
Sensitivity Enhancement for Separation-Based Analytical Techniques Utilizing Solid-Phase Enrichment Approaches and Analyte Derivatization for Trace Analysis in Various Matrices
 , ,
, , 
Abstract
1. Introduction
2. Solid-Phase Extraction/Pre-Concentration Strategies for Drug Analysis
2.1. Offline Solid-Phase Extraction/Enrichment
2.2. Offline Solid-Phase Microextraction
3. Column Switching and Large Volume Sample Injection
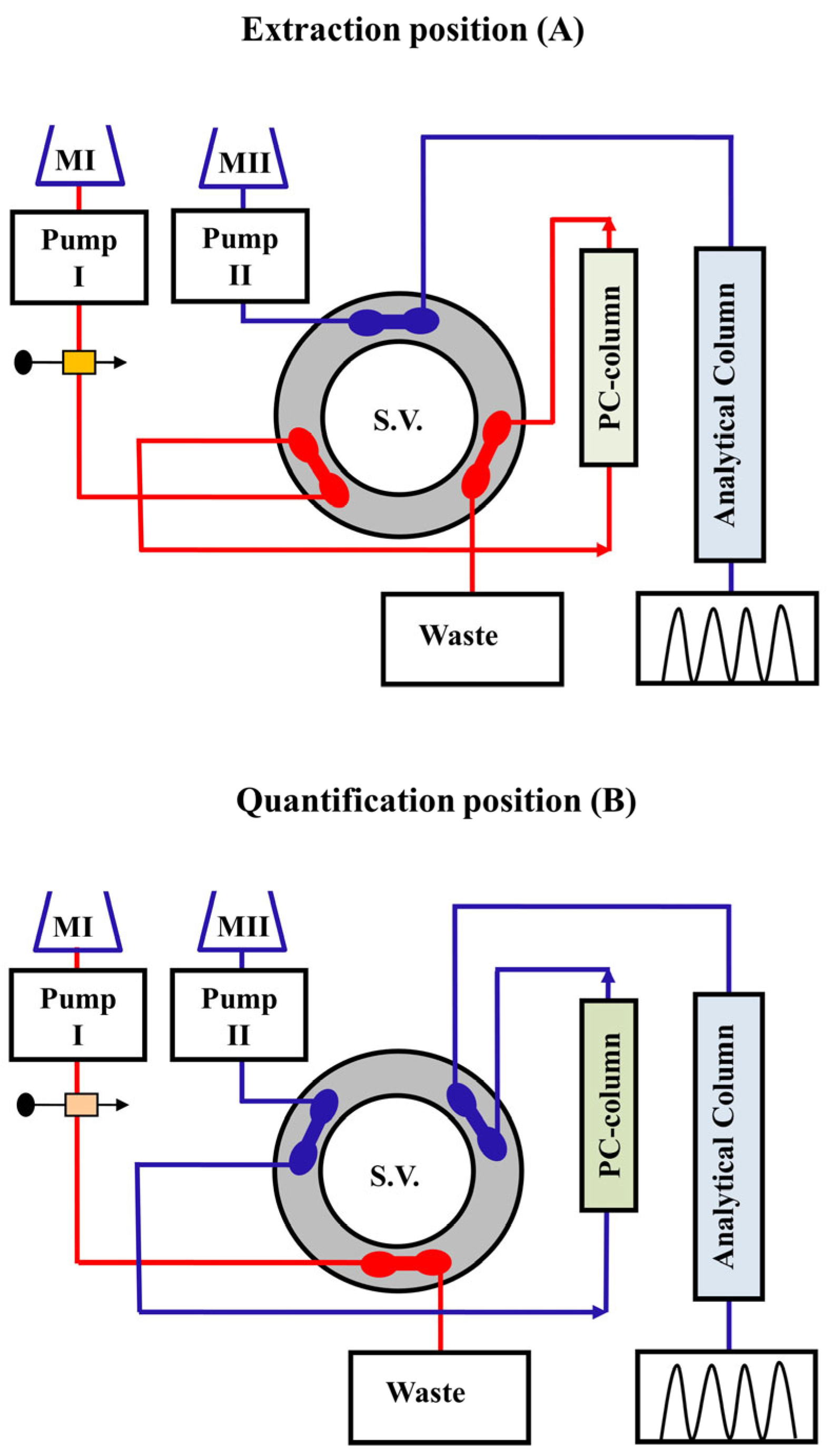
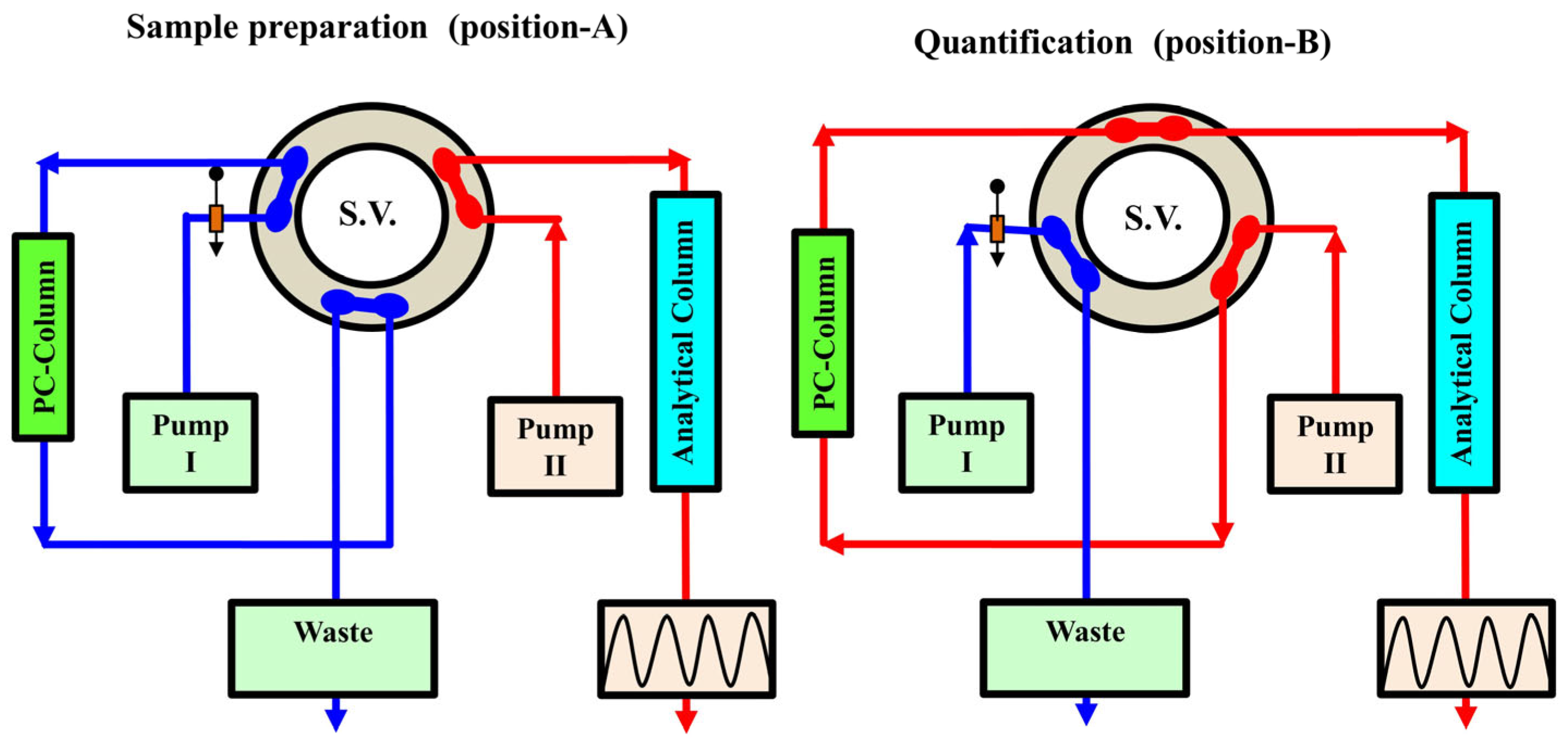
3.1. Online Solid-Phase Extraction and Solid-Phase Enrichment
3.2. Immobilized Metal Affinity Chromatography
3.3. Immobilized Protein Reversed Phase Columns for Solid-Phase Extraction/Enrichment
3.4. Restricted Access Media Solid-Phase Extraction/Enrichment
3.5. Surface Modified Restricted Access Media
3.6. Restricted Access Media Molecularly Imprinted Polymers
3.7. N-Rich Twin-Column Continuous Chromatography
4. Enhancement of Sensitivity through Chemical Modification
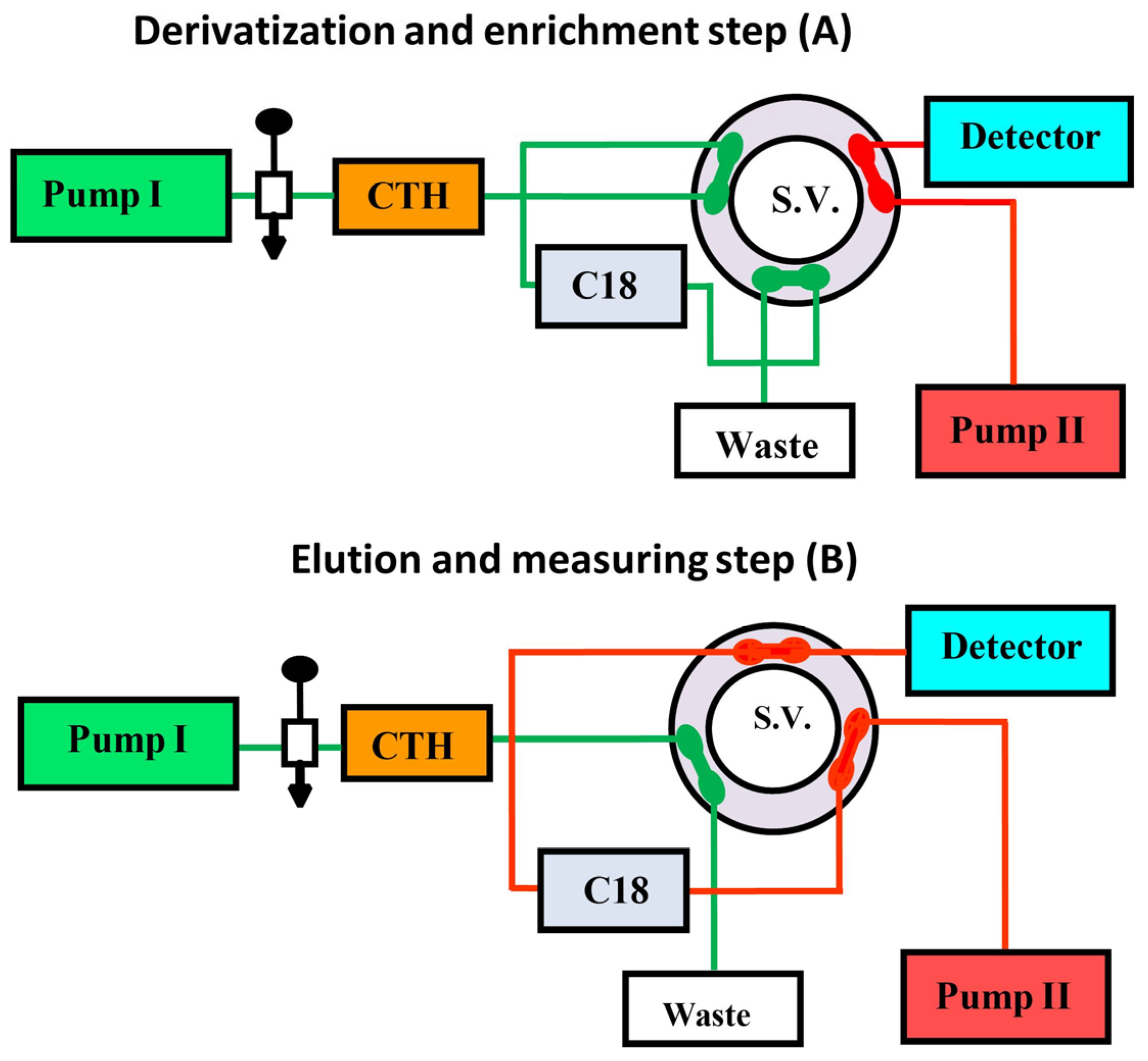
4.1. Solid-Phase Analytical Derivatization
4.2. Solid-Phase Permethylation
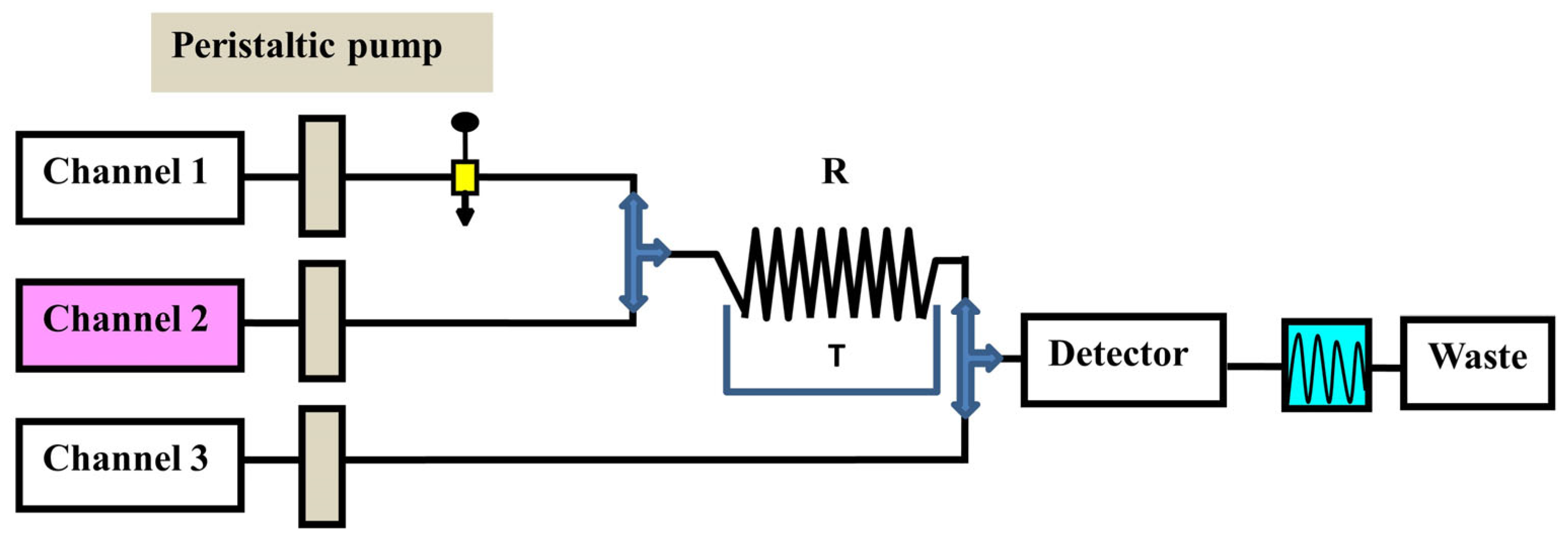
4.3. Packed Oxidant Reactors
5. Conclusions
6. Challenges and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aydin, D.C.; Pineres, J.Z.; Al-Manji, F.; Rijnaarts, H.; Grotenhuis, T. Direct Analysis of Aromatic Pollutants Using an HPLC-FLD/DAD Method for Monitoring Biodegradation Processes. Anal. Methods 2021, 13, 1635–1642. [Google Scholar] [CrossRef]
- Chiriac, U.; Rau, H.; Frey, O.R.; Röhr, A.C.; Klein, S.; Meyer, A.L.; Morath, B. Validation and Application of an HPLC-UV Method for Routine Therapeutic Drug Monitoring of Dalbavancin. Antibiotics 2022, 11, 541. [Google Scholar] [CrossRef]
- Al-Sanea, M.M.; Gamal, M. Critical Analytical Review: Rare and Recent Applications of Refractive Index Detector in HPLC Chromatographic Drug Analysis. Microchem. J. 2022, 178, 107339. [Google Scholar] [CrossRef]
- Marzouk, H.M.; Rezk, M.R.; Gouda, A.S.; Abdel-Megied, A.M. A Novel Stability-Indicating HPLC-DAD Method for Determination of Favipiravir, a Potential Antiviral Drug for COVID-19 Treatment; Application to Degradation Kinetic Studies and In-Vitro Dissolution Profiling. Microchem. J. 2022, 172, 106917. [Google Scholar] [CrossRef] [PubMed]
- El-Gindy, A.; Emara, S.; Mesbah, M.K.; Hadad, G.M. Liquid Chromatography Determination of Citalopram Enantiomers Using Beta-Cyclodextrin as a Chiral Mobile Phase Additive. J. AOAC Int. 2006, 89, 65–70. [Google Scholar]
- El-Gindy, A.; Emara, S.; Mesbah, M.K.; Hadad, G.M. Liquid Chromatography and Chemometric-Assisted Spectrophotometric Methods for the Analysis of Two Multicomponent Mixtures Containing Cough Suppressant Drugs. J. AOAC Int. 2005, 88, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Hadad, G.M.; Emara, S.; Mahmoud, W.M.M. Optimization and Validation of an LC Method for the Determination of Cefdinir in Dosage form and Human Urine. Chromatographia 2009, 70, 1593–1598. [Google Scholar] [CrossRef]
- EL-Gindy, A.; Emara, S.; Shaaban, H. Stability-Indicating Method for Determination of Oxyphenonium Bromide and Its Degradation Product by High-Performance Liquid Chromatography. J. AOAC Int. 2007, 90, 1250–1257. [Google Scholar] [CrossRef]
- Mostafa, A.; El-Gindy, A.; Emara, S. Development, Application and Validation of RP-HPLC Method for the Simultaneous Determination of Butamirate Citrate and its Main Degradation Product in Pharmaceutical Dosage forms. Anal. Methods 2011, 3, 1643–1651. [Google Scholar] [CrossRef]
- El-Gindy, A.; Emara, S.; Mostafa, A. HPLC and Chemometric-Assisted Spectrophotometric Methods for Simultaneous Determination of Atenolol, Amiloride Hydrochloride and Chlorthalidone. Il Farm. 2005, 60, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Hadad, G.M.; Salam, R.A.A.; Emara, S. Validated Stability—Indicating HPTLC and HPLC Methods for Determination of Pipazethate and its Degradant. J. Liq. Chromatogr. Relat. Technol. 2011, 34, 1850–1869. [Google Scholar] [CrossRef]
- Hadad, G.M.; Emara, S.; Mahmoud, W.M.M. Development and Validation of a Stability-Indicating RP-HPLC Method for the Determination of Paracetamol with Dantrolene or/and Cetirizine and Pseudoephedrine in Two Pharmaceutical Dosage forms. Talanta 2009, 79, 1360–1367. [Google Scholar] [CrossRef]
- Rabe, M.; Verdes, D.; Seeger, S. Understanding Protein Adsorption Phenomena at Solid Surfaces. Adv. Colloid Interface Sci. 2011, 162, 87–106. [Google Scholar] [CrossRef]
- Turner, N.W.; Subrahmanyam, S.; Piletsky, S.A. Analytical Methods for Determination of Mycotoxins: A Review. Anal. Chim. Acta 2009, 632, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, E.d. Sample Preparation for Atomic Spectroscopy: Evolution and Future Trends. J. Braz. Chem. Soc. 2003, 14, 174–182. [Google Scholar] [CrossRef]
- Urge, A.Y.; Pampanin, D.M.; Martino, M.E.; Knudsen, D.L.; Brede, C. Salting-Out Assisted Liquid-Liquid Extraction for UPLC-MS/MS Determination of Thyroxine and Steroid Hormones in Human Serum and Fish Plasma. Separations 2023, 10, 240. [Google Scholar] [CrossRef]
- Zygler, A.; Wasik, A.; Namieśnik, J. Retention Behaviour of Some High-Intensity Sweeteners on Different SPE Sorbents. Talanta 2010, 82, 1742–1748. [Google Scholar] [CrossRef]
- Badawy, M.E.I.; El-Nouby, M.A.M.; Kimani, P.K.; Lim, L.W.; Rabea, E.I. Review of the Modern Principles and Applications of Solid-Phase Extraction Techniques in Chromatographic Analysis. Anal. Sci. 2022, 38, 1457–1487. [Google Scholar] [CrossRef] [PubMed]
- Hadad, G.M.; Abdel Salam, R.A.; Emara, S. Validated and Optimized High-Performance Liquid Chromatographic Determination of Tizoxanide, the Main Active Metabolite of Nitazoxanide in Human Urine, Plasma and Breast Milk. J. Chromatogr. Sci. 2012, 50, 509–515. [Google Scholar] [CrossRef]
- Ebrahim, H.; Sonbol, H.; Malak, M.; Ali, A.; Aboulella, Y.; Hadad, G.; Zarad, W.; Emara, S.; Bazan, L. Green Automated Solid Phase Extraction to Measure Levofloxacin in Human Serum via Liquid Chromatography with Fluorescence Detection for Pharmacokinetic Study. Separations 2023, 10, 136. [Google Scholar] [CrossRef]
- Sonbol, H.; Ebrahim, H.; Malak, M.; Ali, A.; Aboulella, Y.; Hadad, G.; Emara, S.; Shawky, A. Application of a Small Protein-Coated Column to Trap, Extract and Enrich Carbamazepine Directly from Human Serum for Direct Chromatographic Analysis. Separations 2023, 10, 71. [Google Scholar] [CrossRef]
- Abughrin, S.E.; Alshana, U.; Bakirdere, S. Magnetic Nanoparticle-Based Dispersive Solid-Phase Microextraction of Three UV Blockers Prior to their Determination by HPLC-DAD. Int. J. Environ. Res. Public Health 2022, 19, 6037. [Google Scholar] [CrossRef]
- Nishida, M.; Namera, A.; Yashiki, M.; Kojima, T. On-Column Derivatization for Determination of Amphetamine and Methamphetamine in Human Blood by Gas Chromatography–Mass Spectrometry. Forensic Sci. Int. 2002, 125, 156–162. [Google Scholar] [CrossRef]
- Malak, M.; Ebrahim, H.; Sonbol, H.; Ali, A.; Aboulella, Y.; Hadad, G.; Emara, S. Highly Sensitive In-Capillary Derivatization and Field Amplified Sample Stacking to Analyze Narcotic Drugs in Human Serum by Capillary Zone Electrophoresis. Separations 2023, 10, 58. [Google Scholar] [CrossRef]
