
Mycotoxins and Maize Value Chain: Multi-Matrix and Multi-Analyte Tools towards Global Feed and Food Safety




Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials, Reagents and Standard Solutions
2.2. Instrumentation
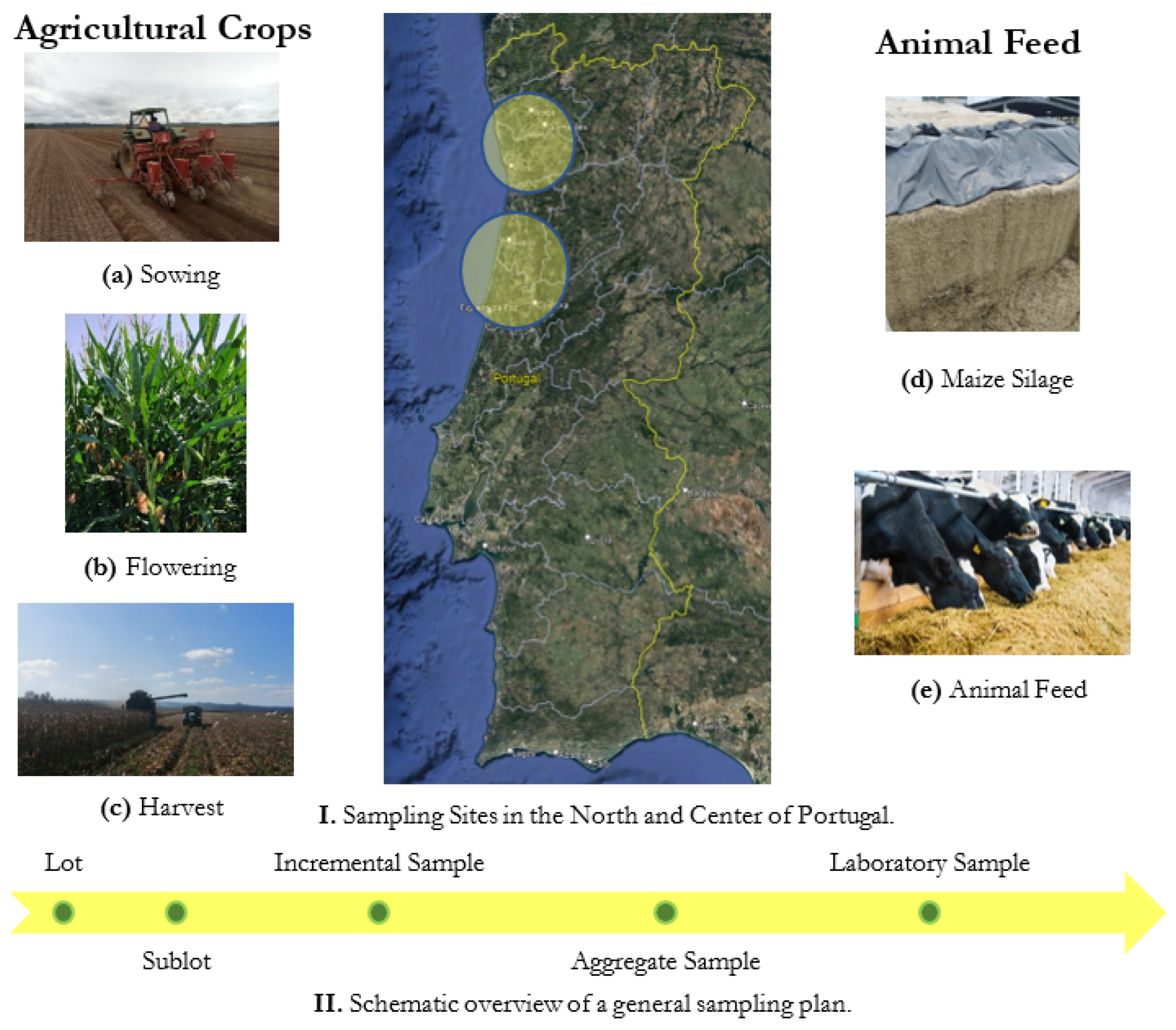
2.3. Samples
2.4. Sample Preparation
2.5. Method Validation
3. Results and Discussion
3.1. Method Validation
3.1.1. Method Specificity and Selectivity
3.1.2. Limits of Detection (LOD) and Limits of Quantification (LOQ)
3.1.3. Calibration and Linearity
3.1.4. Precision and Recovery
3.2. Application to Real Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grote, U.; Fasse, A.; Nguyen, T.T.; Erenstein, O. Food Security and the Dynamics of Wheat and Maize Value Chains in Africa and Asia. Front. Sustain. Food Syst. 2021, 4, 617009. [Google Scholar] [CrossRef]
- Leite, M.; Freitas, A.; Silva, A.S.; Barbosa, J.; Ramos, F. Maize Food Chain and Mycotoxins: A Review on Occurrence Studies. Trends Food Sci. Technol. 2021, 115, 307–331. [Google Scholar] [CrossRef]
- Food and Agriculture Organization (FAO). FAO World Food and Agriculture—Statistical Yearbook 2021; FAO: Rome, Italy, 2021; ISBN 9789251343326. [Google Scholar]
- Munkvold, G.P.; Arias, S.; Taschl, I.; Gruber-Dorninger, C. Mycotoxins in Corn: Occurrence, Impacts, and Management, 3rd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2019; ISBN 9780128119716. [Google Scholar]
- Neme, K.; Mohammed, A. Mycotoxin Occurrence in Grains and the Role of Postharvest Management as a Mitigation Strategies. A Review. Food Control 2017, 78, 412–425. [Google Scholar] [CrossRef]
- European Commission (EC). Consolidated Text: Commission Regulation (EC) No 1881/2006 of 19 December 2006 Setting Maximum Levels for Certain Contaminants in Foodstuffs. Off. J. Eur. Union 2020, 364, 5–24. [Google Scholar]
- European Commission. Commission Regulation (EC) No 1126/2007 of 28 September 2007 Amending Regulation (EC) No 1881/2006 Setting Maximum Levels for Certain Contaminants in Foodstuffs as Regards Fusarium Toxins in Maize and Maize Products. Off. J. Eur. Union 2007, 255, 14–17. [Google Scholar]
- European Commission (EC). Commission Recommendation No 2013/165/EU of 27 March 2013 on the Presence of T-2 and HT-2 Toxin in Cereals and Cereal Products. Off. J. Eur. Union 2013, 91, 12–15. [Google Scholar]
