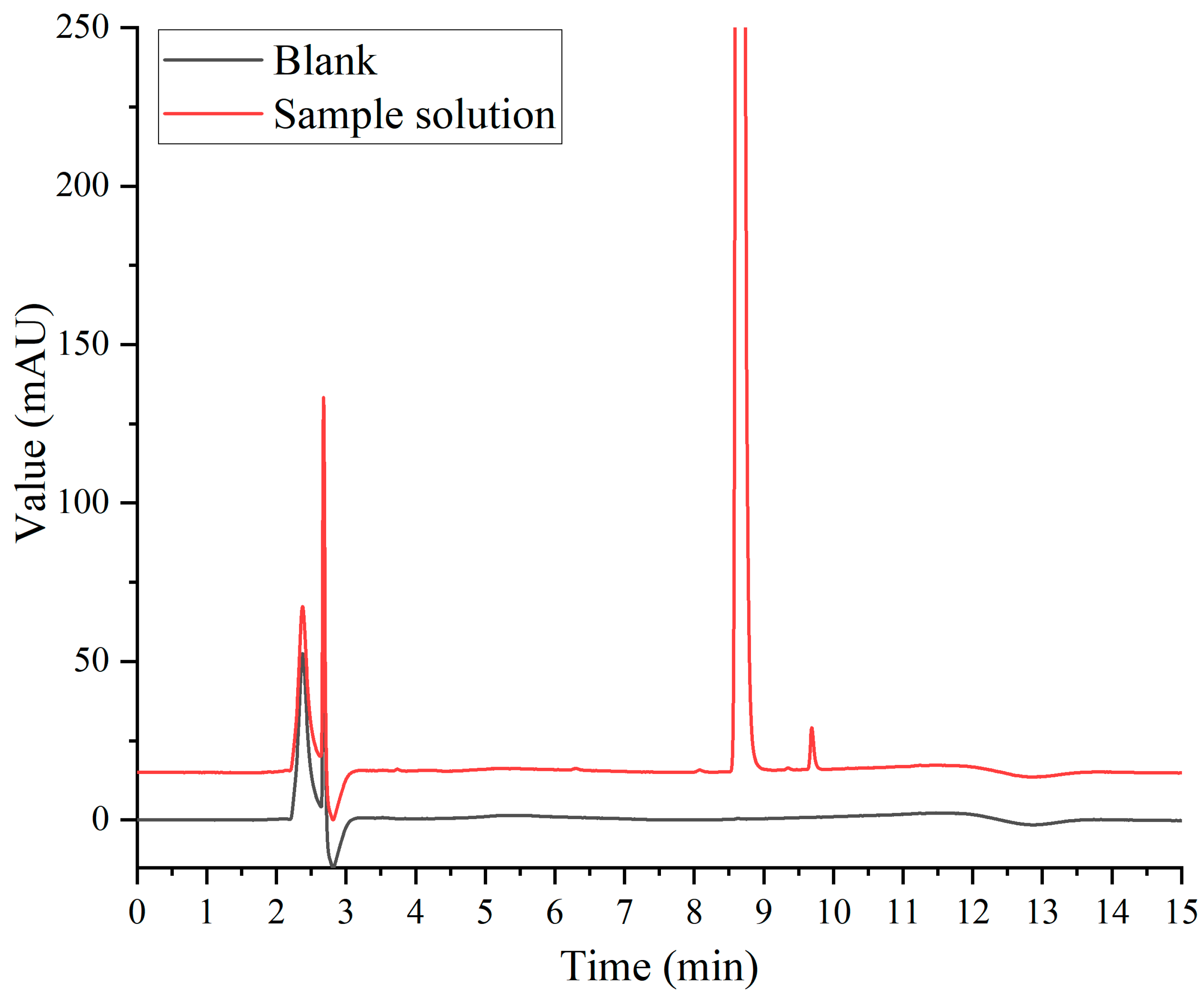
In the Chinese Pharmacopoeia (Chp2020), the detection method for paclitaxel-related substances has an analysis time of 80 min. Along with this long duration, this analytical method has low analysis efficiency and is not suitable for the determination of large quantities of samples. Based on the Chp2020 method, we optimized and adjusted our chromatographic method in the early stage of this study, such as the terms of flow, injection volume, gradient elution procedure, and column temperature, to ensure successful separation and shorten the detection time [20]. According to the results of the previous study, HPLC with two pumps was used to combine the SPE separation method with the pre-optimized chromatographic method for the removal of human albumin and the analysis of paclitaxel-related substances. As can be seen from Figure 3, the interference of human albumin in the detection of paclitaxel-related substances was effectively prevented. Paclitaxel was separated well with impurity I and II, and the signal-to-noise ratio (S/N) of the sensitivity solution was greater than 10. As shown in Figure 4, the blank solution did not interfere with the determination of principal components and paclitaxel-related substances during the investigation of the method’s specificity, which was found to be good.