- Archana, G.; Dhodapkar, R.; Kumar, A. Offline Solid-Phase Extraction for Preconcentration of Pharmaceuticals and Personal Care Products in Environmental Water and their Simultaneous Determination Using the Reversed Phase High-Performance Liquid Chromatography Method. Environ. Monit. Assess. 2016, 188, 512. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.; Messina, S.M.; Stokes, C.; Salyani, S.; Alcalay, N.; De Fiebre, N.C.; De Fiebre, C.M. Solid-Phase Extraction and HPLC Assay of Nicotine and Cotinine in Plasma and Brain. Toxicol. Mech. Methods 2002, 12, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.P.; Sepúlveda, C.M.J.; Von Plessing, R.C. Pharmacokinetic Study of Risperidone: Application of an HPLC Method with Solid Phase Extraction. J. Chil. Chem. Soc. 2011, 56, 606–609. [Google Scholar] [CrossRef]
- Sun, H.; Wang, L.; Ai, L.; Liang, S.; Wu, H. A Sensitive and Validated Method for Determination of Melamine Residue in Liquid Milk by Reversed Phase High-Performance Liquid Chromatography with Solid-Phase Extraction. Food Control 2010, 21, 686–691. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, L.; Wang, H.; Zhang, X.; Zeng, Q.; Xu, H.; Sun, L.; Zhao, Q.; Ding, L. Preparation of Magnetic Strong Cation Exchange Resin for the Extraction of Melamine from Egg Samples Followed by Liquid Chromatography–Tandem Mass Spectrometry. Anal. Chim. Acta 2010, 661, 35–41. [Google Scholar] [CrossRef]
- He, L.; Su, Y.; Shen, X.; Zheng, Y.; Guo, H.; Zeng, Z. Solid-Phase Extraction of Melamine from Aqueous Samples Using Water-Compatible Molecularly Imprinted Polymers. J. Sep. Sci. 2009, 32, 3310–3318. [Google Scholar] [CrossRef]
- Wang, T.; Zhu, Y.; Ma, J.; Xuan, R.; Gao, H.; Liang, Z.; Zhang, L.; Zhang, Y. Hydrophilic Solid-Phase Extraction of Melamine with Ampholine-Modified Hybrid Organic–Inorganic Silica Material. J. Sep. Sci. 2015, 38, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Braus, H.; Middleton, F.; Walton, G. Organic Chemical Compounds in Raw and Filtered Surface Waters. Anal. Chem. 1951, 23, 1160–1164. [Google Scholar] [CrossRef]
- Theodoridis, G.; Koster, E.H.M.; de Jong, G.J. Solid-Phase Microextraction for the Analysis of Biological Samples. J. Chromatogr. B 2000, 745, 49–82. [Google Scholar] [CrossRef]
- Zheng, J.; Huang, J.; Yang, Q.; Ni, C.; Xie, X.; Shi, Y.; Sun, J.; Zhu, F.; Ouyang, G. Fabrications of Novel Solid Phase Microextraction Fiber Coatings Based on New Materials for High Enrichment Capability. Trends Anal. Chem. 2018, 108, 135–153. [Google Scholar] [CrossRef]
- Gao, Y.; Sheng, K.; Bao, T.; Wang, S. Recent Applications of Organic Molecule-Based Framework Porous Materials in Solid-Phase Microextraction for Pharmaceutical Analysis. J. Pharm. Biomed. Anal. 2022, 221, 115040. [Google Scholar] [CrossRef] [PubMed]
- Arrebola, F.J.; Martínez-Vidal, J.L.; Fernández-Gutiérrez, A.; Akhtar, M.H. Monitoring of Pyrethroid Metabolites in Human Urine Using Solid-Phase Extraction Followed by Gas Chromatography-Tandem Mass Spectrometry. Anal. Chim. Acta 1999, 401, 45–54. [Google Scholar] [CrossRef]
- Le Grand, R.; Dulaurent, S.; Gaulier, J.M.; Saint-Marcoux, F.; Moesch, C.; Lachâtre, G. Simultaneous Determination of Five Synthetic Pyrethroid Metabolites in Urine by Liquid Chromatography–Tandem Mass Spectrometry: Application to 39 Persons without Known Exposure to Pyrethroids. Toxicol. Lett. 2012, 210, 248–253. [Google Scholar] [CrossRef]
- Wielgomas, B.; Nahorski, W.; Czarnowski, W. Urinary Concentrations of Pyrethroid Metabolites in the Convenience Sample ofan Urban Population of Northern Poland. Int. J. Hyg. Environ. Health 2013, 216, 295–300. [Google Scholar] [CrossRef]
- Lin, C.-H.; Yan, C.-T.; Kumar, P.V.; Li, H.-P.; Jen, J.-F. Determination of Pyrethroid Metabolites in Human Urine Using Liquid Phase Microextraction Coupled In-Syringe Derivatization Followed by Gas Chromatography/Electron Capture Detection. Anal. Bioanal. Chem. 2011, 401, 927–937. [Google Scholar] [CrossRef]
- Bartosz, W.; Marcin, W.; Wojciech, C. Development of Hollow Fiber-Supported Liquid-Phase Microextraction and HPLC-DAD Method for the Determination of Pyrethroid Metabolites in Human and Rat Urine. Biomed. Chromatogr. 2014, 28, 708–716. [Google Scholar] [CrossRef]
- Klimowska, A.; Wielgomas, B. Off-Line Microextraction by Packed Sorbent Combined with on Solid Support Derivatization and GC-MS: Application for the Analysis of Five Pyrethroid Metabolites in Urine Samples. Talanta 2018, 176, 165–171. [Google Scholar] [CrossRef]
- Bernardo, R.A.; da Silva, L.C.; Queiroz, M.E.C.; Vaz, B.G.; Chaves, A.R. Lab-Made Solid Phase Microextraction Phases for off Line Extraction and Direct Mass Spectrometry Analysis: Evaluating the Extraction Parameters. J. Chromatogr. A 2019, 1603, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Theodoridis, G.; Lontou, M.A.; Michopoulos, F.; Sucha, M.; Gondova, T. Study of Multiple Solid-Phase Microextraction Combined Off-Line with High Performance Liquid Chromatography: Application in the Analysis of Pharmaceuticals in Urine. Anal. Chim. Acta 2004, 516, 197–204. [Google Scholar] [CrossRef]
- Cantú, M.D.; Toso, D.R.; Lacerda, C.A.; Lanças, F.M.; Carrilho, E.; Queiroz, M.E.C. Optimization of Solid-Phase Microextraction Procedures for the Determination of Tricyclic Antidepressants and Anticonvulsants in Plasma Samples by Liquid Chromatography. Anal. Bioanal. Chem. 2006, 386, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Slíž, K.; Olešová, D.; Piešt’anský, J.; Mikuš, P. Simple and Sensitive Analysis of Clenbuterol in Urine Matrices by UHPLC-MS/MS Method with Online-SPE Sample Preparation. Separations 2022, 9, 440. [Google Scholar] [CrossRef]
- Kovačić, J.; Jeličić, M.-L.; Klarić, D.A.; Mornar, A. Green Solid-Phase (Micro)Extraction of Andrographolides from Human Plasma Samples Followed by UHPLC-DAD-QqQ-MS/MS Analysis. Separations 2023, 10, 69. [Google Scholar] [CrossRef]
- Haque, A.; Iqbal, M.; Alamoudi, M.K.; Alam, P. A Selective and Accurate LC-MS/MS Method for Simultaneous Quantification of Valsartan and Hydrochlorothiazide in Human Plasma. Separations 2023, 10, 119. [Google Scholar] [CrossRef]
- Haq, N.; Iqbal, M.; Hussain, A.; Shakeel, F.; Ahmad, A.; Ibrahim, A.; Alsarra, I.A.; AlAjmi, M.F.; Mahfooz, A.; Abouzadeh, M.A. Utilization of Waste Biomaterial as an Efficient and Eco-Friendly Adsorbent for Solid-Phase Extraction of Pantoprazole Contaminants in Wastewater. Separations 2023, 10, 253. [Google Scholar] [CrossRef]
- Dziurkowska, E.; Kosinska, S.; Plenis, A.; Wesolowski, M. A New Method for the Determination of Amisulpride in a Small Volume (200 µL) of Human Saliva Using LC-DAD Supported by SPE. Separations 2023, 10, 277. [Google Scholar] [CrossRef]
- Abdel-Gawad, S.A.; Altharawi, A. Simultaneous Quantification of Some Fluoroquinolone Residues in Real Wastewater Effluents Using CZE. Separations 2023, 10, 292. [Google Scholar] [CrossRef]
- Díaz-Corona, L.R.; Parra-Saavedra, K.J.; Mora-Alonzo, R.S.; Macías-Rodríguez, M.E.; Martínez-Preciado, A.H.; Guevara-Martínez, S.J.; Zamudio-Ojeda, A.; Macias-Lamas, A.M. HPLC-DAD Development and Validation Method for Short-Chain Fatty Acids Quantification from Chicken Feces by Solid-Phase Extraction. Separations 2023, 10, 308. [Google Scholar] [CrossRef]
- Pascual-Caro, S.; Borrull, F.; Aguilar, C.; Calull, M. Comparison of different chiral selectors for the enantiomeric determination of amphetamine-type substances in human urine by solid-phase extraction followed by capillary electrophoresis-tandem mass spectrometry. Electrophoresis 2022, 43, 437–445. [Google Scholar] [CrossRef]
- Yang, J.; Chen, L.; Wang, Q.; Mei, X.; Yang, X.; Huo, F. Determination of nitroimidazole antibiotics based on dispersive solid-phase extraction combined with capillary electrophoresis. Electrophoresis 2023, 44, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.H.; Nguyen, T.D.; Vu, M.T.; Duong, H.A.; Pham, H.V. Determination of Glyphosate, Glufosinate, and Their Major Metabolites in Tea Infusions by Dual-Channel Capillary Electrophoresis following Solid-Phase Extraction. J. Anal. Methods Chem. 2022, 2022, 5687025. [Google Scholar] [CrossRef]