- European Commission (EC). Consolidated Text: Commission Recommendation of 17 August 2006 on the Presence of Deoxynivalenol, Zearalenone, Ochratoxin A, T-2 and HT-2 and Fumonisins in Products Intended for Animal Feeding (2006/576/EC). Off. J. Eur. Union 2016, 229, 7–10. [Google Scholar]
- European Commission (EC). Consolidated Text: Directive 2002/32/EC of the European Parliament and of the Council of 7 May 2002 on Undesirable Substances in Animal Feed. Off. J. Eur. Union 2019, 140, 10–22. [Google Scholar]
- Čolović, R.; Puvača, N.; Cheli, F.; Avantaggiato, G.; Greco, D.; Đuragić, O.; Kos, J.; Pinotti, L. Decontamination of Mycotoxin-Contaminated Feedstuffs and Compound Feed. Toxins 2019, 11, 617. [Google Scholar] [CrossRef]
- Ismaiel, A.A.; Papenbrock, J. Mycotoxins: Producing Fungi and Mechanisms of Phytotoxicity. Agriculture 2015, 5, 493–537. [Google Scholar] [CrossRef]
- Leite, M.; Freitas, A.; Sanches, A.; Barbosa, J.; Ramos, F. Maize (Zea mays L.) and Mycotoxins: A Review on Optimization and Validation of Analytical Methods by Liquid Chromatography Coupled to Mass Spectrometry. Trends Food Sci. Technol. 2020, 99, 542–565. [Google Scholar] [CrossRef]
- Leite, M.; Freitas, A.; Barbosa, J.; Ramos, F. Comprehensive Assessment of Different Extraction Methodologies for Optimization and Validation of an Analytical Multi-Method for Determination of Emerging and Regulated Mycotoxins in Maize by UHPLC-MS/MS. Food Chem. Adv. 2023, 2, 100145. [Google Scholar] [CrossRef]
- European Commission (EC). Commission Regulation (EC) No 401/2006 of 23 February 2006 Laying down the Methods of Sampling and Analysis for the Official Control of the Levels of Mycotoxins in Foodstuffs. Off. J. Eur. Union 2006, 70, 12–34. [Google Scholar]
- Food and Drug Administration (FDA). Bioanalytical Method Validation: Guidance for Industry; Food and Drug Administration (FDA): Silverspring, MD, USA, 2018. [Google Scholar]
- European Medicines Agency (EMA). Guideline on Bioanalytical Method Validation; European Medicines Agency (EMA): Amsterdam, The Netherlands, 2012. [Google Scholar]
- European Commission (EC). Commission Regulation (EU) No 519/2014 of 16 May 2014 Amending Regulation (EC) No 401/2006 as Regards Methods of Sampling of Large Lots, Spices and Food Supplements, Performance Criteria for T-2, HT-2 Toxin and Citrinin and Screening Methods of Analysis. Off. J. Eur. Union 2014, L147, 29–43. [Google Scholar]