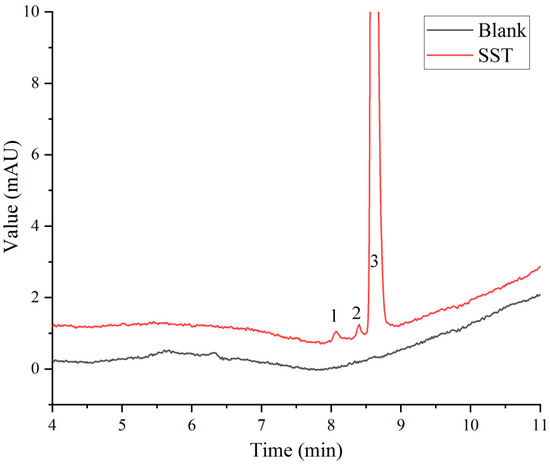
Figure 3.
System suitability solution (SST) chromatogram: 1, 2, and 3 refer to impurity I (cephalomannine), impurity II (7-epi-10-deacetylpaclitaxel), and paclitaxel, respectively.
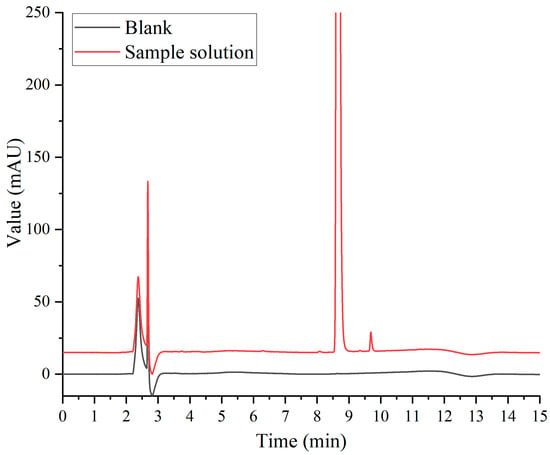
Figure 4.
Specific chromatogram.
3.2. Method Validation for the Analysis of Impurities
The 2D-HPLC-UV method for the analysis of impurities was validated by estimating the linearity, LOQ, LOD, precision, repeatability, solution stability, robustness, and accuracy according to the ICH guideline Q2 (R1). According to ChP2020, the related substances in the paclitaxel powder were quantified via the principal component control method.
3.2.1. Forced-Degradation Studies
Paclitaxel is a natural extract with a complex structure. Its diterpenoid parent ring has 11 chiral centers and many hydroxyl and amino groups. Paclitaxel contains more impurities of the same series, so it is more difficult to control impurities. The sample solution was degraded under acid, alkali, oxidation, and other experimental conditions to understand the possible degradation pathway for paclitaxel and predict its degradation and stability; additionally, the applicability of the detection method for related substances was investigated.