- Islas, G.; Rodriguez, J.A.; Perez-Silva, I.; Jose, M.; Miranda, J.M.; Ibarra, I.S. Solid-Phase Extraction and Large-Volume Sample Stacking-Capillary Electrophoresis for Determination of Tetracycline Residues in Milk. J. Anal. Methods Chem. 2018, 2018, 5394527. [Google Scholar] [CrossRef]
- An, J.; Wang, X.; Ming, M.; Li, J.; Ye, N. Determination of sulfonamides in milk by capillary electrophoresis with PEG@MoS2 as a dispersive solid-phase extraction sorbent. R. Soc. Open Sci. 2018, 5, 172104. [Google Scholar] [CrossRef]
- Chik, Z.; Mustafa, A.M.; Mohamed, Z.; Lee, T.C. Analysis of Captopril in Human Plasma Using Gas Chromatography-Mass Spectrometry (GCMS) with Solid-Phase Extraction (SPE). Curr. Anal. Chem. 2010, 6, 329–333. [Google Scholar] [CrossRef]
- Fan, Y.; Yu, R.; Chen, Y.; Sun, Y.; Waterhouse, G.I.N.; Xu, Z. A Capillary Electrophoresis Method Based on Molecularly Imprinted Solid-Phase Extraction for Selective and Sensitive Detection of Histamine in Foods. Molecules 2022, 27, 6987. [Google Scholar] [CrossRef]
- Majors, R.E. Multidimensional High Performance Liquid Chromatography. J. Chromatogr. Sci. 1980, 18, 571–579. [Google Scholar] [CrossRef]
- El-Gendy, H.; Zarad, W.; Bazan, L.; Ali, A.; Aboulella, Y.; Kamal, M.; Emara, S.; Shawky, A. Rapid Back Flushed Direct Sample Injection Bio-Analytical HPLC-UV Method for therapeutic Drug Monitoring of Terbinafine. Anal. Biochem. 2022, 659, 114951. [Google Scholar] [CrossRef] [PubMed]
- Emara, S.; Kamal, M.; Hadad, G.; ZaaZaa, H.; Kawi, M.A. Back-Flush Column-Switching Technique for On-Line Sample Cleanup and Enrichment to Determine Guaiphenesin in Human Serum. J. Liq. Chromatogr. Relat. Technol. 2012, 35, 15–27. [Google Scholar] [CrossRef]
- Karger, B.L.; Martin, M.; Guiochon, G. Role of Column Parameters and Injection Volume on Detection Limits in Liquid Chromatography. Anal. Chem. 1974, 46, 1640–1647. [Google Scholar] [CrossRef]
- Little, J.N.; Fallick, G.J. New Considerations in Detector-Application Relationships. J. Chromatogr. A 1975, 112, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Poole, C.F.; Evans, N.J.; Wibberley, D.G. Determination of Selenium in Biological Samples by Gas—Liquid Chromatography with Electron-Capture Detection. J. Chromatogr. A 1977, 136, 63–72. [Google Scholar] [CrossRef]
- Huber, J.F.K.; Becker, R.R. Enrichment of Trace Components from Liquids by Displacement Column Liquid Chromatography. J. Chromatogr. A 1977, 142, 765–776. [Google Scholar] [CrossRef]
- Lankelma, J.; Poppe, H. Determination of Methotrexate in Plasma by on-Column Concentration and Ion-Exchange Chromatography. J. Chromatogr. A 1978, 149, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Bristow, R.L.; Haworth-Duff, A.; Young, I.S.; Myers, P.; Hampson, M.R.; Williams, J.; Maher, S. An Automated Micro Solid Phase Extraction Gas Chromatography-Mass Spectrometry (μSPE-GC-MS) Detection Method for Geosmin and 2-Methylisoborneol in Drinking Water. Sci. Rep. 2023, 13, 1768. [Google Scholar] [CrossRef]
- Chafi, S.; Ballesteros, E. A Sensitive, Robust Method for Determining Natural and Synthetic Hormones in Surface and Wastewaters by Continuous Solid-Phase Extraction–Gas Chromatography–Mass Spectrometry. Sci. Pollut. Res. 2022, 29, 53619–53632. [Google Scholar] [CrossRef]
- Lee, H.S.; Kim, E.J.; Zee, O.P.; Lee, Y.J. On-Line Trace Enrichment in High Performance Liquid Chromatography Using XAD-2 Precolumn for the Determination of Lonazolac in Human Plasma. Arch. Pharm. Res. 1989, 12, 108–113. [Google Scholar] [CrossRef]
- Maris, F.A.; Stäb, J.A.; de Jong, G.J.; Brinkman, U.A.T. On-Line Trace Enrichment on a Reversed-Phase Pre-Column for Normal-Phase Liquid Chromatography with Electron-Capture Detection. J. Chromatogr. A 1988, 445, 129–138. [Google Scholar] [CrossRef]
- Wang, H.; Guo, B.; Pang, X.; Yu, H.; Liu, H.; Yan, H.; Han, D.; Guo, H.; Bai, L. Simultaneous Determination and Enrichment of β-Sitosterol from Edible Oil Samples Using Poly(NMA-ST-Co-TAIC-Co-EDMA) Monolith as Sorbent with On-Line SPE-HPLC. Chromatographia 2019, 82, 1285–1293. [Google Scholar] [CrossRef]
- Woo, C.M.; Iavarone, A.T.; Spiciarich, D.R.; Palaniappan, K.K.; Bertozzi, C.R. Isotope-Targeted Glycoproteomics (Isotag): A Mass-Independent Platform for Intact N- and O-Glycopeptide Discovery and Analysis. Nat. Methods 2015, 12, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Hägglund, P.; Bunkenborg, J.; Elortza, F.; Jensen, O.N.; Roepstorff, P. A New Strategy for Identification of N-Glycosylated Proteins and Unambiguous Assignment of their Glycosylation Sites Using HILIC Enrichment and Partial Deglycosylation. J. Proteome Res. 2004, 3, 556–566. [Google Scholar] [CrossRef]
- Zauner, G.; Deelder, A.M.; Wuhrer, M. Recent Advances in Hydrophilic Interaction Liquid Chromatography (HILIC) for Structural Glycomics. Electrophoresis 2011, 32, 3456–3466. [Google Scholar] [CrossRef]
- Shah, P.; Wang, X.; Yang, W.; Eshghi, S.T.; Sun, S.; Hoti, N.; Chen, L.; Yang, S.; Pasay, J.; Rubin, A. Integrated Proteomic and Glycoproteomic Analyses of Prostate Cancer Cells Reveal Glycoprotein Alteration in Protein Abundance and Glycosylation. Mol. Cell. Proteom. 2015, 14, 2753–2763. [Google Scholar] [CrossRef]
- Darula, Z.; Sarnyai, F.; Medzihradszky, K.F. O-Glycosylation Sites Identified from Mucin Core-1 Type Glycopeptides from Human Serum. Glycoconj. J. 2016, 33, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Cheng, K.; Zhu, J.; Mao, J.; Wang, F.; Dong, M.; Chen, R.; Guo, Z.; Liang, X.; Ye, M.; et al. Proteomics Analysis of O-Galnac Glycosylation in Human Serum by an Integrated Strategy. Anal. Chem. 2017, 89, 1469–1476. [Google Scholar] [CrossRef]
- Yang, W.; Shah, P.; Hu, Y.; Eshghi, S.T.; Sun, S.; Liu, Y.; Zhang, H. Comparison of Enrichment Methods for Intact N- and O-Linked Glycopeptides Using Strong Anion Exchange and Hydrophilic Interaction Liquid Chromatography. Anal. Chem. 2017, 89, 11193–11197. [Google Scholar] [CrossRef]
- Chaga, G.; Hopp, J.; Nelson, P. Immobilized Metal Ion Affinity Chromatography on Co2+-Carboxymethylaspartate–Agarose Superflow, as Demonstrated by One-Step Purification of Lactate Dehydrogenase from Chicken Breast Muscle. Biotechnol. Appl. Biochem. 1999, 29, 19–24. [Google Scholar]
- Hochuli, E.; Döbeli, H.; Schacher, A. New Metal Chelate Adsorbent Selective for Proteins and Peptides Containing Neighbouring Histidine Residues. J. Chromatogr. A 1987, 411, 177–184. [Google Scholar] [CrossRef]
- Andersson, L.; Porath, J. Isolation of Phosphoproteins by Immobilized Metal (Fe3+) Affinity Chromatography. Anal. Biochem. 1986, 154, 250–254. [Google Scholar] [CrossRef]
- Neville, D.C.; Townsend, R.R.; Rozanas, C.R.; Verkman, A.S.; Price, E.M.; Gruis, D.B. Evidence for Phosphorylation of Serine 753 in CFTR Using a Novel Metal-Ion Affinity Resin and Matrix-Assisted Laser Desorption Mass Spectrometry. Protein Sci. 1997, 6, 2436–2445. [Google Scholar] [CrossRef]
- Figeys, D.; Gygi, S.P.; McKinnon, G.; Aebersold, R. An Integrated Microfluidics-Tandem Mass Spectrometry System for Automated Protein Analysis. Anal. Chem. 1998, 70, 3728–3734. [Google Scholar] [CrossRef]
- Li, S.; Dass, C. Iron (III)-Immobilized Metal Ion Affinity Chromatography and Mass Spectrometry forthe Purification and Characterization of Synthetic Phosphopeptides. Anal. Biochem. 1999, 270, 9–14. [Google Scholar] [CrossRef]
- Nuhse, T.S.; Stensballe, A.; Jensen, O.N.; Peck, S.C. Large-Scale Analysis ofin Vivo Phosphorylated Membrane Proteins by Immobilized Metal Ion Affinity Chromatography and Mass Spectrometry. Mol. Cell. Proteom. 2003, 2, 1234–1243. [Google Scholar] [CrossRef] [PubMed]
