


Strategies for Natural Product Discovery by Unlocking Cryptic Biosynthetic Gene Clusters in Fungi
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Organization of Biosynthetic Gene Clusters of Fungi and Their Regulation
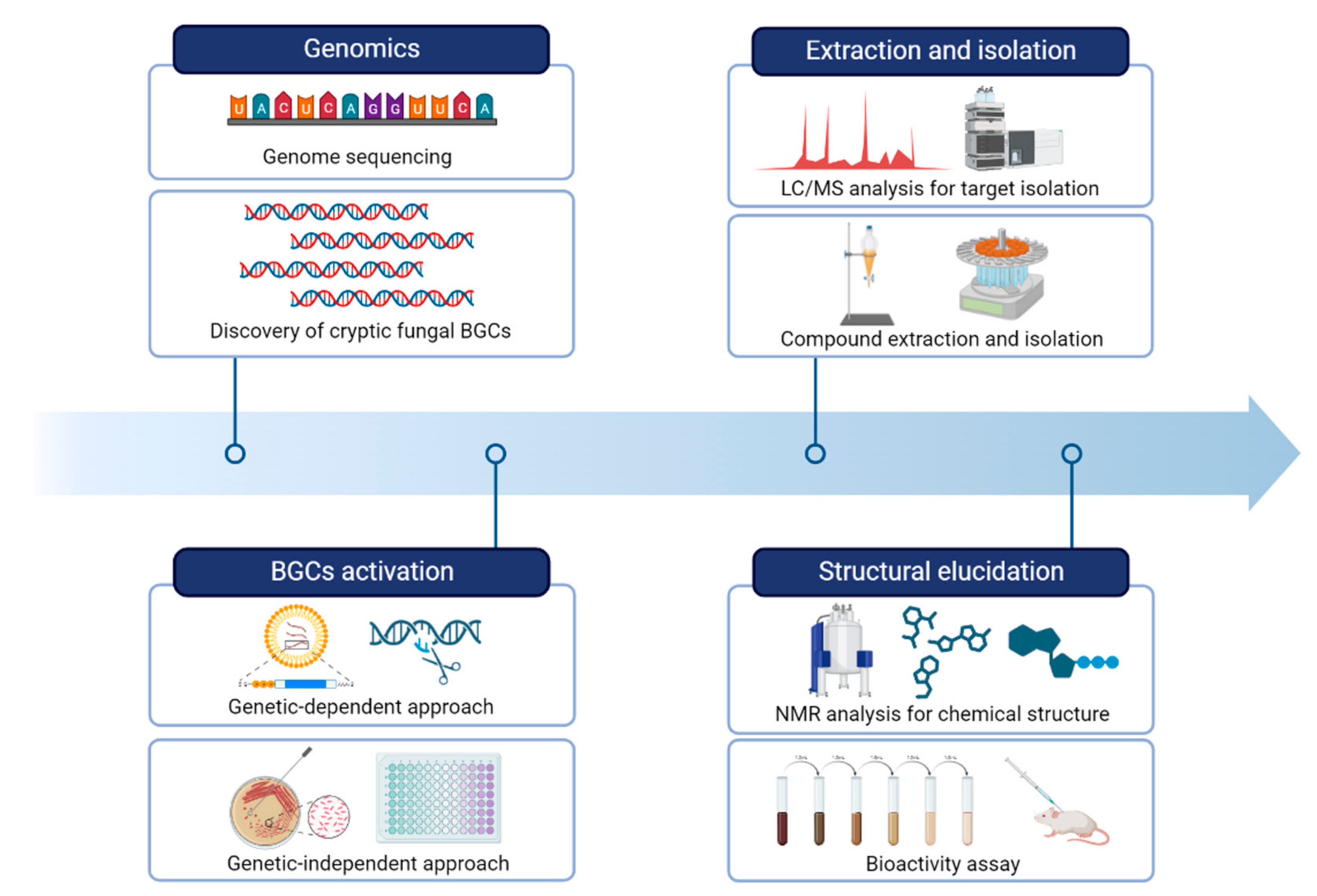
3. Characterization of Biosynthetic Gene Clusters and Natural Product Discovery
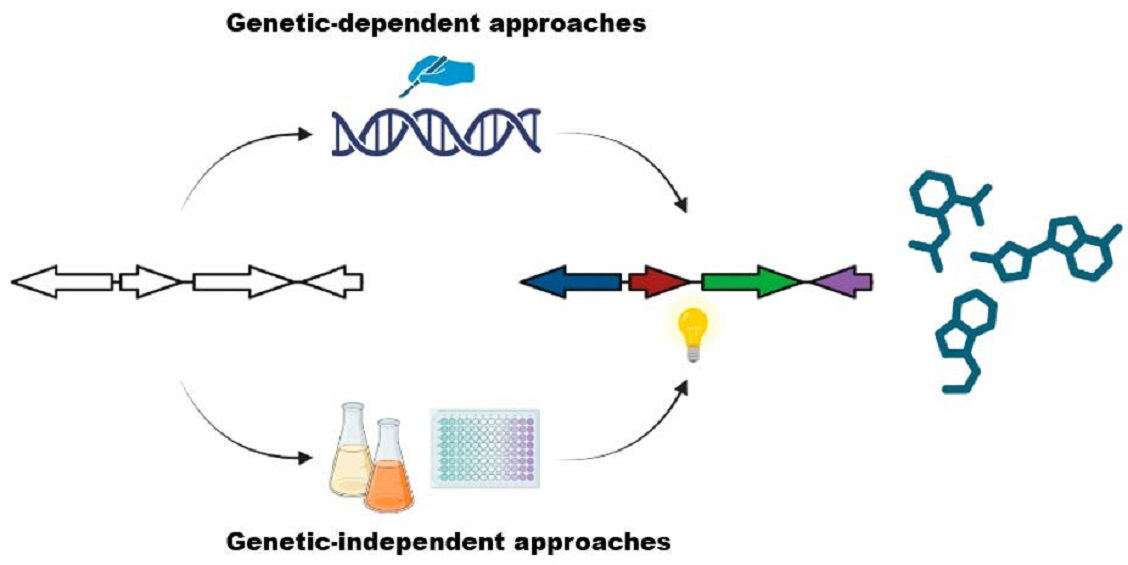
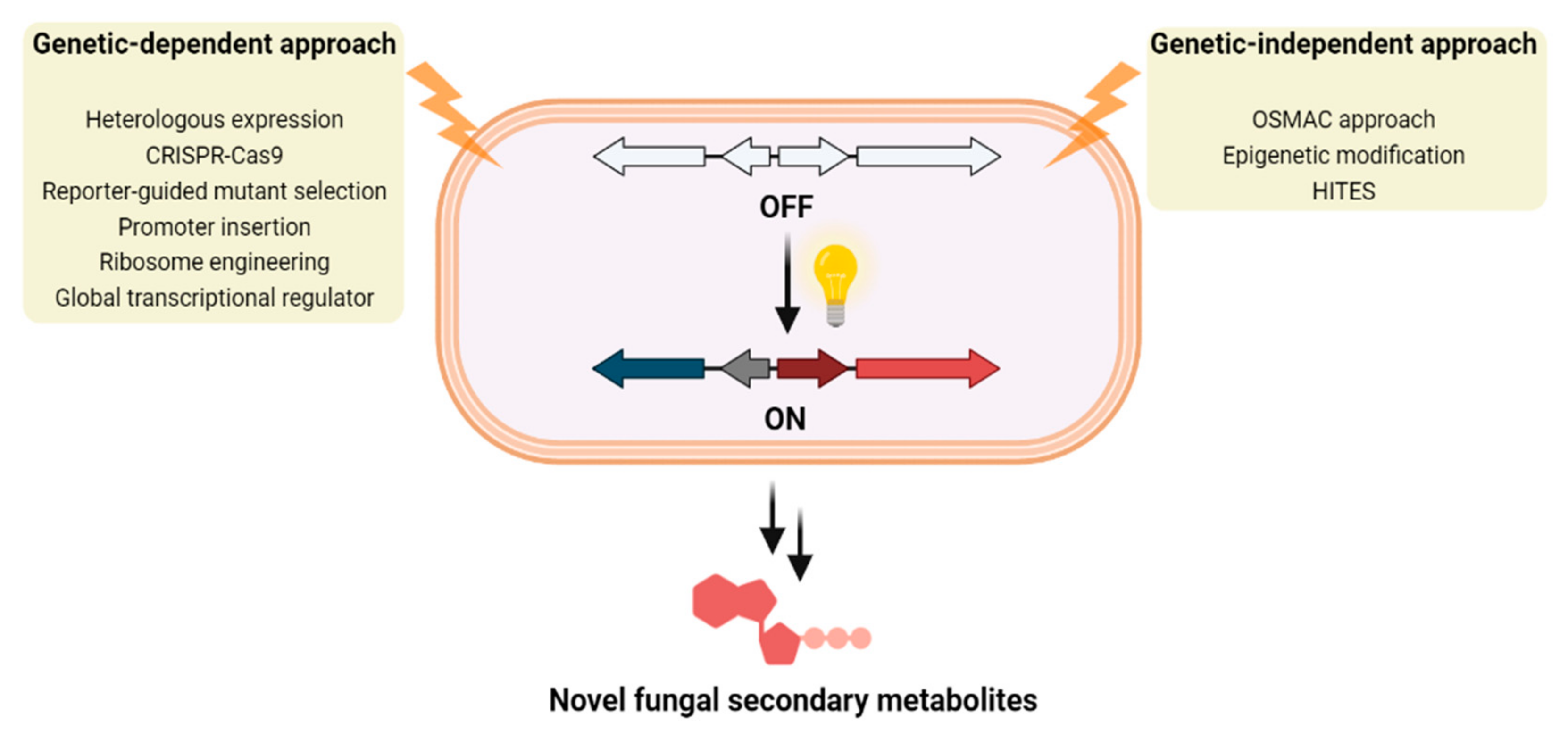
4. Genetics-Dependent Approach
4.1. Heterologous Expression
4.2. CRISPR-Cas9
4.3. Reporter-Guided Mutant Selection (RGMS)
4.4. Promoter Insertion
4.5. Ribosome Engineering
4.6. Global Transcriptional Regulator
5. Genetics-Independent Approach
5.1. OSMAC Approach
5.2. Epigenetic Modification
5.3. High-Throughput Elicitor Screening (HiTES)
6. Techniques for the Extraction, Purification, and Identification of Secondary Metabolites
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Helaly, S.E.; Thongbai, B.; Stadler, M. Diversity of biologically active secondary metabolites from endophytic and saprotrophic fungi of the ascomycete order Xylariales. Nat. Prod. Rep. 2018, 35, 992–1014. [Google Scholar] [CrossRef] [PubMed]
- Demain, A.L. Valuable secondary metabolites from fungi. In Biosynthesis and Molecular Genetics of Fungal Secondary Metabolites; Springer: New York, NY, USA, 2014; pp. 1–15. [Google Scholar]
