
A One-Step Sample Processing Method in Combination with HPLC-MS/MS for the Simultaneous Quantification of Atorvastatin, Ezetimibe and Three Metabolites including o-Hydroxyl Atorvastatin, p-Hydroxyl Atorvastatin, and Ezetimibe-Glucuronide in Human Plasma

Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
2.2. Liquid Chromatography and Mass Spectrometry Conditions
2.3. Calibrators and Quality Control Samples
2.4. Extraction Efficiency and Matrix Factor
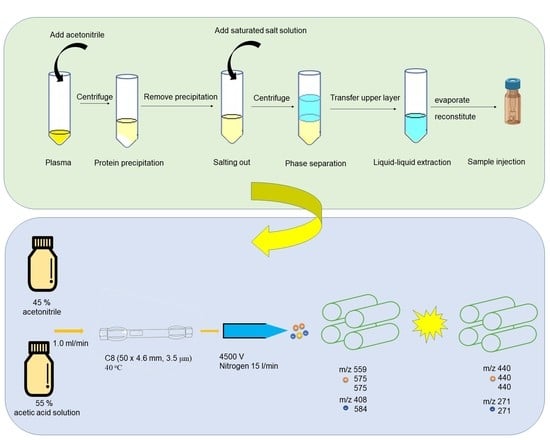
2.5. Protocol for Sample Preparation
2.6. Method Validation
3. Results and Discussion
3.1. LC-ESI-MS/MS Method Development
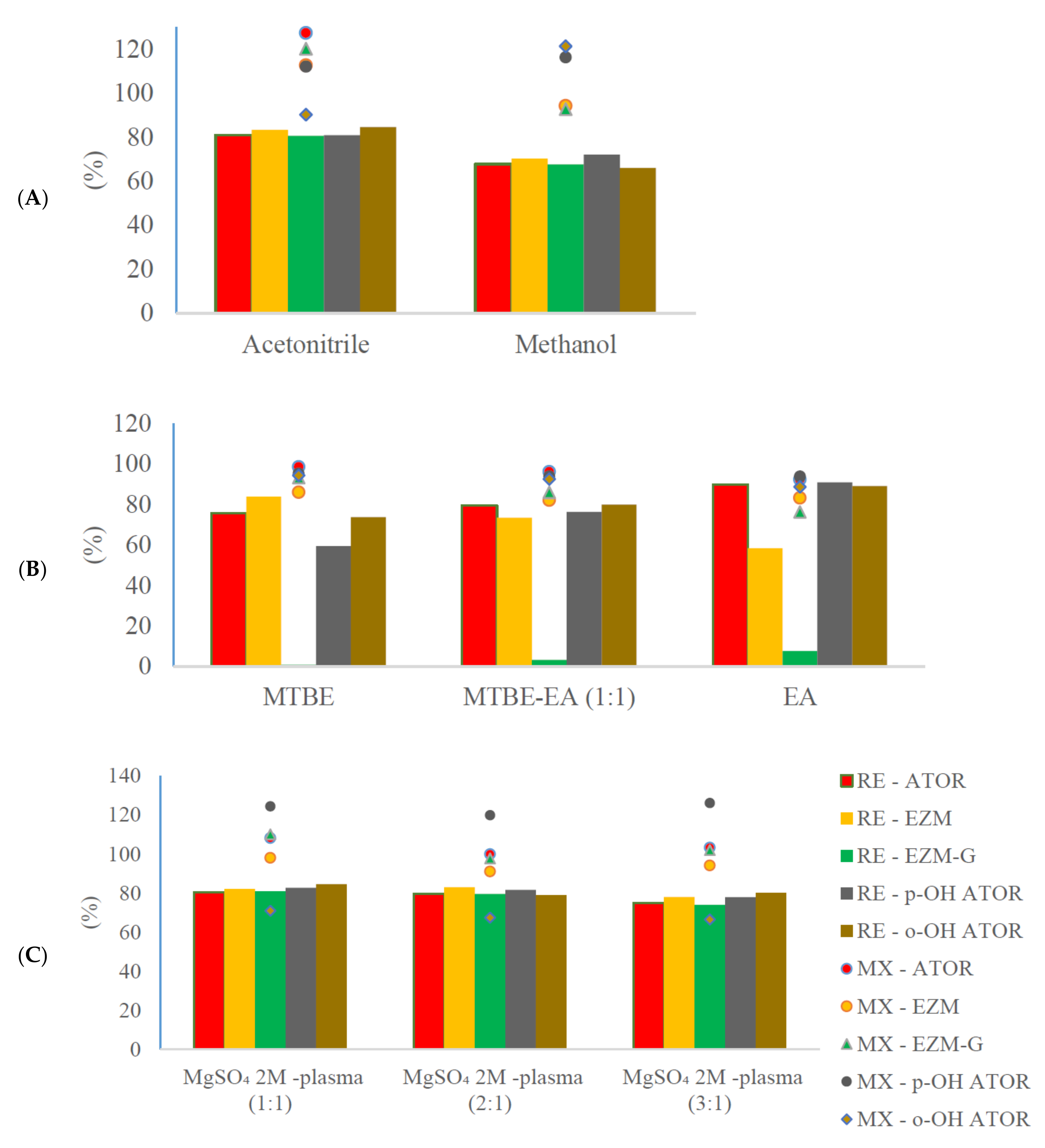
3.2. Optimization of Sample Preparation