- Posewitz, M.C.; Tempst, P. Immobilized Gallium (III) Affinity Chromatography of Phosphopeptides. Anal. Chem. 1999, 71, 2883–2892. [Google Scholar] [CrossRef]
- Stensballe, A.; Andersen, S.; Jensen, O.N. Characterization of Phosphoproteins from Electrophoretic Gels by Nanoscale Fe (III) Affinity Chromatography with off-Line Mass Spectrometry Analysis. Proteomics 2001, 1, 207–222. [Google Scholar] [CrossRef]
- Zhou, H.; Ye, M.; Dong, J.; Han, G.; Jiang, X.; Wu, R.; Zou, H. Specific Phosphopeptide Enrichment with Immobilized Titanium Ion Affinity Chromatography Adsorbent for Phosphoproteome Analysis. J. Proteome Res. 2008, 7, 3957–3967. [Google Scholar] [CrossRef]
- Feng, S.; Ye, M.; Zhou, H.; Jiang, X.; Jiang, X.; Zou, H.; Gong, B. Immobilized Zirconium Ion Affinity Chromatography for Specific Enrichment of Phosphopeptides in Phosphoproteome Analysis. Mol. Cell. Proteom. 2007, 6, 1656–1665. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Ye, M.; Dong, J.; Corradini, E.; Cristobal, A.; Heck, A.J.; Zou, H.; Mohammed, S. Robust Phosphoproteome Enrichment Using Monodisperse Microsphere–Based Immobilized Titanium (Iv) Ion Affinity Chromatography. Nat. Protoc. 2013, 8, 461–480. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Qian, L.; Ji, L.; Liu, S.; Wahid, A.; Jiang, X.; Sohail, A.; Ji, Y.; Zhang, Y.; Wang, P. Affinity Chromatography Assisted Comprehensive Phosphoproteomics Analysis of Human Saliva forLung Cancer. Anal. Chim. Acta 2020, 1111, 103–113. [Google Scholar] [CrossRef]
- Zhang, Q.; Tang, N.; Brock, J.W.; Mottaz, H.M.; Ames, J.M.; Baynes, J.W.; Smith, R.D.; Metz, T.O. Enrichment and Analysis of Nonenzymatically Glycated Peptides: Boronate Affinity Chromatography Coupled with Electron-Transfer Dissociation Mass Spectrometry. J. Proteome Res. 2007, 6, 2323–2330. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.-S.; Ding, X.-Y.; Min, H.-P.; Li, B.; Su, M.-X.; Niu, M.-M.; Di, B.; Yan, F. Design andSynthesis ofan Immobilized Metal Affinity Chromatography and Metal Oxide Affinity Chromatography Hybrid Material for Improved Phosphopeptide Enrichment. J. Chromatogr. A 2017, 1505, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Wolschin, F.; Wienkoop, S.; Weckwerth, W. Enrichment of Phosphorylated Proteins andPeptides from Complex Mixtures Using Metal Oxide/Hydroxide Affinity Chromatography (MOAC). Proteomics 2005, 5, 4389–4397. [Google Scholar] [CrossRef]
- Zarad, W.; El-Gendy, H.; Bazan, L.; Ali, A.; Aboulella, Y.; Kamal, M.; Emara, S.; Shawky, A. Bio-Analytical Liquid Chromatographic-Based Method with a Mixed Mode Online Solid Phase Extraction for Drug Monitoring of Fluconazole in Human Serum. J. Chromatogr. B 2021, 1187, 123045. [Google Scholar] [CrossRef] [PubMed]
- Emara, S.; Kamal, M.; Abdel Kawi, M. On-Line Sample Cleanup and Enrichment Chromatographic Technique for the Determination of Ambroxol in Human Serum. J. Chromatogr. Sci. 2012, 50, 91–96. [Google Scholar] [CrossRef][Green Version]
- Emara, S. Simultaneous Determination of Caffeine, theophylline andtheobromine in Human Plasma by On-Line Solid-Phase Extraction Coupled to Reversed-Phase Chromatography. Biomed. Chromatogr. 2004, 18, 479–485. [Google Scholar] [CrossRef]
- Emara, S.; El-Gindy, A.; Mesbah, M.K.; Hadad, G.M. Direct Injection Liquid Chromatographic Technique for Simultaneous Determination of Two Antihistaminic Drugs and their Main Metabolites in Serum. J. AOAC Int. 2007, 90, 384–390. [Google Scholar] [CrossRef]
- Emara, S.; Khedr, A.; Askal, H. Rapid and Specific Precolumn Extraction High-Performance Liquid Chromatographic Assay for Bupivacaine in Human Serum. Biomed. Chromatogr. 1996, 10, 131–134. [Google Scholar] [CrossRef]
- Emara, S.; Hussien, S.A.; Mohamed, F.A. Determination of Cholesterol in Egg Yolk by High Performance Liquid Chromatography Using an Automated Precolumn-Switching Procedure. J. Liq. Chromatogr. Relat. Technol. 1999, 22, 1225–1246. [Google Scholar] [CrossRef]
- Zarad, W.; El-Gendy, H.; Ali, A.; Aboulella, Y.; Emara, S. Integration of Solid-Phase Extraction and Reversed-Phase Chromatography in Single Protein-Coated Columns for Direct Injection of Bupivacaine in Human Serum. J. Cchromatogr. Sci. 2020, 58, 535–541. [Google Scholar] [CrossRef]
- Desilets, C.P.; Rounds, M.A.; Regnier, F.E. Semipermeable-Surface Reversed-Phase Media for High-Performance Liquid Chromatography. J. Chromatogr. A 1991, 544, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Pinkerton, T.C.; Koeplinger, K.A. Determination of Warfarin-Human Serum Albumin Protein Binding Parameters by an Improved Hummel-Dreyer High-Performance Liquid Chromatographic Method Using Internal Surface Reversed-Phase Columns. Anal. Chem. 1990, 62, 2114–2122. [Google Scholar] [CrossRef]
- Haginaka, J.; Yasuda, N.; Wakai, J.; Matsunaga, H.; Yasuda, H.; Kimura, Y. Internal-Surface Reversed-Phase Silica Support for Direct-Injection Determination of Drugs in Biological Fluids by Liquid Chromatography. Anal. Chem. 1989, 61, 2445–2448. [Google Scholar] [CrossRef]
- Haginaka, J.; Wakai, J.; Yasuda, H. Synthesis of Mixed-Functional-Phase Silica Supports forLiquid Chromatography and their Applications to Assays of Drugs in Serum. J. Chromatogr. A 1990, 535, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Kimata, K.; Tsuboi, R.; Hosoya, K.; Tanaka, N.; Araki, T. Method for the Preparation of Internal-Surface Reversed-Phase Packing Materials Starting from Alkylsilylated Silica Gels. J. Chromatogr. A 1990, 515, 73–84. [Google Scholar] [CrossRef]
- Vielhauer, S.; Rudolphi, A.; Boos, K.S.; Seidel, D. Evaluation and Routine Application of the Novel Restricted-Access Precolumn Packing Material Alkyl-Diol Silica: Coupled-Column High-Performance Liquid Chromatographic Analysis of the Photoreactive Drug 8-Methoxypsoralen in Plasma. J. Chromatogr. B 1995, 666, 315–322. [Google Scholar] [CrossRef]
- Račaityt, K.; Lutz, E.S.M.; Unger, K.K.; Lubda, D.; Boos, K.S. Analysis of Neuropeptide Y and Its Metabolites by High-Performance Liquid Chromatography–Electrospray Ionization Mass Spectrometry and Integrated Sample Clean-Up with a Novel Restricted-Access Sulphonic Acid Cation Exchanger. J. Chromatogr. A 2000, 890, 135–144. [Google Scholar] [CrossRef]
- Boos, K.-S.; Rudolphi, A. The Use of Restricted-Access Media in HPLC, Part I—Classification and Review. LC GC 1997, 15, 602–611. [Google Scholar]
- Boos, K.-S.; Grimm, C.-H. High-Performance Liquid Chromatography Integrated Solid-Phase Extraction inBioanalysis Using Restricted Access Precolumn Packings. Trends Anal. Chem. 1999, 18, 175–180. [Google Scholar] [CrossRef]
- Souverain, S.; Rudaz, S.; Veuthey, J.-L. Restricted Access Materials and Large Particle Supports for On-Line Sample Preparation: An Attractive Approach for Biological Fluids Analysis. J. Chromatogr. B 2004, 801, 141–156. [Google Scholar] [CrossRef]
- Cassiano, N.M.; Lima, V.V.; Oliveira, R.V.; De Pietro, A.C.; Cass, Q.B. Development of Restricted-Access Media Supports and their Application to the Direct Analysis of Biological Fluid Samples via High-Performance Liquid Chromatography. Anal. Bioanal. Chem. 2006, 384, 1462–1469. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Gonzalo, E.; Domínguez-Álvarez, J.; García-Gómez, D.; García-Jiménez, M.-G.; Carabias-Martínez, R. Determination ofEndocrine Disruptors in Honey by CZE-MS Using Restricted Access Materials for Matrix Cleanup. Electrophoresis 2010, 31, 2279–2288. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, R.; Xie, H.; Yin, Q.; Li, X.; Jia, Z.; Wu, X.; Zhang, J.; Li, W. Online Enrichment Ability of Restricted-Access Column Coupled with High Performance Liquid Chromatography by Column Switching Technique for Benazepril Hydrochloride. Se Pu 2013, 31, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, R.; Xie, H.; Jia, Z.; Li, W.; Zhang, J.; Wang, Y. Determination of Rifampicin in Rat Plasma by Modified Large-Volume Direct Injection RAM-HPLC and its Application to a Pharmcokinetic Study. Biomed. Chromatogr. 2015, 29, 475–480. [Google Scholar] [CrossRef]