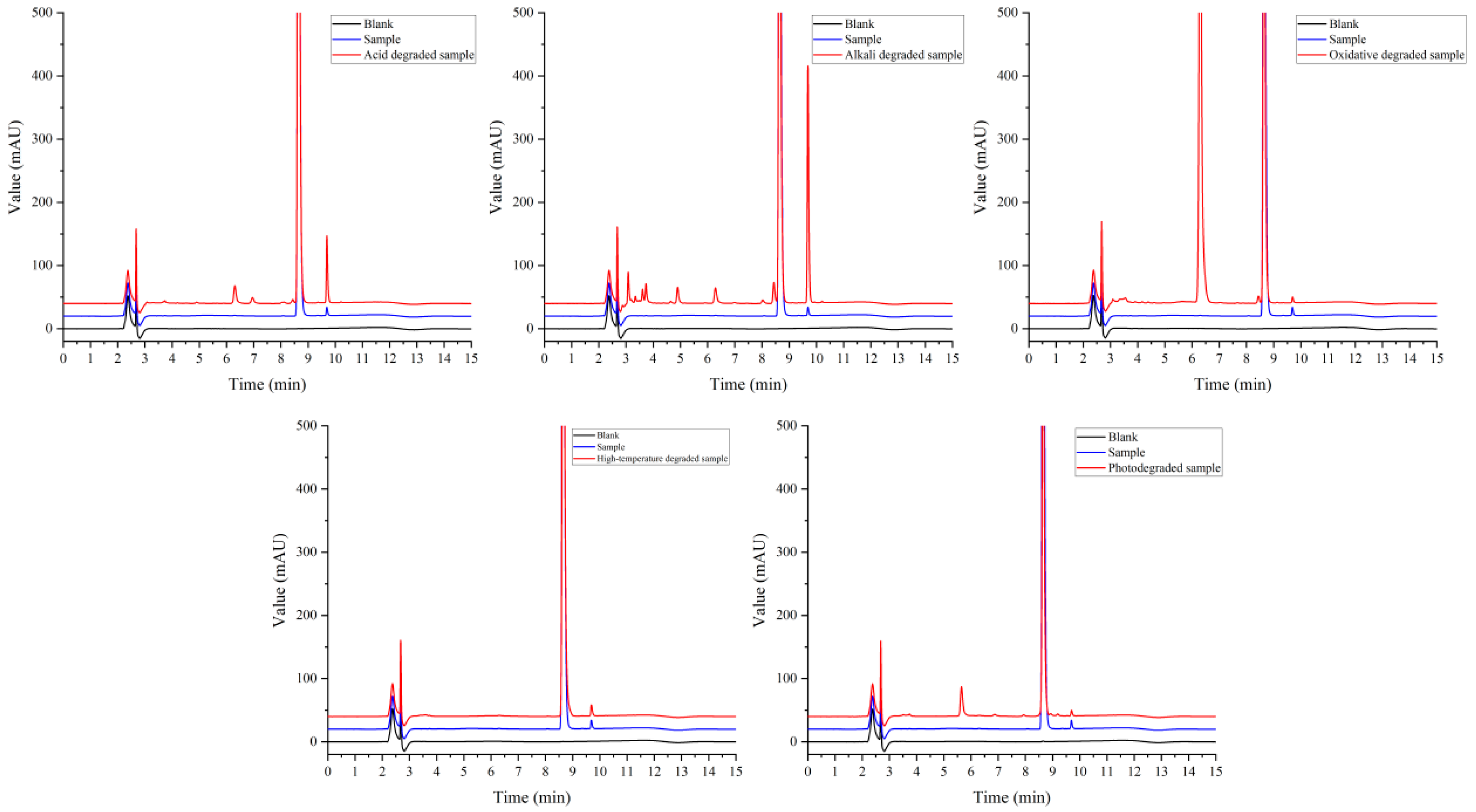
Figure 5 shows that the principal component was well separated from the impurities under each condition. The sample solution was degraded to different degrees under acid, alkali, oxidation, and light conditions in addition to high-temperature damage. Under the condition of acid-and-base degradation, the unknown impurity peak at a retention time of 9.690 min in the sample increased, and the impurity was more easily produced under alkaline conditions. In addition, the content of impurity II (7-epi-10-deacetylpaclitaxel) also increased under this condition. This may be due to the instability of paclitaxel under acid and base conditions; it is easy for degradation impurities to form under acid or base conditions, such as those resulting from deacylation and hydrolysis [21,22]. Under oxidation conditions, large impurity peaks were produced at a retention time of 6.297 min. Kevin J. Volk et al. [23] showed that treatment with hydrogen peroxide produced 10-deacetylpaclitaxel. Further research is needed to confirm the structure of the impurity. Impurity peaks were produced at a retention time of 5.650 min under exposure to high-intensity light. Zhang et al. [24] showed that paclitaxel may produce cephalomannine, 7-epi-paclitaxel, 10-deacetylpaclitaxel, and paclitaxel isomer under light conditions. This provides a reference and basis for subsequent studies of the impurity spectrum.