3.3. Method Validation
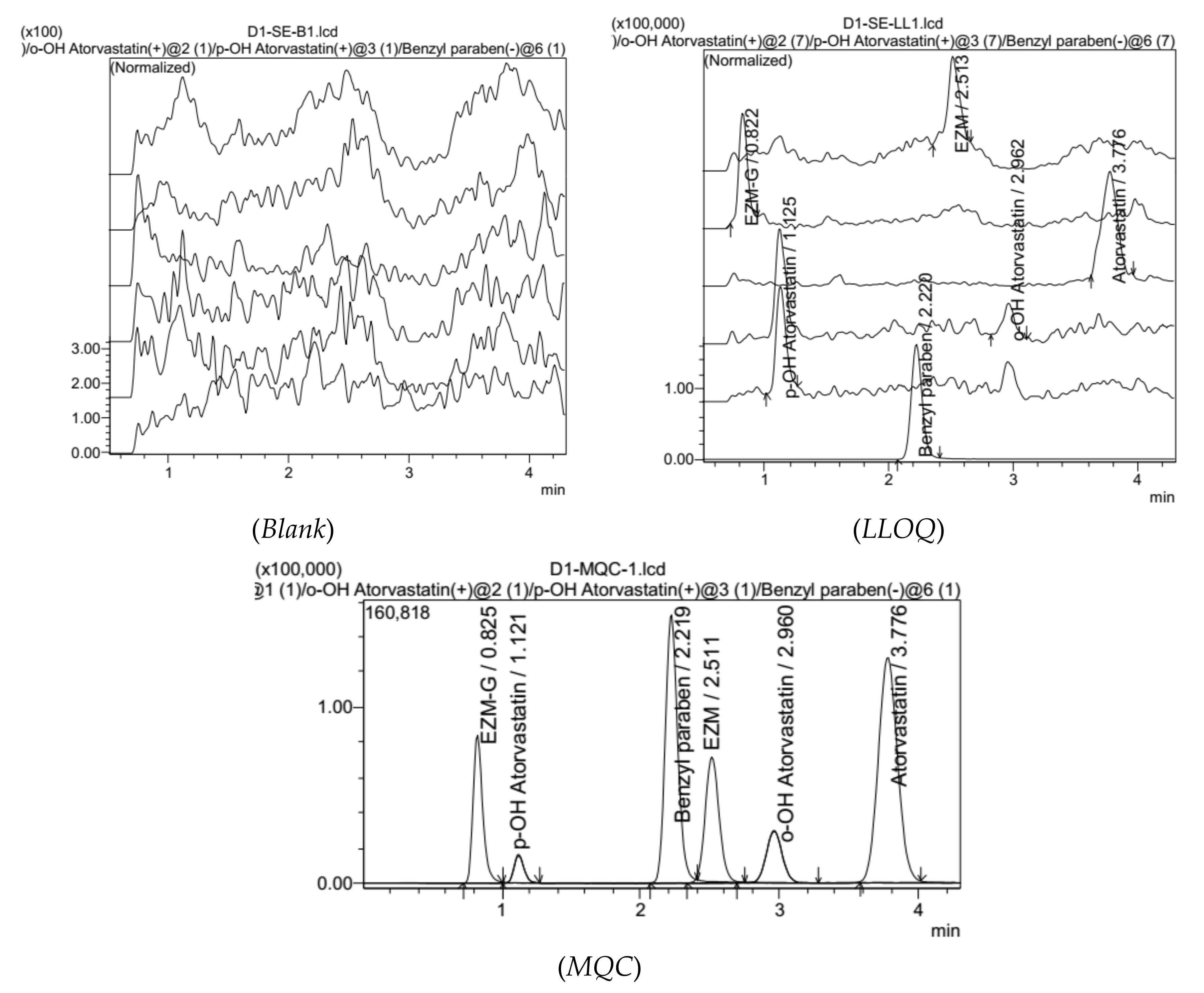
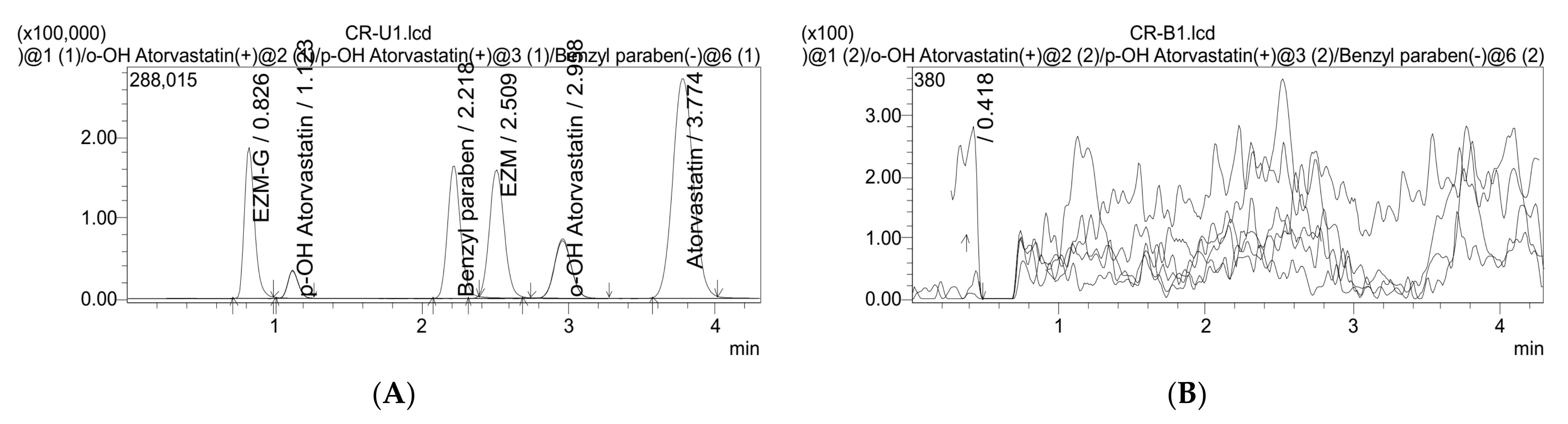
3.3.1. Specifications, Selectivity
3.3.2. Linearity
3.3.3. Recovery and Matrix Effect
3.3.4. Accuracy and Precision
3.3.5. Low Limit of Qualification
3.3.6. Carry-Over
3.3.7. Stability
3.4. Comparison to Previous Analytical Methods
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Lynne, T.; De Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, e1082–e1143. [Google Scholar]
- Lennernäs, H. Clinical pharmacokinetics of atorvastatin. Clin. Pharmacokinet. 2003, 42, 1141–1160. [Google Scholar] [CrossRef] [PubMed]
- FDA. Draft Guidance on Atorvastatin Calcium and Ezetimibe; FSA: Silver Spring, ML, USA, 2014.