- Wu, X.; Wang, R.; Xie, H.; Wang, J.; Yang, P.; Jia, Z.; Zhang, Q.; Wang, X. Preparation and Application of Internal Surface Reversed-Phase Restricted-Access Material. Se Pu 2012, 30, 810–815. [Google Scholar] [CrossRef]
- Denadai, M.; Cass, Q.B. Simultaneous Determination of Fluoroquinolones in Environmental Water by Liquid Chromatography–Tandem Mass Spectrometry with Direct Injection: A Green Approach. J. Chromatogr. A 2015, 1418, 177–184. [Google Scholar] [CrossRef]
- Yang, S.H.; Fan, H.; Classon, R.J.; Schug, K.A. Restricted Access Media as a Streamlined Approach toward On-Line Sample Preparation: Recent Advancements and Applications. J. Sep. Sci. 2013, 36, 2922–2938. [Google Scholar] [CrossRef]
- Wa, C.; Mallik, R.; Hage, D.S. Development of Immunoaffinity Restricted Access Media for Rapid Extractions of Low-Mass Analytes. Anal. Chem. 2008, 80, 8751–8762. [Google Scholar] [CrossRef]
- Sato, Y.; Yamamoto, E.; Takakuwa, S.; Kato, T.; Asakawa, N. Weak Cation-Exchange Restricted-Access Material for On-Line Purification of Basic Drugs in Plasma. J. Chromatogr. A 2008, 1190, 8–13. [Google Scholar] [CrossRef]
- Gasparrini, F.; Ciogli, A.; D’Acquarica, I.; Misiti, D.; Badaloni, E.; Giorgi, F.; Vigevani, A. Synthesis and Characterization of Novel Internal Surface Reversed-Phase Silica Supports for High-Performance Liquid Chromatography. J. Chromatogr. A 2007, 1176, 79–88. [Google Scholar] [CrossRef]
- González-Ortega, O.; Porath, J.; Guzmán, R. Adsorption of Peptides and Small Proteins with Control Access Polymer Permeation to Affinity Binding Sites. Part I: Polymer Permeation-Immobilized Metal Ion Affinity Chromatography Separation Adsorbents with Polyethylene Glycol andImmobilized Metal Ions. J. Chromatogr. A 2012, 1227, 115–125. [Google Scholar] [CrossRef]
- Pereira, A.V.; Cass, Q.B. High-Performance Liquid Chromatography Method for the Simultaneous Determination of Sulfamethoxazole and Trimethoprim in Bovine Milk Using an On-Line Clean-Up Column. J. Chromatogr. B 2005, 826, 139–146. [Google Scholar] [CrossRef]
- Barreiro, J.C.; Vanzolini, K.L.; Madureira, T.V.; Tiritan, M.E.; Cass, Q.B. A Column-Switching Method for Quantification of the Enantiomers of Omeprazole in Native Matrices of Waste and Estuarine Water Samples. Talanta 2010, 82, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Cass, Q.B.; Lima, V.V.; Oliveira, R.V.; Cassiano, N.M.; Degani, A.L.G.; Pedrazzoli, J., Jr. Enantiomeric Determination of the Plasma Levels of Omeprazole by Direct Plasma Injection Using High-Performance Liquid Chromatography with a chiral–Chiral Column-Switching. J. Chromatogr. B 2003, 798, 275–281. [Google Scholar] [CrossRef]
- Cass, Q.B.; Gomes, R.F.; Calafatti, S.A.; Pedrazolli, J., Jr. Determination of Amoxycillin in Human Plasma by Direct Injection and Coupled-Column High-Performance Liquid Chromatography. J. Chromatogr. A 2003, 987, 235–241. [Google Scholar] [CrossRef]
- Fagundes, V.F.; Leite, C.P.; Pianetti, G.A.; Fernandes, C. Rapid and Direct Analysis of Statins in Human Plasma by Column-Switching Liquid Chromatography with Restricted-Access Material. J. Chromatogr. B 2014, 947, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Lopes, B.R.; Barreiro, J.C.; Baraldi, P.T.; Cass, Q.B. Quantification of Carbamazepine and Its Active Metabolite by Direct Injection of Human Milk Serum Using Liquid Chromatography Tandem Ion Trap Mass Spectrometry. J. Chromatogr. B 2012, 889, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Mendiara, I.; Bentayeb, K.; Nerín, C.; Domeño, C. Online Solid-Phase Extraction–Liquid Chromatography–Mass Spectrometry to Determine Free Sterols in Human Serum. Talanta 2015, 132, 690–697. [Google Scholar] [CrossRef]
- De Paula, F.; De Pietro, A.C.; Cass, Q.B. Simultaneous Quantification of Sulfamethoxazole and Trimethoprim in Whole Egg Samples by Column-Switching High-Performance Liquid Chromatography Using Restricted Access Media Column for On-Line Sample Clean-Up. J. Chromatogr. A 2008, 1189, 221–226. [Google Scholar] [CrossRef]
- Rosa, M.A.; Mendes, T.; Figueiredo, E.C. Restricted Access Molecularly Imprinted Polymers. Methods Mol. Biol. 2021, 2359, 53–70. [Google Scholar]
- Silveira, A.T.; Barbosa, A.M.C.; Faria, H.D.; Marciano, L.P.A.; Eduardo Costa Figueiredo, E.C.; Martins, I. Online Restricted Access Molecularly Imprinted Solid-Phase Extraction Coupled with Liquid Chromatography-Mass Spectrometry for the Selective Determination of Serum Bile Acids. Analyst 2022, 147, 2779–2792. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.G.; Tavares, I.M.C.; Barbosa, A.F.; Bettini, J.; Figueiredo, E.C. Analysis of Tricyclic Antidepressants in Human Plasma Using Online-Restricted Access Molecularly Imprinted Solid Phase Extraction Followed by Direct Mass Spectrometry Identification/Quantification. Talanta 2017, 163, 8–16. [Google Scholar] [CrossRef]
- Papp, R.; Mullett, W.M.; Kwong, E.A. Method for the Direct Analysis of Drug Compounds in Plasma Using a Single Restricted Access Material (RAM) Column. J. Pharm. Biomed. Anal. 2004, 36, 457–464. [Google Scholar] [CrossRef]
- Bigelow, E.; Song, Y.; Chen, J.; Holstein, M.; Huang, Y.; Duhamel, L.; Stone, K.; Furman, R.; Li, Z.J.; Ghose, S. Using Continuous Chromatography Methodology to Achieve High-Productivity and High-Purity Enrichment of Charge Variants for Analytical Characterization. J. Chromatogr. A 2021, 1643, 462008. [Google Scholar] [CrossRef]
- Weldon, R.; Lill, J.; Olbrich, M.; Schmidt, P.; Müller-Späth, T. Purification of A Galnac-Cluster-Conjugated Oligonucleotide by Reversed-Phase Twin-Column Continuous Chromatography. J. Chromatogr. A 2022, 1663, 462734. [Google Scholar] [CrossRef] [PubMed]
- De Luca, C.; Felletti, S.; Lievore, G.; Buratti, A.; Vogg, S.; Morbidelli, M.; Cavazzini, A.; Catani, M.; Macis, M.; Ricci, A. From Batch to Continuous Chromatographic Purification of a therapeutic Peptide through Multicolumn Countercurrent Solvent Gradient Purification. J. Chromatogr. A 2020, 1625, 461304. [Google Scholar] [CrossRef]
- Aumann, L.; Morbidelli, M. A Continuous Multicolumn Countercurrent Solvent Gradient Purification (Mcsgp) Process. Biotechnol. Bioeng. 2007, 98, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Steinebach, F.; Ulmer, N.; Decker, L.; Aumann, L.; Morbidelli, M. Experimental Design of a Twin-Column Countercurrent Gradient Purification Process. J. Chromatogr. A 2017, 1492, 19–26. [Google Scholar] [CrossRef]
- Kim, T.K.; Botti, C.; Angelo, J.; Xu, X.; Ghose, S.; Li, Z.J.; Morbidelli, M.; Sponchioni, M. Experimental Design of the Multicolumn Countercurrent Solvent Gradient Purification (MCSGP) Unit for the Separation of Pegylated Proteins. Ind. Eng. Chem. Res. 2021, 60, 10764–10776. [Google Scholar] [CrossRef]
- De Luca, C.; Felletti, S.; Lievore, G.; Chenet, T.; Morbidelli, M.; Sponchioni, M.; Cavazzini, A.; Catani, M. Modern Trends in Downstream Processing of Biotherapeutics through Continuous Chromatography: The Potential of Multicolumn Countercurrent Solvent Gradient Purification. Trends Analyt. Chem. 2020, 132, 116051. [Google Scholar] [CrossRef] [PubMed]
- Zydney, A.L. Continuous Downstream Processing forHigh Value Biological Products: A Review. Biotechnol. Bioeng. 2016, 113, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Challener, C.A. Making the Move to Continuous Chromatography. BioPharm. Int. 2018, 31, 14–18. [Google Scholar]