- European Commission (EC). Commission Implementing Regulation (EU) 2021/808 of 22 March 2021 on the Performance of Analytical Methods for Residues of Pharmacologically Active Substances Used in Food-Producing Animals and on the Interpretation of Results as Well as on the Methods To. Off. J. Eur. Union 2021, L180, 84–109. [Google Scholar]
- Shephard, G.S. Current Status of Mycotoxin Analysis: A Critical Review. J. AOAC Int. 2016, 99, 842–848. [Google Scholar] [CrossRef]
- Rausch, A.; Brockmeyer, R.; Schwerdtle, T. Development and Validation of a QuEChERS-Based Liquid Chromatography Tandem Mass Spectrometry Multi-Method for the Determination of 38 Native and Modified Mycotoxins in Cereals. J. Agric. Food Chem. 2020, 68, 4657–4669. [Google Scholar] [CrossRef]
- Garon, D.; Richard, E.; Sage, L.; Bouchart, V.; Pottier, D.; Lebailly, P. Mycoflora and Multimycotoxin Detection in Corn Silage: Experimental Study. J. Agric. Food Chem. 2006, 54, 3479–3484. [Google Scholar] [CrossRef]
- Lattanzio, V.M.; Solfrizzo, M.; Powers, S.; Visconti, A. Simultaneous Determination of Aflatoxins, Ochratoxin A and Fusarium Toxins in Maize by Liquid Chromatography/ Tandem Mass Spectrometry after Multitoxin Immunoaffinity Cleanup. Rapid Commun. Mass Spectrom. 2007, 21, 3253–3261. [Google Scholar] [CrossRef] [PubMed]
- Sulyok, M.; Berthiller, F.; Krska, R.; Schuhmacher, R. Development and Validation of a Liquid Chromatography/Tandem Mass Spectrometric Method for the Determination of 39 Mycotoxins in Wheat and Maize. Rapid Commun. Mass Spectrom. 2006, 24, 2649–2659. [Google Scholar] [CrossRef]
- Abdallah, M.F.; Girgin, G.; Baydar, T. Mycotoxin Detection in Maize, Commercial Feed, and Raw Dairy Milk Samples from Assiut City, Egypt. Vet. Sci. 2019, 6, 57. [Google Scholar] [CrossRef]
- Panasiuk, L.; Jedziniak, P.; Pietruszka, K.; Piatkowska, M.; Bocian, L. Frequency and Levels of Regulated and Emerging Mycotoxins in Silage in Poland. Mycotoxin Res. 2019, 35, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Scarpino, V.; Reyneri, A.; Blandino, M. Development and Comparison of Two Multiresidue Methods for the Determination of 17 Aspergillus and Fusarium Mycotoxins in Cereals Using HPLC-ESI-TQ-MS/MS. Front. Microbiol. 2019, 10, 361. [Google Scholar] [CrossRef] [PubMed]