- Kosoglou, T.; Statkevich, P.; Johnson-Levonas, A.; Paolini, J.; Bergman, A.; Alton, K. Ezetimibe: A review of its metabolism, pharmacokinetics and drug interactions. Clin. Pharmacokinet. 2005, 44, 467–494. [Google Scholar] [CrossRef] [PubMed]
- EMA. Ezetimibe Tablet 10 mg Product Specific Bioequivalence Guidance; EMA: Amsterdam, The Netherlands, 2019.
- Macwan, J.S.; Ionita, I.; Dostalek, M.; Akhlaghi, F. Development and validation of a sensitive, simple, and rapid method for simultaneous quantitation of atorvastatin and its acid and lactone metabolites by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Anal. Bioanal. Chem. 2011, 400, 423–433. [Google Scholar] [CrossRef]
- Ghosh, C.; Jain, I.; Gaur, S.; Patel, N.; Upadhyay, A.; Chakraborty, B. Simultaneous estimation of atorvastatin and its two metabolites from human plasma by ESI-LC-MS/MS. Drug Test. Anal. 2011, 3, 352–362. [Google Scholar] [CrossRef]
- El-Zailik, A.; Cheung, L.; Wang, Y.; Sherman, V.; Chow, D.S.-L. Simultaneous LC–MS/MS analysis of simvastatin, atorvastatin, rosuvastatin and their active metabolites for plasma samples of obese patients underwent gastric bypass surgery. J. Pharm. Biomed. Anal. 2019, 164, 258–267. [Google Scholar] [CrossRef]
- Hermann, M.; Christensen, H.; Reubsaet, J.L.E. Determination of atorvastatin and metabolites in human plasma with solid-phase extraction followed by LC–tandem MS. Anal. Bioanal. Chem. 2005, 382, 1242–1249. [Google Scholar] [CrossRef]
- Nirogi, R.V.S.; Kandikere, V.N.; Shukla, M.; Mudigonda, K.; Maurya, S.; Boosi, R.; Anjaneyulu, Y. Simultaneous quantification of atorvastatin and active metabolites in human plasma by liquid chromatography–tandem mass spectrometry using rosuvastatin as internal standard. Biomed. Chromatogr. 2006, 3, 924–936. [Google Scholar] [CrossRef]
- Partani, P.; Verma, M.; Gurule, S.; Khuroo, A.; Monif, T. Simultaneous quantitation of atorvastatin and its two active metabolites in human plasma by liquid chromatography/(–) electrospray tandem mass spectrometry. J. Pharm. Anal. 2014, 4, 26–36. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, J.; He, X.; Jia, M.; Liu, M.; Li, H.; Xiong, Z.; Fan, Y.; Li, W. Development and validation of a liquid chromatography–tandem mass spectrometry method for simultaneous determination of amlodipine, atorvastatin and its metabolites ortho-hydroxy atorvastatin and para-hydroxy atorvastatin in human plasma and its application in a bioequivalence study. J. Pharm. Biomed. Anal. 2013, 83, 101–107. [Google Scholar] [CrossRef]
- Borek-Dohalský, V.; Huclová, J.; Barrett, B.; Nemec, B.; Ulc, I.; Jelínek, I. Validated HPLC–MS–MS method for simultaneous determination of atorvastatin and 2-hydroxyatorvastatin in human plasma—Pharmacokinetic study. Anal. Bioanal. Chem. 2006, 386, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Crevar-Sakač, M.; Vujić, Z.; Brborić, J.; Kuntić, V.; Uskoković-Marković, S. An improved HPLC method with the aid of a chemometric protocol: Simultaneous determination of atorvastatin and its metabolites in plasma. Molecules 2013, 18, 2469–2482. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Zheng, Z.; Wang, X.; Tang, L.; Mai, L.; He, G.; Lei, H.; Zhong, S. Simultaneous determination of atorvastatin and its metabolites in human plasma by UPLC-MS/MS. Anal. Methods 2017, 9, 1038–1045. [Google Scholar] [CrossRef]
- Yang, Y.; Xu, Q.; Zhou, L.; Zhong, D.; Chen, X. High-throughput salting-out-assisted liquid–liquid extraction for the simultaneous determination of atorvastatin, ortho-hydroxyatorvastatin, and para-hydroxyatorvastatin in human plasma using ultrafast liquid chromatography with tandem mass spectrometry. J. Sep. Sci. 2015, 38, 1026–1034. [Google Scholar] [CrossRef]