- Bills, G.F.; Gloer, J.B. Biologically active secondary metabolites from the fungi. Microbiol. Spectr. 2017, 4, 1087–1119. [Google Scholar]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Fischer, M.S.; Glass, N.L. Communicate and fuse: How filamentous fungi establish and maintain an interconnected mycelial network. Front. Microbiol. 2019, 10, 619. [Google Scholar] [CrossRef]
- Dias, D.A.; Urban, S.; Roessner, U. A historical overview of natural products in drug discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef]
- Robey, M.T.; Caesar, L.K.; Drott, M.T.; Keller, N.P.; Kelleher, N.L. An interpreted atlas of biosynthetic gene clusters from 1000 fungal genomes. Proc. Natl. Acad. Sci. USA 2021, 118, e2020230118. [Google Scholar] [CrossRef]
- Clevenger, K.D.; Bok, J.W.; Ye, R.; Miley, G.P.; Verdan, M.H.; Velk, T.; Chen, C.; Yang, K.; Robey, M.T.; Gao, P. A scalable platform to identify fungal secondary metabolites and their gene clusters. Nat. Chem. Biol. 2017, 13, 895–901. [Google Scholar] [CrossRef]
- Hoskisson, P.A.; Seipke, R.F. Cryptic or silent? The known unknowns, unknown knowns, and unknown unknowns of secondary metabolism. MBio 2020, 11, e02642-20. [Google Scholar] [CrossRef]
- Amos, G.C.; Awakawa, T.; Tuttle, R.N.; Letzel, A.-C.; Kim, M.C.; Kudo, Y.; Fenical, W.; Moore, B.S.; Jensen, P.R. Comparative transcriptomics as a guide to natural product discovery and biosynthetic gene cluster functionality. Proc. Natl. Acad. Sci. USA 2017, 114, E11121–E11130. [Google Scholar] [PubMed]