- Malachová, A.; Stránská, M.; Václavíková, M.; Elliott, C.T.; Black, C.; Meneely, J.; Hajšlová, J.; Ezekiel, C.N.; Schuhmacher, R.; Krska, R. Advanced LC–MS-Based Methods to Study the Co-Occurrence and Metabolization of Multiple Mycotoxins in Cereals and Cereal-Based Food. Anal. Bioanal. Chem. 2018, 410, 801–825. [Google Scholar] [CrossRef] [PubMed]
- Jettanajit, A.; Nhujak, T. Determination of Mycotoxins in Brown Rice Using QuEChERS Sample Preparation and UHPLC–MS-MS. J. Chromatogr. Sci. 2016, 54, 720–729. [Google Scholar] [CrossRef]
- European Medicines Agency (EMA). ICH Guideline Q2(R2) on Validation of Analytical Procedures; European Medicines Agency: Amsterdam, The Netherlands, 2022; Volume 2. [Google Scholar]
- Van Pamel, E.; Verbeken, A.; Vlaemynck, G.; De Boever, J.; Daeseleire, E. Ultrahigh-performance liquid chromatographic-tandem mass spectrometric multimycotoxin method for quantitating 26 mycotoxins in maize silage. J. Agric. Food Chem. 2011, 59, 9747. [Google Scholar] [CrossRef]
- Dagnac, T.; Latorre, A.; Fernández Lorenzo, B.; Llompart, M. Validation and application of a liquid chromatography-tandem mass spectrometry-based method for the assessment of the co-occurrence of mycotoxins in maize silages from dairy farms in NW Spain. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2016, 33, 1850. [Google Scholar] [CrossRef]
- De Beer, J.O.; Van Poucke, C. Ensuring the Quality of Results from Food Control Laboratories: Laboratory Accreditation, Method Validation and Measurement Uncertainty; Woodhead Publishing Limited: Sawston, UK, 2011; ISBN 9781845696740. [Google Scholar]
- Debevere, S.; De Baere, S.; Haesaert, G.; Rychlik, M.; Fievez, V.; Croubels, S. Development of an UPLC-MS/MS Method for the Analysis of Mycotoxins in Rumen Fluid with and without Maize Silage Emphasizes the Importance of Using Matrix-Matched Calibration. Toxins 2019, 11, 519. [Google Scholar] [CrossRef]
- Biemond, P.C.; Stomph, T.J.; Kumar, P.L.; Struik, P.C. How Maize Seed Systems Can Contribute to the Control of Mycotoxigenic Fungal Infection: A Perspective. Agronomy 2021, 11, 2168. [Google Scholar] [CrossRef]

{kind=link}
| Mycotoxin | Range (ng g−1) | Linearity | Concentration Level (ng g−1) | Precision (%) | Recovery (%) | LOD (ng g−1) | LOQ (ng g−1) |
|---|---|---|---|---|---|---|---|
| AFB1 | 5 to 25 | 0.994 | 20 | 9.50 | 102.50 | 1.31 | 1.58 |
| AFB2 | 5 to 25 | 0.997 | 20 | 1.13 | 102.00 | 5.23 | 13.33 |