- Bahrami, G.; Mohammadi, B.; Khatabi, P.M.; Farzaei, M.H.; Majnooni, M.B.; Bahoosh, S.R. Application of one-step liquid chromatography–electrospray tandem MS/MS and collision-induced dissociation to quantification of ezetimibe and identification of its glucuronated metabolite in human serum: A pharmacokinetic study. J. Chromatogr. B 2010, 878, 2789–2795. [Google Scholar] [CrossRef]
- Bae, J.-W.; Choi, C.-I.; Park, S.-H.; Jang, C.-G.; Lee, S.-Y. Analytical LC-MS/MS method for ezetimibe and its application for pharmacokinetic study. J. Liq. Chromatogr. Relat. Technol. 2012, 35, 141–152. [Google Scholar] [CrossRef]
- Danafar, H.; Hamidi, M. A rapid and sensitive LC–MS method for determination of ezetimibe concentration in human plasma: Application to a bioequivalence study. Chromatographia 2013, 76, 1667–1675. [Google Scholar] [CrossRef]
- Guo, L.; Wang, M.-M.; He, M.; Qiu, F.-R.; Jiang, J. Simultaneous determination of ezetimibe and its glucuronide metabolite in human plasma by solid phase extraction and liquid chromatography–tandem mass spectrometry. J. Chromatogr. B 2015, 986, 108–114. [Google Scholar] [CrossRef]
- Abdelbary, G.; Nebsen, M. Application of a novel UPLC-MS/MS method for the pharmacokinetic/bioequivalence determination of atorvastatin and ezetimibe in human plasma. J. Pharm. Res. 2013, 7, 24–32. [Google Scholar] [CrossRef]
- Gowda, K.V.; Rajan, D.S.; Mandal, U.; Solomon, W.D.S.; Selvan, P.S.; Bose, A.; Sarkar, A.K.; Chattaraj, T.K.; Pal, T.K. Bioequivalence Study of Fixed Dose Combination of Atorvastatin and Ezetimibe Tablet in Healthy Volunteers by LC-MS/MS Method. Asian J. Chem. 2007, 19, 1293–1300. [Google Scholar]
- El-Bagary, R.I.; Elkady, E.F.; El-Sherif, Z.A.; Kadry, A.M. LC–MS–MS Simultaneous Determination of Atorvastatin and Ezetimibe in Human Plasma. J. Chromatogr. Sci. 2013, 52, 773–780. [Google Scholar] [CrossRef]
- Munaga, S.B.; Valluru, R.K.; Bonga, P.B.R.; Rao, S.; Sharma, H.K. Development and validation of an LC–MS-MS method for the simultaneous determination of simvastatin, simvastatin acid and ezetimibe in human plasma and its application to pharmacokinetic study in the Indian population. J. Chromatogr. Sci. 2016, 54, 985–996. [Google Scholar] [CrossRef]
- Elawady, T.; Ibrahim, F.; Khedr, A.; Belal, F. Simultaneous determination of ezetimibe, atorvastatin and simvastatin using quadrupole LC-MS: Application to combined tablets and plasma after SPE. Acta Chromatogr. 2021, 33, 245–252. [Google Scholar] [CrossRef]
- Ashfaq, M.; Ahmad, H.; Khan, I.U.; Mustafa, G. LC determination of rosuvastatin and ezetimibe in human plasma. J. Chil. Chem. Soc. 2013, 58, 2177–2181. [Google Scholar] [CrossRef]
- Bhadoriya, A.; Sanyal, M.; Shah, P.A.; Shrivastav, P.S. Simultaneous quantitation of rosuvastatin and ezetimibe in human plasma by LC–MS/MS: Pharmacokinetic study of fixed-dose formulation and separate tablets. Biomed. Chromatogr. 2018, 32, e4291. [Google Scholar] [CrossRef] [PubMed]
- Varghese, S.J.; Kochupappy, R.T. Development and validation of a liquid chromatography/mass spectrometry method for the simultaneous quantitation of rosuvastatin and ezetimibe in human plasma. J. AOAC Int. 2013, 96, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Elkady, E.F.; Jaadan, B.M.; Ramadan, E.; Elmaaty, A.A. Bioanalytical LC–MS/MS method for simultaneous estimation of atorvastatin, its major active metabolites and ezetimibe. Bioanalysis 2022, 14, 1349–1363. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, H.; Kim, E.; El-Shourbagy, T. Salting-out assisted liquid/liquid extraction with acetonitrile: A new high throughput sample preparation technique for good laboratory practice bioanalysis using liquid chromatography–mass spectrometry. Biomed. Chromatogr. 2009, 23, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.Q.; Weng, N. Salting-out assisted liquid–liquid extraction for bioanalysis. Bioanalysis 2013, 5, 1583–1598. [Google Scholar] [CrossRef] [PubMed]
- FDA. Guidance for Industry: Bioanalytical Method Validation; FSA: Silver Spring, ML, USA, 2018.