- Honda, S.; Eusebio-Cope, A.; Miyashita, S.; Yokoyama, A.; Aulia, A.; Shahi, S.; Kondo, H.; Suzuki, N. Establishment of Neurospora crassa as a model organism for fungal virology. Nat. Commun. 2020, 11, 5627. [Google Scholar]
- Keller, N.P. Fungal secondary metabolism: Regulation, function and drug discovery. Nat. Rev. Microbiol. 2019, 17, 167–180. [Google Scholar]
- Dunlap, J.C.; Borkovich, K.A.; Henn, M.R.; Turner, G.E.; Sachs, M.S.; Glass, N.L.; McCluskey, K.; Plamann, M.; Galagan, J.E.; Birren, B.W. Enabling a community to dissect an organism: Overview of the Neurospora functional genomics project. Adv. Genet. 2007, 57, 49–96. [Google Scholar]
- Liu, Z.; Lin, Z.; Nielsen, J. Expression of fungal biosynthetic gene clusters in S. cerevisiae for natural product discovery. Synth. Syst. Biotechnol. 2021, 6, 20–22. [Google Scholar] [CrossRef]
- Reen, F.J.; Romano, S.; Dobson, A.D.; O’Gara, F. The sound of silence: Activating silent biosynthetic gene clusters in marine microorganisms. Mar. Drugs 2015, 13, 4754–4783. [Google Scholar]
- Almeida, H.; Tsang, A.; Diallo, A.B. Improving candidate Biosynthetic Gene Clusters in fungi through reinforcement learning. Bioinformatics 2022, 38, 3984–3991. [Google Scholar] [CrossRef]
- Mózsik, L.; Iacovelli, R.; Bovenberg, R.A.; Driessen, A.J. Transcriptional activation of biosynthetic gene clusters in filamentous fungi. Front. Bioeng. Biotechnol. 2022, 10, 1199. [Google Scholar]