| AFG1 | 5 to 25 | 0.983 | 15 | 1.83 | 97.23 | 3.12 | 4.13 |
| AFG2 | 5 to 25 | 0.978 | 15 | 4.83 | 98.01 | 1.90 | 4.04 |
| BEA | 100 to 500 | 0.950 | 200 | 11.83 | 89.00 | 8.16 | 10.99 |
| CIT | 250 to 1250 | 0.981 | 750 | 1.30 | 103.77 | 1.74 | 4.37 |
| DON | 375 to 1875 | 0.996 | 1500 | 2.05 | 103.60 | 40.30 | 42.13 |
| ENNA | 75 to 375 | 0.997 | 300 | 3.18 | 98.80 | 0.84 | 1.00 |
| ENNB | 75 to 375 | 0.965 | 300 | 1.54 | 97.10 | 0.53 | 1.31 |
| FB1 | 250 to 1250 | 0.991 | 750 | 1.48 | 100.75 | 2.29 | 4.72 |
| FB2 | 250 to 1250 | 0.964 | 750 | 3.01 | 92.30 | 4.30 | 7.45 |
| HT-2 | 50 to 250 | 0.988 | 200 | 20.52 | 106.40 | 0.83 | 2.06 |
| MPA | 100 to 500 | 0.993 | 300 | 0.31 | 98.13 | 13.76 | 13.85 |
| MON | 250 to 1250 | 0.976 | 750 | 1.53 | 110.84 | 4.85 | 10.90 |
| NIV | 200 to 1000 | 0.983 | 500 | 1.92 | 105.51 | 20.41 | 39.00 |
| OTA | 10 to 50 | 0.956 | 40 | 18.23 | 90.20 | 1.25 | 1.35 |
| PAT | 12.5 to 62.5 | 0.920 | 37.5 | 7.04 | 110.25 | 3.80 | 8.03 |
| PA | 150 to 750 | 0.993 | 450 | 3.76 | 102.64 | 4.46 | 8.71 |
| T-2 | 50 to 250 | 0.998 | 200 | 17.42 | 97.10 | 4.65 | 9.24 |
| TEA | 51 to 250 | 0.968 | 150 | 4.06 | 101.32 | 10.64 | 19.25 |
| TTX | 12.5 to 62.5 | 0.986 | 37.5 | 2.14 | 86.52 | 2.41 | 2.46 |
| ZEA | 100 to 500 | 0.941 | 400 | 0.66 | 102.10 | 0.74 | 1.24 |
| Mycotoxin | Range (ng g−1) | Linearity | Concentration Level (ng g−1) | Precision (%) | Recovery (%) | LOD (ng g−1) | LOQ (ng g−1) |
|---|---|---|---|---|---|---|---|
| AFB1 | 5 to 25 | 0.997 | 20 | 1.83 | 104.18 | 0.14 | 0.32 |
| AFB2 | 5 to 25 | 0.993 | 20 | 4.74 | 98.89 | 0.47 | 1.04 |
| AFG1 | 5 to 25 | 0.989 | 20 | 5.55 | 104.40 | 3.14 | 6.42 |
| AFG2 | 5 to 25 | 0.990 | 20 | 0.15 | 96.20 | 2.71 | 3.18 |
| BEA | 100 to 500 | 0.967 | 400 | 7.20 | 104.80 | 0.09 | 0.21 |
| CIT | 250 to 1250 | 0.981 | 750 | 2.10 | 86.88 | 4.33 | 10.55 |
| DON | 375 to 1875 | 0.985 | 1500 | 14.52 | 102.40 | 1.82 | 3.82 |
| ENNA | 75 to 375 | 0.985 | 300 | 0.07 | 101.50 | 1.07 | 1.93 |
| ENNB | 75 to 375 | 0.990 | 300 | 2.47 | 104.00 | 0.22 | 0.57 |
| FB1 | 250 to 1250 | 0.998 | 750 | 2.09 | 102.24 | 8.77 | 26.31 |
| FB2 | 250 to 1250 | 0.978 | 750 | 1.99 | 90.10 | 11.72 | 26.01 |
| HT-2 | 50 to 250 | 0.999 | 200 | 1.56 | 100.20 | 9.90 | 20.88 |
| MPA | 100 to 500 | 0.992 | 300 | 1.17 | 98.79 | 0.78 | 1.66 |
| MON | 250 to 1250 | 0.989 | 750 | 5.36 | 99.11 | 5.60 | 12.07 |