- EMA. Guideline on Bioanalytical Method Validation; EMA: Amsterdam, The Netherlands, 2015.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | Sources | Parent Ions (m/z) | CE (V) | Daughter Ions (m/z) | |
|---|---|---|---|---|---|
| Quantitative Ions | Reference Ions | ||||
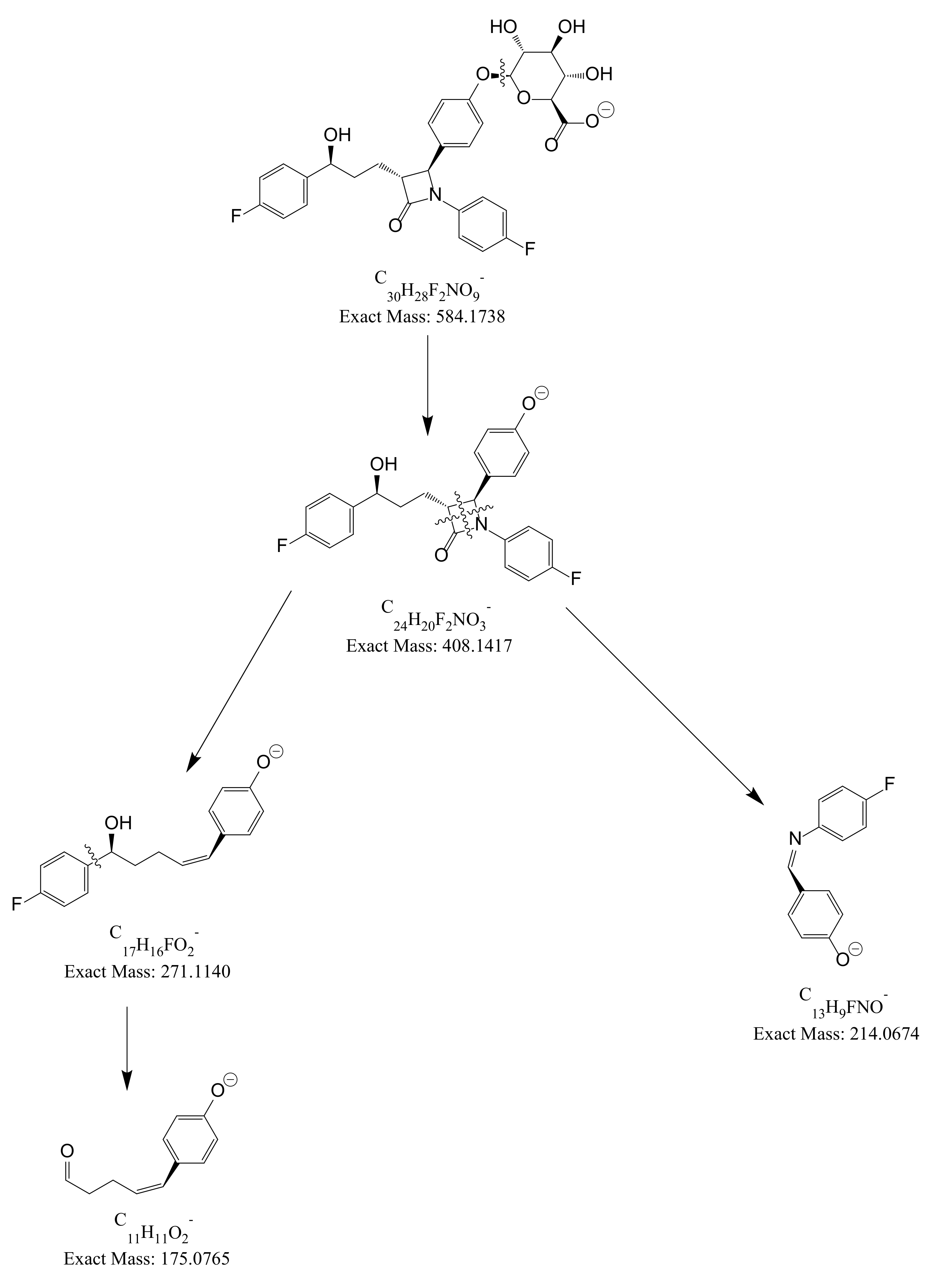
| EZM | ESI (−) | 408 | 17 | 271 | 284, 214, 175 |
| EZM-G | ESI (−) | 584 | 31 | 271 | 284, 214, 175 |
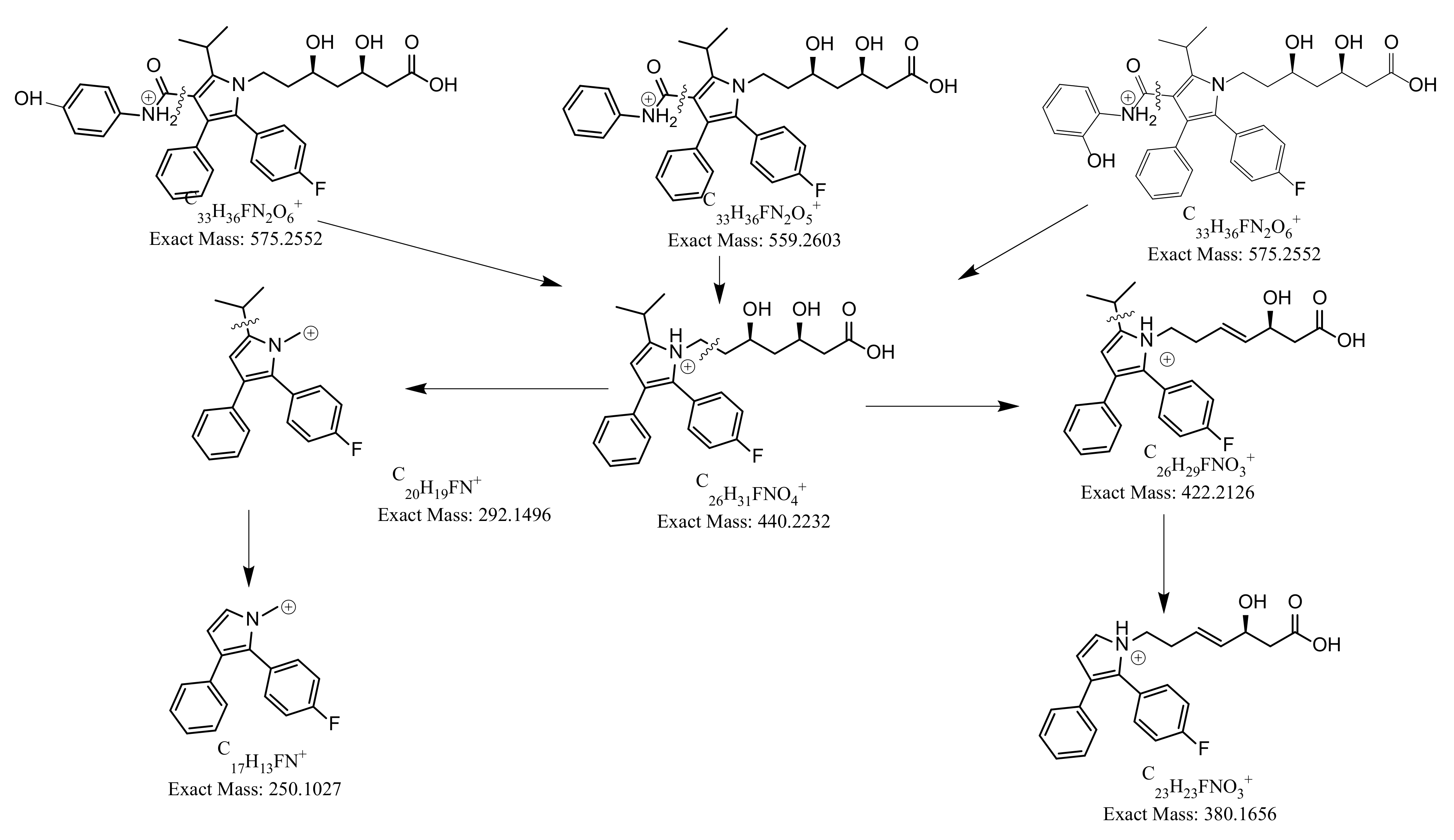
| ATOR | ESI (+) | 559 | −23 | 440 | 380, 292, 250 |
| o-OH ATOR | ESI (+) | 575 | −24 | 440 | 380, 292, 250 |
| p-OH ATOR | ESI (+) | 575 | −24 | 440 | 380, 292, 250 |
| Analyte | Range (ng/mL) | Equation = ax + b, Weighting Factor 1/x2 | ||
|---|---|---|---|---|
| a | b | R2 | ||
| EZM | 0.06–15 | 0.0688 | 0.0007 | 0.9974 |
| EZM-G | 0.6–150 | 0.0060 | −0.0001 | 0.9941 |
| ATOR | 0.4–100 | 0.0265 | −0.0008 | 0.9951 |
| o-OH ATOR | 0.12–30 | 0.0147 | −0.0003 | 0.9959 |
| p-OH ATOR | 0.05–3 | 0.0595 | −0.0001 | 0.9932 |
| Analyte | Conc. Added (ng/mL) | Accuracy (%) Mean (CV) | Recovery (n = 6) (%) | Matrix Effect (n = 6) | ||
|---|---|---|---|---|---|---|
| Intraday (n = 6) | Interday (3 days, n = 18) | MF (%) | IS-Normalized MFs (%) | |||
| EZM | 0.061 | 111.69 (5.86) | 98.77 (11.79) | - | - | - |
| 0.184 | 105.77 (4.69) | 103.09 (6.01) | 68.57 ± 1.91 | 106.58 ± 9.47 | 113.5 ± 9.46 | |
| 7.664 | 103.17 (1.66) | 101.49 (2.20) | 82.62 ± 3.86 | - | - | |
| 12.263 | 102.17 (1.89) | 101.28 (3.61) | 82.07 ± 3.28 | 90.98 ± 1.82 | 100.56 ± 2.19 | |
| EZM-G | 0.600 | 102.32 (6.65) | 97.09 (8.84) | - | ||
| 1.801 | 96.13 (3.33) | 99.91 (5.76) | 91.50 ± 5.19 | 85.94 ± 1.13 | 75.72 ± 5.99 | |
| 75.029 | 96.19 (2.41) | 95.29 (2.33) | 85.61 ± 4.80 | - | ||
| 120.046 | 96.42 (2.99) | 94.45 (2.78) | 87.10 ± 2.68 | 91.30 ± 1.88 | 90.79 ± 2.35 | |
| ATOR | 0.400 | 112.68 (4.05) | 104.52 (7.73) | - | - | - |
| 1.200 | 99.00 (3.86) | 103.06 (5.66) | 82.09 ± 4.23 | 104.03 ± 1.95 | 110.78 ± 2.95 | |
| 50.000 | 98.53 (1.62) | 102.79 (3.33) | 81.48 ± 4.26 | - | - | |
| 79.999 | 96.56 (2.81) | 100.98 (5.39) | 81.39 ± 3.09 | 98.24 ± 1.74 | 108.59 ± 2.25 | |
| o-OH ATOR | 0.122 | 103.51 (14.18) | 99.17 (11.50) | - | - | - |
| 0.366 | 107.71 (4.47) | 102.80 (7.43) | 74.37 ± 3.27 | 103.11 ± 3.58 | 93.07 ± 3.08 | |