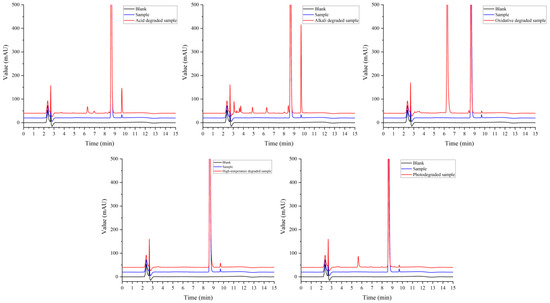
Figure 5.
Chromatogram of sample degradation experiment.
These results showed that paclitaxel was degraded to different degrees under acid, alkali, oxidation, and light conditions. It is relatively stable under high-temperature conditions. The results provided a useful reference for the formulation, process design, and product storage conditions.
3.2.2. Linearity and Range, LOD, and LOQ
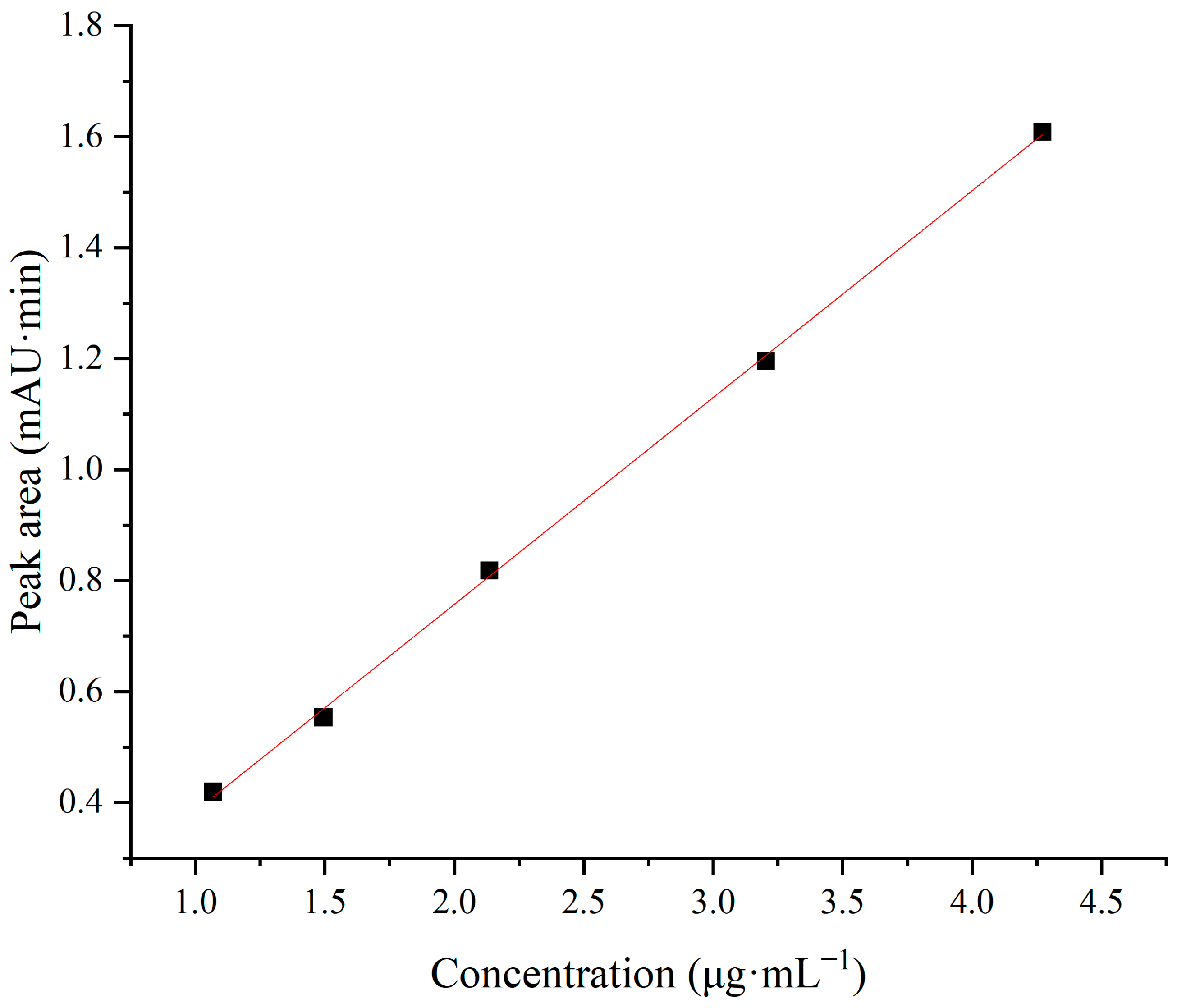
The linear range refers to the range of concentrations within which the signal shows a linear relationship with concentration. It is based on the premise that the method can enable quantitative calculations in which the signal can be converted into the concentration based on a linear relationship. In this study, the concentration of the reference solution was taken as the X-axis and the measured peak area was taken as the Y-axis to fit the calibration curve at five points. The results are shown in Table 2 and Figure 6, with a linear curve equation coefficient R2 of 0.9994, which indicates a good linear relationship between the sample concentration and the measured peak area. To assess the fit of the model and examine the difference between the data and the predictions of the regression model, we used the residual sum of squares (RSS) to quantify the model’s error and fit quality. The RSS of the model is 0.0006, which indicates that the model error is small enough and the model fits the data well.
Table 2.
The results of validation.
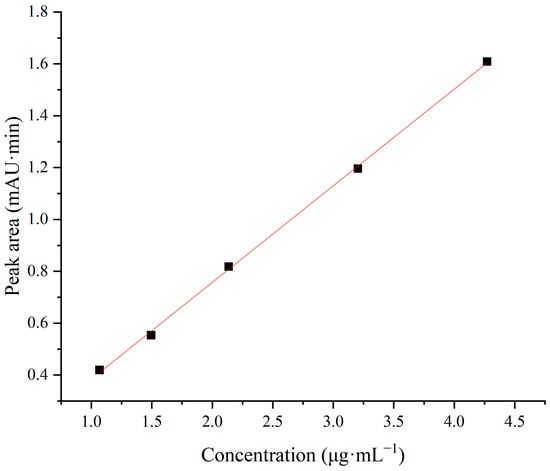
Figure 6.
Linear fitting graph.