| NIV | 200 to 1000 | 0.986 | 500 | 2.35 | 106.75 | 17.90 | 23.35 |
| OTA | 10 to 50 | 0.979 | 40 | 19.07 | 94.20 | 14.44 | 19.35 |
| PAT | 12.5 to 62.5 | 0.982 | 37.5 | 6.66 | 99.04 | 2.60 | 5.08 |
| PA | 150 to 750 | 0.992 | 450 | 1.24 | 103.70 | 2.63 | 4.52 |
| T-2 | 50 to 250 | 0.983 | 200 | 2.88 | 93.80 | 8.10 | 14.56 |
| TEA | 51 to 250 | 0.982 | 150 | 11.94 | 109.10 | 11.38 | 15.17 |
| TTX | 12.5 to 62.5 | 0.994 | 37.5 | 2.35 | 98.92 | 1.67 | 1.71 |
| ZEA | 100 to 500 | 0.987 | 400 | 2.21 | 94.30 | 2.07 | 4.06 |
| Mycotoxin | Range (ng g−1) | Linearity | Concentration Level (ng g−1) | Precision (%) | Recovery (%) | LOD (ng g−1) | LOQ (ng g−1) |
|---|---|---|---|---|---|---|---|
| AFB1 | 5 to 25 | 0.991 | 20 | 4.78 | 104.10 | 2.66 | 7.77 |
| AFB2 | 5 to 25 | 0.984 | 20 | 0.51 | 98.10 | 2.46 | 9.04 |
| AFG1 | 5 to 25 | 0.987 | 15 | 1.41 | 96.15 | 2.69 | 3.19 |
| AFG2 | 5 to 25 | 0.985 | 15 | 4.85 | 107.33 | 1.62 | 7.88 |
| BEA | 100 to 500 | 0.964 | 400 | 10.23 | 108.90 | 2.98 | 4.93 |
| CIT | 250 to 1250 | 0.968 | 750 | 2.71 | 80.65 | 4.65 | 11.00 |
| DON | 375 to 1875 | 0.989 | 1500 | 8.87 | 106.30 | 14.82 | 22.75 |
| ENNA | 75 to 375 | 0.973 | 300 | 5.04 | 99.90 | 0.97 | 1.22 |
| ENNB | 75 to 375 | 0.990 | 300 | 1.03 | 103.30 | 0.03 | 0.05 |
| FB1 | 250 to 1250 | 0.996 | 750 | 2.98 | 99.79 | 3.11 | 11.52 |
| FB2 | 250 to 1250 | 0.974 | 750 | 3.67 | 89.76 | 6.90 | 12.60 |
| HT-2 | 50 to 250 | 0.992 | 200 | 8.49 | 96.00 | 11.05 | 11.49 |
| MPA | 100 to 500 | 0.992 | 300 | 1.09 | 98.14 | 0.69 | 1.45 |
| MON | 250 to 1250 | 0.992 | 750 | 2.34 | 80.65 | 4.65 | 11.00 |
| NIV | 200 to 1000 | 0.947 | 500 | 1.55 | 118.06 | 11.73 | 14.14 |
| OTA | 10 to 50 | 0.916 | 40 | 0.28 | 98.30 | 1.35 | 1.38 |
| PAT | 12.5 to 62.5 | 0.978 | 37.5 | 1.97 | 97.87 | 4.90 | 7.87 |
| PA | 150 to 750 | 0.994 | 450 | 2.19 | 102.16 | 4.22 | 8.21 |
| T-2 | 50 to 250 | 0.977 | 200 | 1.12 | 108.20 | 2.52 | 2.94 |
| TEA | 51 to 250 | 0.902 | 150 | 10.91 | 87.98 | 3.82 | 5.21 |
| TTX | 12.5 to 62.5 | 0.994 | 37.5 | 1.36 | 99.79 | 5.33 | 5.39 |
| ZEA | 100 to 500 | 0.994 | 400 | 1.09 | 99.90 | 0.75 | 1.73 |
| Mycotoxin | Range (ng g−1) | Linearity | Concentration Level (ng g−1) | Precision (%) | Recovery (%) | LOD (ng g−1) | LOQ (ng g−1) |
|---|---|---|---|---|---|---|---|
| AFB1 | 5 to 25 | 0.990 | 20 | 2.03 | 94.50 | 1.78 | 2.67 |