- Peuker, T.; Bogner, A. Equipment Design and Facility Layout for Flexible Biomanufacturing Processes. Eng. Life Sci. 2011, 11, 443–451. [Google Scholar] [CrossRef]
- Goyon, A.; Dai, L.; Chen, T.; Wei, B.; Yang, F.; Andersen, N.; Kopf, R.; Leiss, M.; Mølhøj, M.; Guillarme, D. From Proof of Concept to the Routine Use of an Automated and Robust Multi-Dimensional Liquid Chromatography Mass Spectrometry Workflow Applied for the Charge Variant Characterization of Therapeutic Antibodies. J. Chromatogr. A 2020, 1615, 460740. [Google Scholar] [CrossRef] [PubMed]
- Jing, S.-Y.; Shi, C.; Leong, H.Y.; Yuan, J.-J.; Gao, D.; Wang, H.-B.; Yao, S.-J.; Lin, D.-Q. A Novel Twin-Column Continuous Chromatography Approach for Separation and Enrichment of Monoclonal Antibody Charge Variants. Eng. Life Sci. 2021, 21, 382–391. [Google Scholar] [CrossRef]
- Weldon, R.; Müller-Späth, T. Enrichment and Purification of Peptide Impurities Using Twin-Column Continuous Chromatography. J. Chromatogr. A 2022, 1667, 462894. [Google Scholar] [CrossRef]
- Qi, B.-L.; Liu, P.; Wang, Q.-W.; Cai, W.-J.; Yuan, B.-F.; Feng, Y.-Q. Derivatization for Liquid Chromatography–Mass Spectrometry. Trends Anal. Chem. 2014, 59, 121–132. [Google Scholar] [CrossRef]
- Medvedovici, A.; Bacalum, E.; David, V. Sample Preparation for Large-Scale Bioanalytical Studies Based on Liquid Chromatographic Techniques. Biomed. Chromatogr. 2018, 32, e4137. [Google Scholar] [CrossRef]
- Hadad, G.M.; Abdel-Salam, R.A.; Emara, S. Determination of Glucosamine and Carisoprodol in Pharmaceutical formulations by LC with Pre-Column Derivatization and UV Detection. J. Chromatogr. Sci. 2012, 50, 307–315. [Google Scholar] [CrossRef]
- Tang, T.; Qian, K.; Shi, T.; Wang, F.; Li, J.; Cao, Y.; Hu, Q. Monitoring the Contents of Biogenic Amines in Sufu by HPLC with SPE and Pre-Column Derivatization. Food Control 2011, 22, 1203–1208. [Google Scholar] [CrossRef]
- Emara, S. Development of Highly Sensitive and Specific HPLC Assay for Plasma Morphine Using Direct Injection Technique and Post-Column Derivatization. Biomed. Chromatogr. 1998, 12, 15–20. [Google Scholar] [CrossRef]
- Deelder, R.S.; Kroll, M.G.F.; Beeren, A.J.; Van Den Berg, J.H.M. Post-Column Reactor Systems in Liquid Chromatography. J. Chromatogr. A 1978, 149, 669–682. [Google Scholar] [CrossRef]
- Shin, K.-O.; Park, K. A Newly Developed HPLC-UV/Vis Method Using Chemical Derivatization with 2-Naphthalenethiol for Quantitation of Sulforaphane in Rat Plasma. Molecules 2021, 26, 5473. [Google Scholar] [CrossRef] [PubMed]
- Galuszka, A.; Migaszewski, Z.; Namieśnik, J. The 12 Principles of Green Analytical Chemistry and the Significance Mnemonic of Green Analytical Practices. Trends Anal. Chem. 2013, 50, 78–84. [Google Scholar] [CrossRef]
- Poole, C.F. Microreactions in Separation Science: Reagents and Techniques. J. Chromatogr. A 2013, 1296, 1. [Google Scholar] [CrossRef]
- Keith, L.H.; Gron, L.U.; Young, J.L. Green Analytical Methodologies. Chem. Rev. 2007, 107, 2695–2708. [Google Scholar] [CrossRef]
- Razee, S.; Tamura, A.; Emara, S.; Masujima, T. Application of Capillary Electrophoresis for the Determination of Dissolved Gases in Water Samples: I. Determination of Dissolved Oxygen. Anal. Chim. Acta 1997, 356, 1–6. [Google Scholar] [CrossRef]
- Zarad, W.; Shawky, A.; Ali, A.; Aboulella, Y.; Kamal, M.; Masujima, T.; Emara, S.; El-Gendy, H. Field Amplified Sample Stacking and In-Capillary Derivatization for forensic Analysis of Morphine and Morphine-6-Glucuronide in Human Urine by Capillary Electrophoresis. Talanta Open 2021, 3, 100041. [Google Scholar] [CrossRef]
- Emara, S.; Zarad, W.; Kamal, M.; Ali, A.; Aboulella, Y. Sensitivity Enhancement for Direct Injection Capillary Electrophoresis to Determine Morphine in Human Serum via In-Capillary Derivatization. J. Chromatogr. Sci. 2018, 57, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Emara, S.; Masujima, T.; Zarad, W.; Mohamed, K.; Kamal, M.; Fouad, M.; EL-Bagary, R. Field-Amplified Sample Stacking β-Cyclodextrin Modified Capillary Electrophoresis for Quantitative Determination of Diastereomeric Saponins. J. Chromatogr. Sci. 2013, 52, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Lavilla, I.; Romero, V.; Costas, I.; Bendicho, C. Greener Derivatization in Analytical Chemistry. Trends Anal. Chem. 2014, 61, 1–10. [Google Scholar] [CrossRef]
- Marsol-Vall, A.; Balcells, M.; Eras, J.; Canela-Garayoa, R. Injection-Port Derivatization Coupled to GC–MS/MS for the Analysis of Glycosylated and non-Glycosylated Polyphenols in Fruit Samples. Food Chem. 2016, 204, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.M. Before the Injection—Modern Methods of Sample Preparation for Separation Techniques. J. Chromatogr. A 2003, 1000, 3–27. [Google Scholar] [CrossRef]
- Pawliszyn, J.; Lord, H.L. Handbook of Sample Preparation; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp. 225–245. [Google Scholar]
- Rosenfeld, J.M. Solid-Phase Analytical Derivatization: Enhancement of Sensitivity and Selectivity of Analysis. J. Chromatogr. A 1999, 843, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, J.M. Derivatization in the Current Practice of Analytical Chemistry. Trends Anal. Chem. 2003, 22, 785–798. [Google Scholar] [CrossRef]
- Atapattu, S.N.; Rosenfeld, J.M. Solid Phase Analytical Derivatization as a Sample Preparation Method. J. Chromatogr. A 2013, 1296, 204–213. [Google Scholar] [CrossRef]
- Atapattu, S.N.; Rosenfeld, J.M. Micro Scale Analytical Derivatizations on Solid Phase. Trends Anal. Chem. 2019, 113, 351–356. [Google Scholar] [CrossRef]
- Oenning, A.L.; Morés, L.; Dias, A.N.; Carasek, E. A New Configuration for Bar Adsorptive Microextraction (Baμe) for the Quantification of Biomarkers (Hexanal and Heptanal) in Human Urine by HPLC Providing an Alternative for Early Lung Cancer Diagnosis. Anal. Chim. Acta 2017, 965, 54–62. [Google Scholar] [CrossRef]
- Liu, J.-F.; Yuan, B.-F.; Feng, Y.-Q. Determination of Hexanal and Heptanal in Human Urine Using Magnetic Solid Phase Extraction Coupled with In-Situ Derivatization by High Performance Liquid Chromatography. Talanta 2015, 136, 54–59. [Google Scholar] [CrossRef]
- Zhao, S.; Li, L. Chemical Derivatization in LC-MS-Based Metabolomics Study. Trends Anal. Chem. 2020, 131, 115988. [Google Scholar] [CrossRef]
- Baños, C.E.; Silva, M. Liquid Chromatography–Tandem Mass Spectrometry for the Determination of Low-Molecular Mass Aldehydes in Human Urine. J. Chromatogr. B 2010, 878, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Du, Y.; Xiao, X.; Li, G. One-Step Membrane Protected Micro-Solid-Phase Extraction and Derivatization Coupling to High-Performance Liquid Chromatography for Selective Determination of Aliphatic Aldehydes in Cosmetics and Food. Talanta 2019, 202, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Kang, P.; Mechref, Y.; Kyselova, Z.; Goetz, J.A.; Novotny, M.V. Comparative Glycomic Mapping through Quantitative Permethylation and Stable-Isotope Labeling. Anal. Chem. 2007, 79, 6064–6073. [Google Scholar] [CrossRef]
- Kang, P.; Mechref, Y.; Novotny, M.V. High-Throughput Solid-Phase Permethylation of Glycans Prior to Mass Spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 721–734. [Google Scholar] [CrossRef]
- Kang, P.; Mechref, Y.; Klouckova, I.; Novotny, M.V. Solid-Phase Permethylation of Glycans for Mass Spectrometric Analysis. Rapid Commun. Mass Spectrom. 2005, 19, 3421–3428. [Google Scholar] [CrossRef]