| 15.261 | 113.82 (0.38) | 108.33 (6.83) | 75.37 ± 4.13 | - | - | |
| 24.418 | 111.33 (2.31) | 107.12 (5.75) | 77.36 ± 3.55 | 99.76 ± 1.73 | 91.87 ± 1.14 | |
| p-OH ATOR | 0.050 | 98.56 (5.22) | 101.98 (6.18) | - | - | - |
| 0.149 | 98.80 (4.31) | 100.76 (7.85) | 80.69 ± 4.27 | 129.18 ± 5.11 | 137.56 ± 5.67 | |
| 1.548 | 92.51 (1.81) | 101.24 (8.19) | 80.13 ± 4.80 | - | - | |
| 2.476 | 89.56 (0.75) | 101.46 (9.99) | 83.02 ± 2.11 | 129.28 ± 1.32 | 142.9 ± 0.86 | |
| IS | 50.000 | 80.01 ± 3.35 * | 92.19 ± 1.93 ** | - | ||
| Compound | Stock Standard Solution | Plasma | Wet Extraction | |||||
|---|---|---|---|---|---|---|---|---|
| Conc. (μg/mL) | Bench-Top | Long-Term | Conc. (ng/mL) | Bench-Top | Freeze-Thaw | Long-Term | 45 h at 15 °C in an Autosampler | |
| EZM | 187.5 | 100.52 (0.82) | 100.80 (0.92) | 0.184 | 104.07 (4.45) | 106.50 (4.92) | 103.86 (6.74) | 103.78 (3.17) |
| 12.263 | 104.91 (0.75) | 104.54 (1.06) | 100.52 (0.71) | 105.45 (1.34) | ||||
| EZM-G | 100 | 94.23 (2.66) | 106.14 (0.89) | 1.801 | 93.76 (3.47) | 93.96 (5.44) | 98.06 (3.58) | 97.92 (5.02) |
| 120.046 | 93.64 (1.59) | 92.86 (1.49) | 100.70 (2.09) | 92.30 (2.43) | ||||
| ATOR | 500 | 103.16 (2.13) | 99.88 (0.67) | 1.200 | 100.87 (2.86) | 102.03 (2.98) | 98.61 (1.71) | 106.52 (3.98) |
| 79.999 | 105.99 (0.62) | 106.44 (1.03) | 99.80 (4.77) | 107.29 (0.85) | ||||
| o-OH ATOR | 60 | 98.66 (2.76) | 100.81 (0.71) | 0.366 | 94.46 (4.53) | 94.54 (3.64) | 88.88 (91.61) | 92.62 (5.47) |
| 24.418 | 97.09 (0.59) | 96.53 (1.11) | 103.76 (6.70) | 97.36 (1.35) | ||||
| p-OH ATOR | 120 | 101.50 (5.34) | 106.08 (1.36) | 0.149 | 100.57 (6.75) | 92.98 (7.24) | 100.39 (2.51) | 101.55 (8.08) |
| 2.476 | 100.04 (2.30) | 99.54 (1.04) | 108.85 (2.27) | 99.45 (2.16) | ||||
| IS | 100 | 101.88 (2.03) | 98.97 (1.00) | 50 | - | - | - | 97.47 (1.06) |
| Refs. | Run Time | Extraction Technique | Analyte | LLOQ (ng/mL) | Extraction Efficiency (%) | Matrix Effect | ||
|---|---|---|---|---|---|---|---|---|
| MF | MF/IS | CV | ||||||
| [6] | 7 min | PPT | ATOR | 0.05 | 88.6–111 | Not available | ||
| o-OH ATOR | 0.05 | Not available | ||||||
| p-OH ATOR | 0.05 | Not available | ||||||
| [7] | 3.5 min | SPE | ATOR | 0.05 | 66.18 | 97.51–98.7 | - | 0.96–4.22 |
| o-OH ATOR | 0.05 | 45.36 | 98.90–101.12 | - | 1.32–4.10 | |||