| AFB2 | 5 to 25 | 0.977 | 20 | 2.40 | 107.80 | 1.85 | 2.54 |
| AFG1 | 5 to 25 | 0.984 | 15 | 1.02 | 95.01 | 3.23 | 4.41 |
| AFG2 | 5 to 25 | 0.952 | 15 | 7.61 | 98.36 | 1.05 | 5.02 |
| BEA | 100 to 500 | 0.943 | 400 | 1.27 | 95.00 | 0.28 | 0.61 |
| CIT | 250 to 1250 | 0.975 | 750 | 1.30 | 103.90 | 1.83 | 4.63 |
| DON | 375 to 1875 | 0.928 | 1500 | 0.84 | 106.00 | 6.75 | 15.19 |
| ENNA | 75 to 375 | 0.950 | 300 | 1.02 | 103.80 | 3.51 | 6.81 |
| ENNB | 75 to 375 | 0.990 | 300 | 0.52 | 97.80 | 0.03 | 0.04 |
| FB1 | 250 to 1250 | 0.989 | 750 | 3.57 | 106.51 | 5.14 | 15.41 |
| FB2 | 250 to 1250 | 0.975 | 750 | 5.32 | 90.40 | 7.55 | 24.92 |
| HT-2 | 50 to 250 | 0.960 | 200 | 1.03 | 99.80 | 3.03 | 3.34 |
| MPA | 100 to 500 | 0.993 | 300 | 0.31 | 98.13 | 0.75 | 1.46 |
| MON | 250 to 1250 | 0.991 | 750 | 1.81 | 105.66 | 4.35 | 9.22 |
| NIV | 200 to 1000 | 0.952 | 500 | 4.50 | 113.14 | 15.70 | 32.27 |
| OTA | 10 to 50 | 0.995 | 40 | 5.06 | 104.20 | 0.39 | 12.28 |
| PAT | 12.5 to 62.5 | 0.986 | 37.5 | 3.89 | 105.53 | 3.84 | 4.91 |
| PA | 150 to 750 | 0.996 | 450 | 1.88 | 102.83 | 3.75 | 6.37 |
| T-2 | 50 to 250 | 0.959 | 200 | 0.79 | 99.60 | 1.53 | 14.07 |
| TEA | 51 to 250 | 0.982 | 150 | 8.51 | 99.44 | 8.87 | 14.38 |
| TTX | 12.5 to 62.5 | 0.977 | 37.5 | 10.61 | 88.87 | 5.35 | 6.38 |
| ZEA | 100 to 500 | 0.988 | 400 | 0.40 | 95.30 | 0.86 | 2.16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leite, M.; Freitas, A.; Barbosa, J.; Ramos, F. Mycotoxins and Maize Value Chain: Multi-Matrix and Multi-Analyte Tools towards Global Feed and Food Safety. Separations 2023, 10, 486. https://doi.org/10.3390/separations10090486
Leite M, Freitas A, Barbosa J, Ramos F. Mycotoxins and Maize Value Chain: Multi-Matrix and Multi-Analyte Tools towards Global Feed and Food Safety. Separations. 2023; 10(9):486. https://doi.org/10.3390/separations10090486
Chicago/Turabian StyleLeite, Marta, Andreia Freitas, Jorge Barbosa, and Fernando Ramos. 2023. "Mycotoxins and Maize Value Chain: Multi-Matrix and Multi-Analyte Tools towards Global Feed and Food Safety" Separations 10, no. 9: 486. https://doi.org/10.3390/separations10090486
APA StyleLeite, M., Freitas, A., Barbosa, J., & Ramos, F. (2023). Mycotoxins and Maize Value Chain: Multi-Matrix and Multi-Analyte Tools towards Global Feed and Food Safety. Separations, 10(9), 486. https://doi.org/10.3390/separations10090486