The limit of detection (LOD) and limit of quantification (LOQ) are important attributes of the analytical method; the LOD refers to the lowest amount of analyte that can be detected in the sample, and the LOQ refers to the lowest amount of analyte in the samples that can be quantitatively determined. In this study, the LOD and LOQ of the method were investigated using the signal-to-noise (S/N) ratio method, which is suitable for instrumental analysis methods that can display baseline noise. It can be seen from the results that the S/N of three samples of the LOD solution was greater than 3. In the continuous analysis of a six-needle LOQ solution, the S/N was greater than 10, and the relative standard deviation (RSD) of the paclitaxel peak area was 3.47%. For this analytical method, the LOD and LOQ concentrations of the solution were 0.25 μg·mL−1 and 0.5 μg·mL−1, respectively.
3.2.3. Precision and Repeatability
Good precision and repeatability are necessary for the method to have good accuracy, as seen from Table 2 which shows the results of six consecutive analyses of the same paclitaxel standard solution and a paclitaxel peak area RSD of 0.50%. Six identical samples were injected in parallel for analysis, and the maximum impurity content RSD for the sample solution was 1.88%. The above results indicate that the method has good precision and repeatability.
3.2.4. Accuracy, Solution Stability, and Robustness
Accuracy refers to the degree to which the results obtained via the established method are close to the true or reference values. The accuracy of the method was examined using three levels of recovery (80%, 100%, and 120%). It can be seen from Table 2 that the recovery rate of the three levels is between 102.53% and 107.97%, and the RSD (n = 9) is 1.83%, indicating that the accuracy of the method was good.