| p-OH ATOR | 0.05 | 54.01 | 94.42–100.19 | - | 1.12–4.48 | |||
| [8] | 5 min | LLE | ATOR | 0.25 | 96.94–100.37 | 97.45–110.42 | - | - |
| o-OH ATOR | 0.25 | 92.15–97.71 | 96.50–102.93 | - | - | |||
| p-OH ATOR | 0.25 | 96.97–99.17 | 96.60–101.84 | - | - | |||
| [9] | 20 min | SPE | ATOR | 0.5 | 53–78 | Not available | ||
| o-OH ATOR | 1 | Not available | ||||||
| p-OH ATOR | 0.5 | Not available | ||||||
| [10] | 3 min | LLE | ATOR | 0.1 | 51.0–57.3 | Not available | ||
| o-OH ATOR | 0.1 | 46.8–54.3 | Not available | |||||
| p-OH ATOR | 0.1 | 61.6–68.8 | Not available | |||||
| [11] | 6 min | SPE | ATOR | 0.05 | 76.3–78.0 | 92–94 | 101–106 | <4.9 |
| o-OH ATOR | 0.05 | 73.1–75.1 | 92.97 | 99–101 | 4.9 | |||
| p-OH ATOR | 0.05 | 72.6–75.7 | 105–108 | 96–105 | 4.9 | |||
| [12] | 6 min | LLE | ATOR | 0.035 | 77.23–82.69 | 99.34–101.25 | - | 1.44–4.77 |
| o-OH ATOR | 0.02 | 76.39–81.96 | 90.29–97.61 | - | 1.58–8.3 | |||
| p-OH ATOR | 0.015 | 78.24–80.29 | 95.74–97.61 | - | 4.31–5.03 | |||
| [13] | 8 min | LLE | ATOR | 0.02 | 91.5 | - | - | 10.5 |
| o-OH ATOR | 0.07 | 83.7 | - | - | 12.5 | |||
| [14] | 9 min | SPE | ATOR | Not tested | Not tested | Not tested | - | - |
| o-OH ATOR | Not tested | Not tested | Not tested | - | - | |||
| [15] | 4 min | LLE | ATOR | 0.2 | 47.9–49.2 | 102.7–105.5 | - | - |
| o-OH ATOR | 0.2 | 49.2–50.5 | 103.4–107.0 | - | - | |||
| p-OH ATOR | 0.2 | 27.8–31.2 | 101.0–102.9 | - | - | |||
| [16] | 2 min | SALLE | ATOR | 0.02 | 81.1–94.0 | - | 93.9–101 | <7.2 |
| o-OH ATOR | 0.02 | 85.6–89.0 | - | 92.6–101 | <7.2 | |||
| p-OH ATOR | 0.01 | 89.9–102 | - | 93.6–97 | <7.2 | |||
| [17] | 5 min | PPT | EZM | 10 | - | - | - | - |
| EZM-G | Identification | - | - | - | - | |||
| [18] | 4.5 min | LLE | EZM | 0.075 | 61.6 | - | >93.7 | - |
| LLE following deconjugation | Total EZM | 1 | 42.0 | - | >93.7 | - | ||
| [19] | 10 min | LLE | EZM | 0.05 | 96.21–97.27 | Not available | ||
| [20] | 5.5 min | SPE | EZM | 0.1 | 65.3–72.2 | 87.0–104.8 | - | <7.0 |
| EZM-G | 0.5 | 58.6–61.2 | 91.9–102.1 | - | <7.0 | |||
| [21] | 1.2 min | LLE | ATOR | 0.1 | 94.4–95.8 | Not tested | ||
| EZM | 0.1 | 92.6–95.0 | Not tested | |||||
| [22] | - | - | ATOR | - | Not tested | Not tested | ||
| EZM | - | Not tested | Not tested | |||||