- Mechref, Y.; Kang, P.; Novotny, M.V. Solid-Phase Permethylation for Glycomic Analysis. Methods Mol. Biol. 2009, 534, 53–64. [Google Scholar]
- Lei, M.; Mechref, Y.; Novotny, M.V. Structural Analysis of Sulfated Glycans by Sequential Double-Permethylation Using Methyl Iodide and Deuteromethyl Iodide. J. Am. Soc. Mass Spectrom. 2009, 20, 1660–1671. [Google Scholar] [CrossRef]
- Lei, M.; Novotny, M.V.; Mechref, Y. Sequential Enrichment of Sulfated Glycans by Strong Anion-Exchange Chromatography Prior to Mass Spectrometric Measurements. J. Am. Soc. Mass Spectrom. 2010, 21, 348–357. [Google Scholar] [CrossRef]
- Li, L.; Ma, L.; Guo, Y.; Liu, W.; Wang, Y.; Liu, S. Analysis of Oligosaccharides from Panax Ginseng by Using Solid-Phase Permethylation Method Combined with Ultra-High-Performance Liquid Chromatography-Q-Orbitrap/Mass Spectrometry. J. Ginseng Res. 2020, 44, 775–783. [Google Scholar] [CrossRef]
- Desantos-Garcia, J.L.; Khalil, S.I.; Hussein, A.; Hu, Y.; Mechref, Y. Enhanced Sensitivity of LC-MS Analysis of Permethylated N-Glycans Through Online Purification. Electrophoresis 2011, 32, 3516–3525. [Google Scholar] [CrossRef] [PubMed]
- Emara, S. Determination of Methotrexate in Pharmaceutical formulations by Flow Injection Analysis Exploiting the Reaction with Potassium Permanganate. Il Farm. 2004, 59, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Hung, H.C. Simultaneous Determination of Water-Soluble Vitamins in Human Urine by Fluorescence in a Flow-Injection Analysis. J. Liq. Chromatogr. Relat. Technol. 2006, 29, 329–338. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, P.; Liang, X.; Hua, D.; Ma, T.; Pei, G. A New Potassium Tetrabromoaurate (III)–Luminol Chemiluminescence System for the Determination of Folic Acid in Milk Powder. J. Food Sci. 2012, 77, C102–C106. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Wang, L. Chemiluminescence Inhibition Assay for Folic Acid Using Flow Injection Analysis. Phytochem. Anal. 2003, 14, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Hadad, G.; Abdel-Salam, R.; Emara, S. Optimized and Validated Flow-Injection Spectrophotometric Analysis of Topiramate, Piracetam and Levetiracetam in Pharmaceutical formulations. Acta Pharm. 2011, 61, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Emara, S.; El-Gindy, A.; El-Shorbagi, A.-N.; Hadad, G. Utility of Copper(II) Oxide as a Packed Reactor in Flow Injection Assembly for Rapid Analysis of Some Angiotensin Converting Enzyme Inhibitors. Anal. Chim. Acta 2003, 489, 115–123. [Google Scholar] [CrossRef]
- Erlandsson, M.; Hällbrink, M. Metallic Zinc Reduction of Disulfide Bonds between Cysteine Residues in Peptides and Proteins. Int. J. Pept. Res. Ther. 2005, 11, 261. [Google Scholar] [CrossRef]
- Jamal, M.; Hadi, H. Determination of Nitrazepamin Pharmaceutical Preparation Using Solid-Phase Reactor–Reverse Flow Injection Analysis. Asian J. Pharm. Clin. Res. 2018, 11, 487–492. [Google Scholar] [CrossRef]
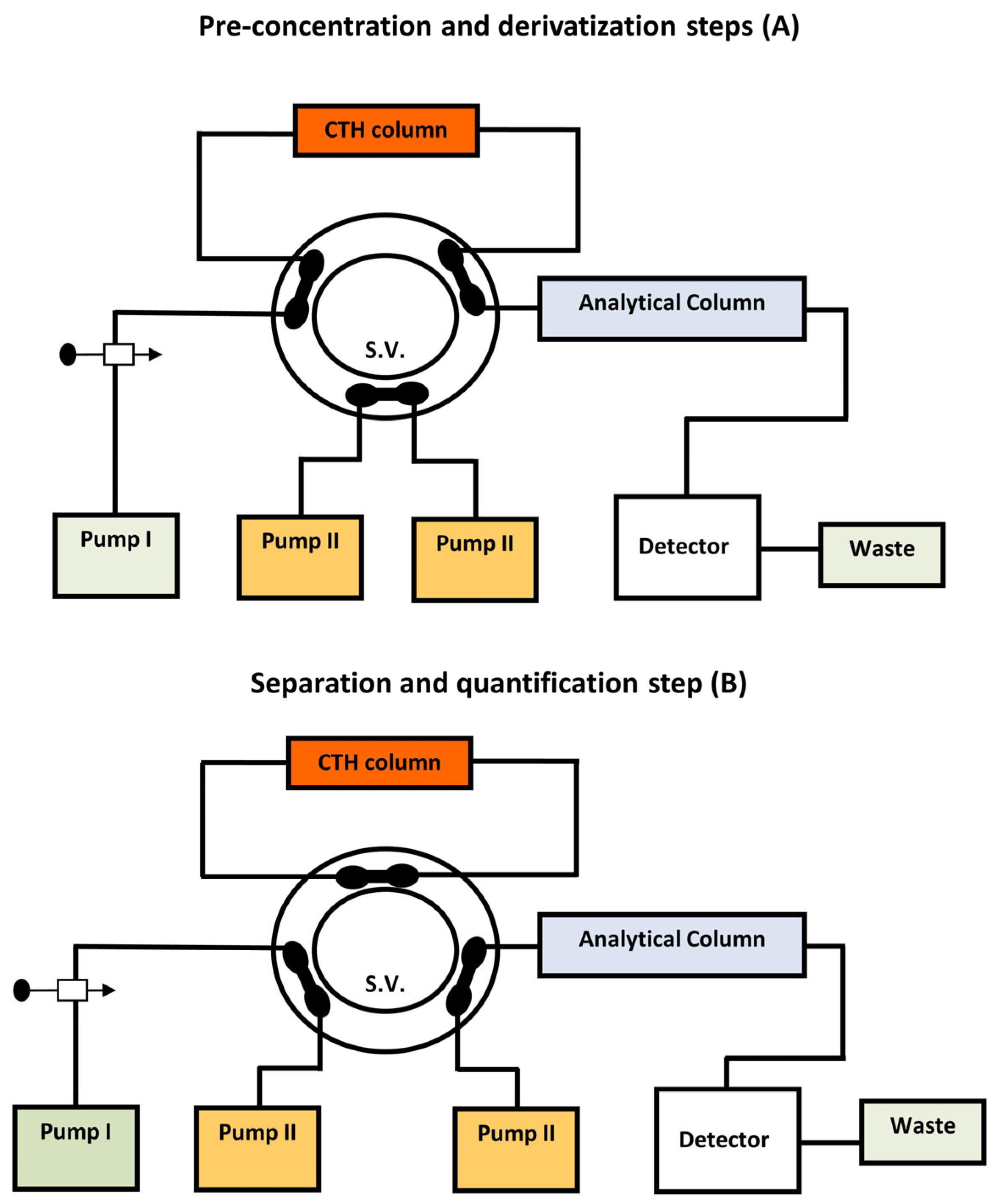
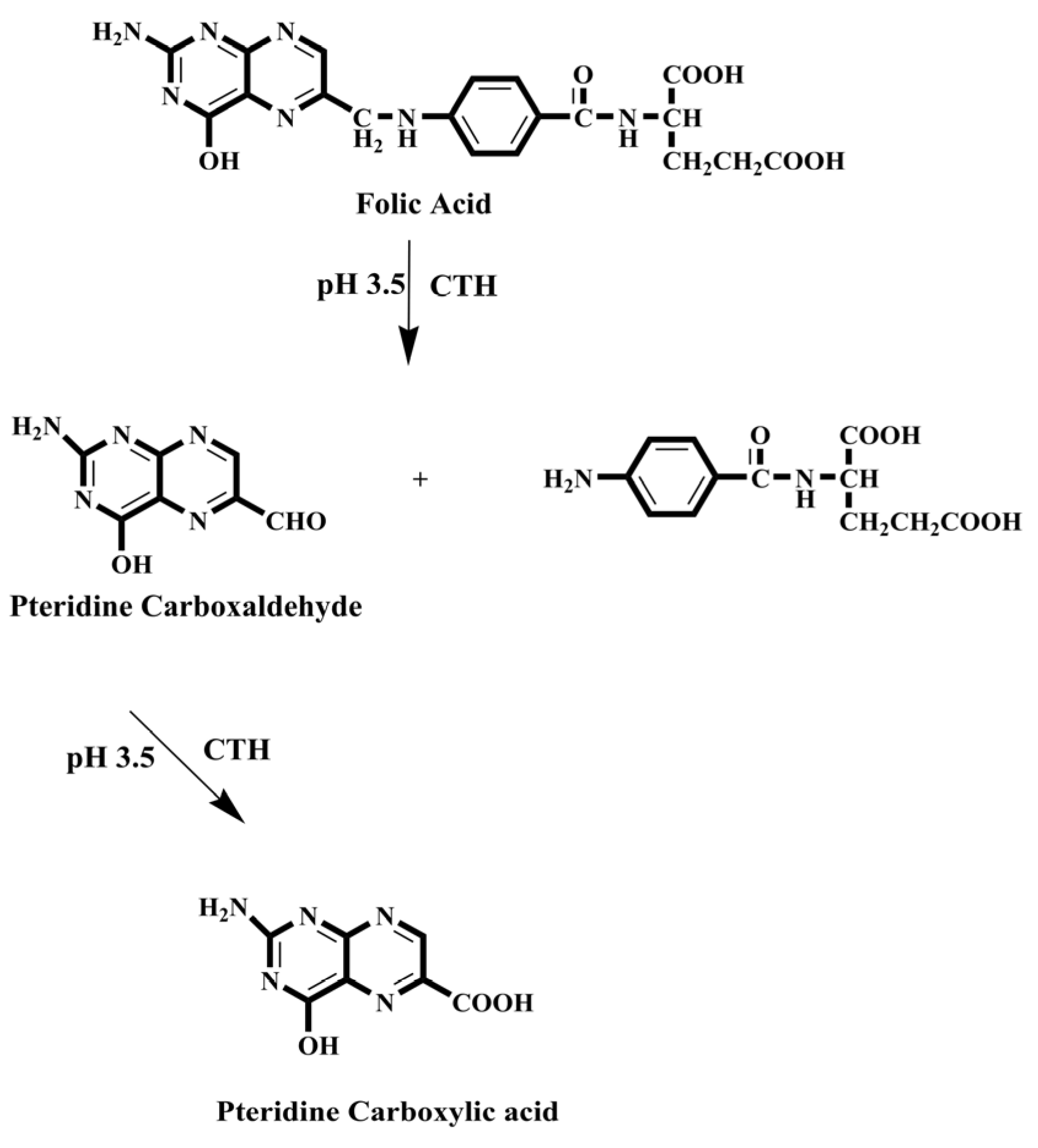
- Emara, S.; Masujima, T.; Zarad, W.; Kamal, M.; El-Bagary, R. On-Line Solid-Phase Enrichment Coupled to Packed Reactor Flow Injection Analysis in a Green Analytical Procedure to Determine Low Levels of Folic Acid Using Fluorescence Detection. Chem. Cent. J. 2012, 6, 155. [Google Scholar] [CrossRef][Green Version]
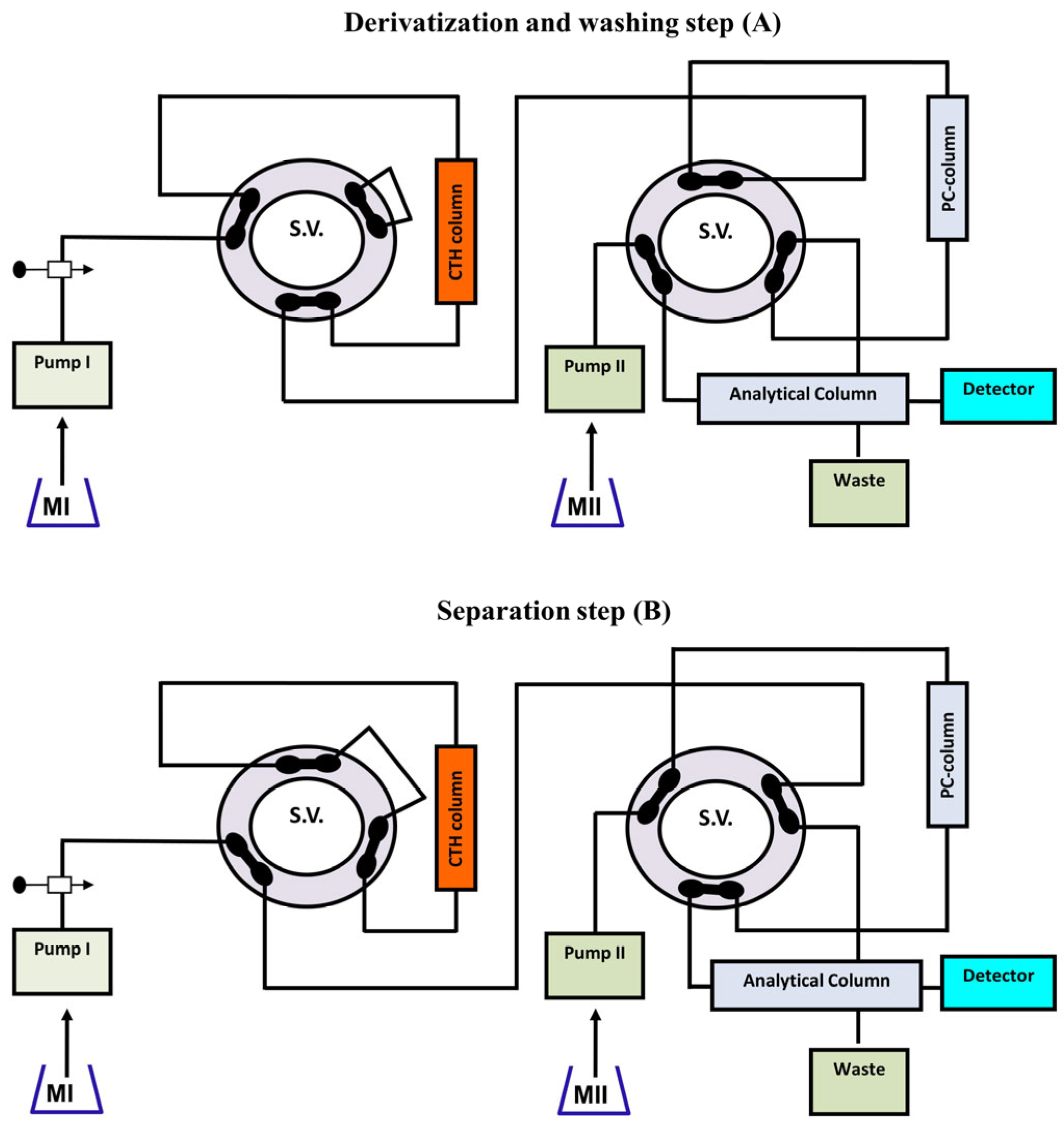
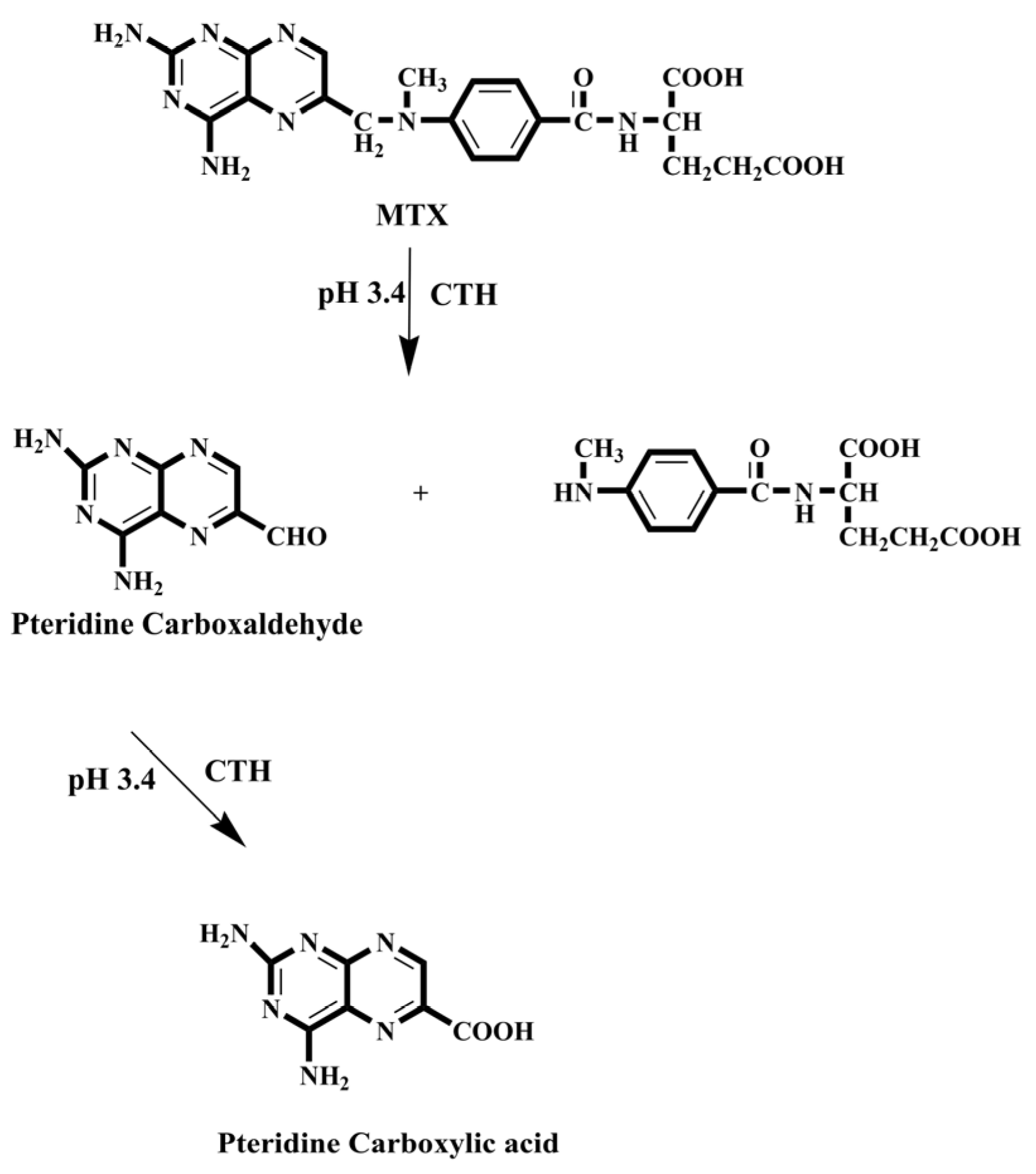
- Emara, S.; Zarad, W.; Kamal, M.; EL-Bagary, R. Coupling of On-Line Pre-Column Oxidative Cleavage and Solid-Phase Enrichment with Liquid Chromatography Using an Eco-Friendly Analytical Procedure to Determine Low Levels of Methotrexate. J. Anal. Methods Chem. 2012, 2, 194–202. [Google Scholar] [CrossRef]
- Emara, S.; Razee, S.; El-Shorbagi, A.-N.; Masujima, T. Flow Injection Method for the Determination of Methotrexate with a Column-Packed Oxidizing Agent. Analyst 1996, 121, 183–188. [Google Scholar] [CrossRef]
- Emara, S.; Masujima, T.; Zarad, W.; Kamal, M.; El-Bagary, R. On-Line Coupling of Derivatization with Pre-Concentration to Determine Trace Levels of Methotrexate. J. Pharm. Anal. 2013, 3, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Emara, S.; Masujima, T.; Zarad, W.; Kamal, M.; EI-Bagary, R. Online Pre-Column Derivatization with Chromatographic Separation to Determine Folic Acid. J. Chromatogr. Sci. 2012, 51, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Emara, S.; Razee, S.; Khedr, A.; Masujima, T. On-Line Precolumn Derivatization for HPLC Determination of Methotrexate Using a Column Packed Oxidant. Biomed. Chromatogr. 1997, 11, 42–46. [Google Scholar]
- Emara, S.; Askal, H.; Masujima, T. Rapid Determination of Methotrexate in Plasma by High-Performance Liquid Chromatography with On-Line Solid-Phase Extraction and Automated Precolumn Derivatization. Biomed. Chromatogr. 1998, 12, 338–342. [Google Scholar] [CrossRef]
- Michaila, K.; Moneeb, M.S. Determination of Methotrexate and Indomethacin in Urine using SPE-LC-DAD after Derivatization. J. Pharm. Biomed. Anal. 2011, 55, 317–324. [Google Scholar] [CrossRef]
- Alahmad, W.R.; Alawi, M.A. HPLC/UV/Fluorescence Detection of Several Pharmaceuticals in Sewage Treatment Plant Wastewaters of Jordan. Fresenius Environ. Bull. 2010, 19, 805–810. [Google Scholar]
- Li, Y.; Li, Y.; Liang, N.; Yang, F.; Kuang, Z. A Reversed-Phase High Performance Liquid Chromatography Method for Quantification of Methotrexate in Cancer Patients’ Serum. J. Chromatogr. B 2015, 1002, 107–112. [Google Scholar] [CrossRef]
- Agrawal, A.; Sharma, M. RP-HPLC Estimation of Methotrexate and Tretinoin in Bulk and Pharmaceutical Dosage Forms. J. Drug Discov. Ther. 2017, 5, 9–20. [Google Scholar]
- Lariya, N.K.; Agrawal, G.P. Development and Validation of RP-HPLC Method for Simultaneous Determination of Methotrexate, Dexamethasone and Indomethacin. Int. J. Pharm. Pharm. Sci. 2014, 7, 443–446. [Google Scholar]
- Wu, C.-S.; Wang, C.-H.; Zhang, J.-L.; Wang, D.-M.; Tong, Y.-F.; Wu, S.; Huang, H.-W.; Ning, B.-M. Separation, Determination of Six Impurities in Methotrexate Drug Substance Using Ultra-Performance Liquid Chromatography. Chin. Chem. Lett. 2014, 25, 447–450. [Google Scholar] [CrossRef]
- Li, H.; Luo, W.; Zeng, Q.; Lin, Z.; Luo, H.; Zhang, Y. Method for the Determination of blood Methotrexate by High Performance Liquid Chromatography with Online Post-column Electrochemical Oxidation and Fluorescence Detection. J. Chromatogr. B 2007, 845, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Merás, I.D.; Mansilla, A.E.; Gómez, M.J.R. Determination of Methotrexate, Several Pteridines, and Creatinine in Human Urine, Previous Oxidation with Potassium Permanganate, Using HPLC with Photometric and Xuorimetric Serial Detection. Anal. Biochem. 2005, 346, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, M.; Matsumoto, T.; Matsumoto, T.; Jimi, S.; Takamatsu, Y.; Tamura, K.; Hara, S. Simple and Sensitive HPLC Method for the Fluorometric Determination of Methotrexate and its Major Metabolites in Human Plasma by Post-Column Photochemical Reaction. Biomed. Chromatogr. 2012, 26, 76–80. [Google Scholar] [CrossRef]
- Nelson, J.A.; Harris, B.A.; Decker, W.J.; Farquhar, D. Analysis of Methotrexate in Human Plasma by High-pressure Liquid Chromatography with Fluorescence Detection. Cancer Res. 1977, 37, 3970–3973. [Google Scholar]
- Espinosa-Mansilla, A.; Durán Merás, I.; Zamora Madera, A.; Pedano, L.; Ferreyra, C. Kinetic Fluorimetric Determination of the Antineoplastic Methotrexate (MTX) in Human Serum. J. Pharm. Biomed. Anal. 2002, 29, 851–858. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Sample Matrix | Separation Technique | Detection System | LOD | LOQ | Ref. |