In this study, we performed solution stability tests to observe changes in the peak area of major impurities, and calculated the relative standard deviation (RSD) to see whether there were new impurities. The freshly prepared sample solution was injected at room temperature at 0, 2, 4, 6, 8, and 10 h to investigate the change in the maximum impurity peak area in the sample solution. The results showed that the maximum impurity peak area varied greatly with time, and the solution should be freshly prepared.
The robustness of the method was investigated by changing the detection wavelength (227 nm ± 2 nm), flow rate (0.4 mL·min−1 ± 0.05 mL·min−1), and column temperature (40 °C ± 2 °C). The results showed that the wavelength, flow rate, and column temperature factors in the method had little effect on the maximum impurity measurement results, and the RSD was less than 6% (n = 7).
4. Conclusions
In this study, two-dimensional high-performance liquid chromatography technology was used to develop a quantitative method for paclitaxel-related substances in the paclitaxel preparation for injection (albumin-bound), which effectively solved the issue of an interference of human albumin in the sample and preserved the sample pretreatment time. In addition, this method is an improved version of the detection method for paclitaxel-related substances in CHP2020, with a shortened analysis time. It has a high degree of automation with good reproducibility, can avoid human error, and is suitable for detecting a large number of samples.
Author Contributions
Formal analysis, H.L. and Q.J.; funding acquisition, G.S.; investigation, L.Z.; methodology, J.Y.; project administration, X.L.; software, X.Q.; validation, X.Z. and Y.J.; writing—original draft, X.Z.; writing—review and editing, H.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the CAMS Initiative for Innovative Medicine, grant number 2021-I2M-1–070. The APC was funded by National Institutes for Food and Drug Control.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Marupudi, N.I.; Han, J.E.; Li, K.W.; Renard, V.M.; Tyler, B.M.; Brem, H. Paclitaxel: A review of adverse toxicities and novel delivery strategies. Expert Opin. Drug Saf. 2007, 6, 609–621. [Google Scholar] [CrossRef]
- Yu, D.-L.; Lou, Z.-P.; Ma, F.-Y.; Najafi, M. The interactions of paclitaxel with tumour microenvironment. Int. Immunopharmacol. 2022, 105, 108555. [Google Scholar] [CrossRef]
- Yang, Y.-H.; Mao, J.-W.; Tan, X.-L. Research progress on the source, production, and anti-cancer mechanisms of paclitaxel. Chin. J. Nat. Med. 2020, 18, 890–897. [Google Scholar] [CrossRef]
- Gallego-Jara, J.; Lozano-Terol, G.; Sola-Martínez, R.A.; Cánovas-Díaz, M.; de Diego Puente, T. A Compressive Review about Taxol®: History and Future Challenges. Molecules 2020, 25, 5986. [Google Scholar] [CrossRef]
- Zhu, L.; Chen, L. Progress in research on paclitaxel and tumor immunotherapy. Cell. Mol. Biol. Lett. 2019, 24, 40. [Google Scholar] [CrossRef]
- Alves, R.C.; Fernandes, R.P.; Eloy, J.O.; Salgado, H.R.N.; Chorilli, M. Characteristics, Properties and Analytical Methods of Paclitaxel: A Review. Crit. Rev. Anal. Chem. 2018, 48, 110–118. [Google Scholar] [CrossRef]
- Nardiello, D.; Melfi, M.T.; Pignatelli, C.; Centonze, D. Enhancing online protein isolation as intact species from soy flour samples by actively modulated two-dimensional liquid chromatography (2D-LC). J. Pharm. Biomed. Anal. 2020, 179, 112976. [Google Scholar] [CrossRef]
- van Beek, F.T.; Edam, R.; Pirok, B.W.J.; Genuit, W.J.L.; Schoenmakers, P.J. Comprehensive two-dimensional liquid chromatography of heavy oil. J. Chromatogr. A 2018, 1564, 110–119. [Google Scholar] [CrossRef]