| [23] | 5 min | LLE | ATOR | 0.2 | 75.07–76.06 | 96.58–97.90 | - | - |
| EZM | 0.2 | 68.89–70.49 | 101.32–103.61 | - | - | |||
| [29] | LLE | ATOR | 0.5 | 84.94 | - | - | 2.43–6.05 | |
| o-OH ATOR | 0.5 | 85.46 | - | - | 0.72–2.42 | |||
| p-OH ATOR | 0.2 | 105.46 | - | - | 0.98–6.99 | |||
| EZM | 0.2 | 85.2 | - | - | 1.63–2.16 | |||
| This study | 4.3 min | SALLE | ATOR | 0.4 | 81.39–82.09 | 98.24–104.03 | 108.59–110.78 | 2.08–2.66 |
| o-OH ATOR | 0.12 | 74.37–77.36 | 99.76–103.11 | 91.87–93.07 | 1.97–4.12 | |||
| p-OH ATOR | 0.05 | 80.13–83.02 | 129.18–129.28 | 137.56–142.9 | 0.60–4.12 | |||
| EZM | 0.06 | 68.57–82.62 | 90.98–106.58 | 100.56–113.5 | 2.18–8.34 | |||
| EZM-G | 0.6 | 85.61–91.50 | 85.94–91.30 | 75.72–90.79 | 1.89–2.27 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, T.N.N.; Chuong, N.N.; Nguyen, T.D. A One-Step Sample Processing Method in Combination with HPLC-MS/MS for the Simultaneous Quantification of Atorvastatin, Ezetimibe and Three Metabolites including o-Hydroxyl Atorvastatin, p-Hydroxyl Atorvastatin, and Ezetimibe-Glucuronide in Human Plasma. Separations 2023, 10, 409. https://doi.org/10.3390/separations10070409
Le TNN, Chuong NN, Nguyen TD. A One-Step Sample Processing Method in Combination with HPLC-MS/MS for the Simultaneous Quantification of Atorvastatin, Ezetimibe and Three Metabolites including o-Hydroxyl Atorvastatin, p-Hydroxyl Atorvastatin, and Ezetimibe-Glucuronide in Human Plasma. Separations. 2023; 10(7):409. https://doi.org/10.3390/separations10070409
Chicago/Turabian StyleLe, T. Nguyen Nguyen, Nai Ngoc Chuong, and Tuan Duc Nguyen. 2023. "A One-Step Sample Processing Method in Combination with HPLC-MS/MS for the Simultaneous Quantification of Atorvastatin, Ezetimibe and Three Metabolites including o-Hydroxyl Atorvastatin, p-Hydroxyl Atorvastatin, and Ezetimibe-Glucuronide in Human Plasma" Separations 10, no. 7: 409. https://doi.org/10.3390/separations10070409
APA StyleLe, T. N. N., Chuong, N. N., & Nguyen, T. D. (2023). A One-Step Sample Processing Method in Combination with HPLC-MS/MS for the Simultaneous Quantification of Atorvastatin, Ezetimibe and Three Metabolites including o-Hydroxyl Atorvastatin, p-Hydroxyl Atorvastatin, and Ezetimibe-Glucuronide in Human Plasma. Separations, 10(7), 409. https://doi.org/10.3390/separations10070409