|---|---|---|---|---|---|---|
| Water | HPLC | UV-Vis | 0.50 ppm 0.09 ppm 0.09 ppm 1.48 ppm 0.65 ppm | 1.69 ppm 0.32 ppm 0.32 ppm 4.96 ppm 2.19 ppm | [25] |
| Plasma | HPLC | UV-Vis | - | 1.25 ng/mL 1.75 ng/mL | [26] |
| Plasma | HPLC | UV-Vis | 1 ng/mL 1 ng/mL | 2 ng/mL 2 ng/mL | [27] |
| Milk | HPLC | DAD | 18 lg/kg | 60 lg/kg | [28] |
| Water | HPLC | UV-Vis | 0.1 ng/mL | 0.5 ng/mL | [30] |
| Milk formula | HPLC | UV-Vis | 0.01 µg/mL | 0.033 µg/mL | [31] |
| Urine | HPLC | MS/MS | 0.015 ng/mL 0.015 ng/mL 0.015 ng/mL 0.015 ng/mL | 0.025 ng/mL 0.025 ng/mL 0.020 ng/mL 0.030 ng/mL | [37] |
| Urine | HPLC | Fluorescence | 0.01 µg/mL 0.02 µg/mL 0.1 µg/mL 0.001 µg/mL 0.05 µg/mL | 1 µg/mL 0.05 µg/mL 0.3 µg/mL 0.003 µg/mL 0.25 µg/mL | [43] |
| Plasma | HPLC | UV-Vis | - | 75 ng/mL 75 ng/mL 75 ng/mL 75 ng/mL 5 ng/mL 6 ng/mL 5 ng/mL 75 ng/mL | [44] |
| Urine | UHPLC | MS/MS | 0.0125 ng/mL | 0.1 ng/mL | [45] |
| Plasma | UPLC | MS/MS | 0.04 µg/mL 01 µg/mL 0.02 µg/mL | 0.15 ug/mL 0.32 ug/mL 0.06 ug/mL | [46] |
| Plasma | HPLC | MS/MS | - | 50.2 ng/mL 1.25 ng/mL | [47] |
| Wastewater | UPLC | MS/MS | 0.02 ng/mL | 0.05 ng/mL | [48] |
| Saliva | UPLC | DAD | 3 ng/mL | 5 ng/mL | [49] |
| Wastewater | CE | UV-Vis | 3 µg/mL 3 µg/mL 3 µg/mL 3 µg/mL | 5 µg/mL 5 µg/mL 5 µg/mL 5 µg/mL | [50] |
| Chicken feces | HPLC | DAD | 0.14 mg/mL 0.14 mg/mL | 0.45 mg/mL 0.43 mg/mL | [51] |
| Urine | CE | MS/MS | 1 ng/mL 1 ng/mL 0.60 ng/mL | 5 ng/mL 8 ng/mL 2 ng/mL | [52] |
| Water | CE | DAD | 0.016 µg/mL 0.040 µg/mL 0.097 µg/mL 0.037 µg/mL 0.037 µg/mL | 0.05 µg/mL 0.14 µg/mL 0.33 µg/mL 0.13 µg/mL 0.13 µg/mL | [53] |
| Tea infusions | CE | Conductivity | 0.80 ng/mL 0.56 ng/mL 1.56 ng/mL 0.54 ng/mL | 2.68 ng/mL 1.87 ng/mL 5.19 ng/mL 1.82 ng/mL | [54] |
| Milk | CE | UV-Vis | 19.93 ng/mL 23.83 ng/mL 18.60 ng/mL | 59.79 ng/mL 71.49 ng/mL 55.8 ng/mL | [55] |
| Milk | CE | UV-Vis | 0.16 µg/mL 0.04 µg/mL 0.03 µg/mL 0.10 µg/mL 0.07 µg/mL 0.03 µg/mL 0.20 µg/mL 0.07 µg/mL | 0.3 µg/mL 0.3 µg/mL 0.3 µg/mL 0.3 µg/mL 0.3 µg/mL 0.3 µg/mL 0.3 µg/mL 0.3 µg/mL | [56] |
| Plasma | GC | MS/MS | 0.5 ng/mL | 1 ng/mL | [57] |
| Foods | CE | UV-Vis | 0.087 ng/mL | 0.29 ng/mL | [58] |
| Analyte | Sample Matrix | Separation Technique | Detection System | LOD | LOQ | Ref. |
|---|---|---|---|---|---|---|
| Serum | HPLC | UV-Vis | 1 ng/mL | 3.5 ng/mL | [60] |
| Serum | HPLC | UV-Vis | 6.04 ng/mL | 18.56 ng/mL | [61] |
| Wastewaters | GC | MS/MS | 0.01 ng/L 0.10 ng/L 0.01 ng/L 0.01 ng/L 0.01 ng/L 0.01 ng/L 0.15 ng/L 0.15 ng/L 0.30 ng/L 0.05 ng/L 0.30 ng/L 0.05 ng/L 0.10 ng/L | 0.04 0.04 0.04 0.04 0.04 0.04 0.50 0.50 1.00 0.18 1.01 0.18 0.35 | [67] |
| Water | GC | MS/MS | 2 ng/L 4.3 ng/L | 10 ng/L | [68] |
| Plasma | HPLC | UV-Vis | - | 0.1 µg/mL | [69] |
| Edible oil | HPLC | UV-Vis | 6 µg/mL | 20 µg/mL | [71] |
| Serum | HPLC | UV-Vis | 50 ng/mL | 180 ng/mL | [95] |
| Serum | HPLC | UV-Vis | 2.60 ng/mL | 8.00 ng/mL | [96] |
| Serum | HPLC | UV-Vis | 22.2 ng/mL 24.1 ng/mL 21.7 ng/mL | 72 ng/mL 79.5 ng/mL 71.8 ng/mL | [97] |
| Serum | HPLC | UV-Vis | 0.045 ng/mL 0.045 ng/mL 0.045 ng/mL 0.045 ng/mL | 0.15 ng/mL 0.15 ng/mL 0.15 ng/mL 0.15 ng/mL | [98] |
| Egg yolk | HPLC | UV-Vis | - | 20 µg/mL | [100] |
| HPLC | UV-Vis | 24.85 ng/mL | 85.36 ng/mL | [101] | |
| Honey | CE | MS | 23 ng/mL 9.5 ng/mL 5.3 ng/mL 7.5 ng/mL 31 ng/mL 7.5 ng/mL | 50 ng/mL 50 ng/mL 50 ng/mL 50 ng/mL 50 ng/mL 50 ng/mL | [113] |
| Plasma | HPLC | UV-Vis | 0.2 µg/mL | 0.2 µg/mL | [115] |
| Water | UHPLC | MS/MS | 10.9 ng/mL 5.3 ng/mL 31.8 ng/mL 11.6 ng/mL 10.5 ng/mL 7.8 ng/mL | 20 ng/mL 20 ng/mL 150 ng/mL 20 ng/mL 20 ng/mL 20 ng/mL | [117] |
| Bovine milk | HPLC | UV-Vis | 15 ng/mL 25 ng/mL | 25 ng/mL 50 ng/mL | [123] |
| Water | HPLC | UV-Vis | 5 µg/mL | 15 µg/mL | [124] |
| Plasma | HPLC | UV-Vis | 0.0063 µg/mL | 0.055 µg/mL | [125] |
| Plasma | HPLC | UV-Vis | 0.02 µg/mL | 0.05 µg/mL | [126] |
| Plasma | HPLC | UV-Vis | - | 125 ng/mL 125 ng/mL 125 ng/mL 500 ng/mL | [127] |
| Milk | HPLC | MS/MS | 20 ng/mL 40 ng/mL | 25 ng/mL 50 ng/mL | [128] |
| Serum | HPLC | MS/MS | 5.0 ng/mL 0.2 ng/mL 7.5 ng/mL 7.5 ng/mL 13 ng/mL | 17 ng/mL 10 ng/mL 25 ng/mL 25 ng/mL 43 ng/mL | [129] |
| Egg | HPLC | UV-Vis | 25 ng/mL 40 ng/mL | 80 ng/mL 80 ng/mL | [130] |
| Serum | HPLC | MS/MS | 4.9 ng/mL 5.7 ng/mL 3.3 ng/mL 5.1 ng/mL 2.0 ng/mL 2.1 ng/mL 5.3 ng/mL 3.2 ng/mL 4.5 ng/mL | 10 ng/mL 10 ng/mL 10 ng/mL 25 ng/mL 25 ng/mL 25 ng/mL 10 ng/mL 10 ng/mL 10 ng/mL | [132] |
| Matrix | Extraction Technique | Analytical Technique | Derivatization Reagent | Derivatization Technique | Detection System | LOD | LOQ | Ref. |
|---|---|---|---|---|---|---|---|---|
| Dosage forms | - | FIA | Potassium permanganate | Online | FL | 1.20 ng/mL | 4.0 ng/mL | [177] |
| Dosage forms | - | FIA | CTH | Online | FL | 1.30 ng/mL | 4.50 ng/mL | [193] |
| Dosage forms | - | HPLC | CTH | Online | FL | 0.06 ng/mL | 0.20 ng/mL | [194] |
| Plasma | Protein precipitation | HPLC | CTH | Online | FL | 2.78 ng/mL | 9.56 ng/mL | [196] |
| Plasma | Online SPE | HPLC | CTH | Online | FL | 2.6 ng/mL | 8 ng/mL | [197] |
| Urine | Offline SPE | HPLC | Carbodiimide coupler and 2-nitrophenylhydrazine | Offline | UV-DAD | 30 ng/mL | 80 ng/mL | [198] |
| Wastewater | Offline SPE | HPLC | - | - | FL | 0.9 µg/mL | 3.0 µg/mL | [199] |
| Serum | Protein precipitation | HPLC | - | - | UV-Vis | 6 ng/mL | 20 ng/mL | [200] |
| Dosage forms | HPLC | - | - | UV-Vis | 0.6 µg/mL | 0.8 µg/mL | [201] | |
| Standard | - | UPLC | - | - | UV-Vis | 3.3 ng/mL | 10.9 ng/mL | [202] |
| Standard | - | HPLC | - | - | UV-Vis | 0.77 µg/mL | 1.03 µg/mL | [203] |
| Blood | Protein precipitation | HPLC | Electrochemical oxidation | Online | FL | 1.2 ng/mL | 2.6 ng/mL | [204] |
| Urine | - | HPLC | Potassium permanganate | Offline | FL | 10.6 ng/mL | 12 ng/mL | [205] |
| Plasma | Protein precipitation | HPLC | Electrochemical oxidation | Online | FL | - | 3 ng/mL | [206] |
| Plasma | Protein precipitation | HPLC | Photochemical reaction | Offline | FL | - | 10 ng/mL | [207] |
| Plasma | - | Fluorimetry | Potassium permanganate | Offline | FL | 0.17 µM | 2.0 µM | [208] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farouk, H.; Ebrahim, H.; Sonbol, H.; Malak, M.; Kamal, M.; Ibrahim, N.; Shawky, A.; Zarad, W.; Emad, A.; Emara, S. Sensitivity Enhancement for Separation-Based Analytical Techniques Utilizing Solid-Phase Enrichment Approaches and Analyte Derivatization for Trace Analysis in Various Matrices. Separations 2023, 10, 351. https://doi.org/10.3390/separations10060351
Farouk H, Ebrahim H, Sonbol H, Malak M, Kamal M, Ibrahim N, Shawky A, Zarad W, Emad A, Emara S. Sensitivity Enhancement for Separation-Based Analytical Techniques Utilizing Solid-Phase Enrichment Approaches and Analyte Derivatization for Trace Analysis in Various Matrices. Separations. 2023; 10(6):351. https://doi.org/10.3390/separations10060351
Chicago/Turabian StyleFarouk, Hanan, Hager Ebrahim, Heba Sonbol, Monika Malak, Maha Kamal, Noha Ibrahim, Ahmed Shawky, Walaa Zarad, Ahmed Emad, and Samy Emara. 2023. "Sensitivity Enhancement for Separation-Based Analytical Techniques Utilizing Solid-Phase Enrichment Approaches and Analyte Derivatization for Trace Analysis in Various Matrices" Separations 10, no. 6: 351. https://doi.org/10.3390/separations10060351
APA StyleFarouk, H., Ebrahim, H., Sonbol, H., Malak, M., Kamal, M., Ibrahim, N., Shawky, A., Zarad, W., Emad, A., & Emara, S. (2023). Sensitivity Enhancement for Separation-Based Analytical Techniques Utilizing Solid-Phase Enrichment Approaches and Analyte Derivatization for Trace Analysis in Various Matrices. Separations, 10(6), 351. https://doi.org/10.3390/separations10060351