- Luo, H.; Zhong, W.; Yang, J.; Zhuang, P.; Meng, F.; Caldwell, J.; Mao, B.; Welch, C.J. 2D-LC as an on-line desalting tool allowing peptide identification directly from MS unfriendly HPLC methods. J. Pharm. Biomed. Anal. 2017, 137, 139–145. [Google Scholar] [CrossRef]
- Jakobsen, S.S.; Christensen, J.H.; Verdier, S.; Mallet, C.R.; Nielsen, N.J. Increasing Flexibility in Two-Dimensional Liquid Chromatography by Pulsed Elution of the First Dimension: A Proof of Concept. Anal. Chem. 2017, 89, 8723–8730. [Google Scholar] [CrossRef]
- Kula, M.; Głód, D.; Krauze-Baranowska, M. Two-dimensional liquid chromatography (LC) of phenolic compounds from the shoots of Rubus idaeus ‘Glen Ample’ cultivar variety. J. Pharm. Biomed. Anal. 2016, 121, 99–106. [Google Scholar] [CrossRef]
- Nägele, E.; Vollmer, M.; Hörth, P.; Vad, C. 2D-LC/MS techniques for the identification of proteins in highly complex mixtures. Expert Rev. Proteom. 2014, 1, 37–46. [Google Scholar] [CrossRef]
- Cao, J.-L.; Wei, J.-C.; Chen, M.-W.; Su, H.-X.; Wan, J.-B.; Wang, Y.-T.; Li, P. Application of two-dimensional chromatography in the analysis of Chinese herbal medicines. J. Chromatogr. A 2014, 1371, 1–14. [Google Scholar] [CrossRef]
- Stoll, D.R.; Li, X.; Wang, X.; Carr, P.W.; Porter, S.E.G.; Rutan, S.C. Fast, comprehensive two-dimensional liquid chromatography. J. Chromatogr. A 2007, 1168, 3–43. [Google Scholar] [CrossRef]
- Chen, Z.; Gao, Y.; Zhong, D. Technologies to improve the sensitivity of existing chromatographic methods used for bioanalytical studies. Biomed. Chromatogr. 2020, 34, e4798. [Google Scholar] [CrossRef]
- Iguiniz, M.; Heinisch, S. Two-dimensional liquid chromatography in pharmaceutical analysis. Instrumental aspects, trends and applications. J. Pharm. Biomed. Anal. 2017, 145, 482–503. [Google Scholar] [CrossRef]
- Stoll, D.R.; Carr, P.W. Two-Dimensional Liquid Chromatography: A State of the Art Tutorial. Anal. Chem. 2016, 89, 519–531. [Google Scholar] [CrossRef]
- Guiochon, G.; Marchetti, N.; Mriziq, K.; Shalliker, R.A. Implementations of two-dimensional liquid chromatography. J. Chromatogr. A 2008, 1189, 109–168. [Google Scholar] [CrossRef]
- Rogeberg, M.; Malerod, H.; Roberg-Larsen, H.; Aass, C.; Wilson, S.R. On-line solid phase extraction–liquid chromatography, with emphasis on modern bioanalysis and miniaturized systems. J. Pharm. Biomed. Anal. 2014, 87, 120–129. [Google Scholar] [CrossRef]
- Jiang, Y.-F.; Yao, J.; Li, Y.-R.; Lian, X.-F.; Shan, G.-Z. Determination of related substances in paclitaxel injection by ultra-high performance liquid chromatography. Chin. Med. Biotechnol. 2023, 18, 116–121. [Google Scholar] [CrossRef]
- Tian, J.; Stella, V.J. Degradation of Paclitaxel and Related Compounds in Aqueous Solutions II: Nonepimerization Degradation under Neutral to Basic pH Conditions. J. Pharm. Sci. 2008, 97, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Stella, V.J. Degradation of Paclitaxel and Related Compounds in Aqueous Solutions III: Degradation under Acidic pH Conditions and Overall Kinetics. J. Pharm. Sci. 2010, 99, 1288–1298. [Google Scholar] [CrossRef]
- Volk, K.J.; Hill, S.E.; Kerns, E.H.; Lee, M.S. Profiling degradants of paclitaxel using liquid chromatography-mass spectrometry and liquid chromatography-tandem mass spectrometry substructural techniques. J. Chromatogr. B 1997, 696, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-Y.; Jie, L.; Gao, J.-M.; Zhang, Q.-M.; Shi, Y.-Q. UPLC-MS/MS method research of taxol and its injection impurity profile. Acta Pharm. Sin. 2016, 51, 965–971. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).