- Chiang, Y.-M.; Szewczyk, E.; Nayak, T.; Davidson, A.D.; Sanchez, J.F.; Lo, H.-C.; Ho, W.-Y.; Simityan, H.; Kuo, E.; Praseuth, A. Molecular genetic mining of the Aspergillus secondary metabolome: Discovery of the emericellamide biosynthetic pathway. Chem. Biol. 2008, 15, 527–532. [Google Scholar] [CrossRef]
- Drott, M.; Bastos, R.; Rokas, A.; Ries, L.; Gabaldón, T.; Goldman, G.; Keller, N.; Greco, C. Diversity of secondary metabolism in Aspergillus nidulans clinical isolates. Msphere 2020, 5, e00156-20. [Google Scholar] [CrossRef] [PubMed]
- Scherlach, K.; Hertweck, C. Mining and unearthing hidden biosynthetic potential. Nat Commun. 2021, 12, 3864. [Google Scholar]
- Machado, H.; Tuttle, R.N.; Jensen, P.R. Omics-based natural product discovery and the lexicon of genome mining. Curr. Opin. Microbiol. 2017, 39, 136–142. [Google Scholar]
- Kwon, M.J.; Steiniger, C.; Cairns, T.C.; Wisecaver, J.H.; Lind, A.L.; Pohl, C.; Regner, C.; Rokas, A.; Meyer, V. Beyond the biosynthetic gene cluster paradigm: Genome-wide coexpression networks connect clustered and unclustered transcription factors to secondary metabolic pathways. Microbiol. Spectr. 2021, 9, e00898-21. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Cimermancic, P.; Sali, A.; Takano, E.; Fischbach, M.A. A systematic computational analysis of biosynthetic gene cluster evolution: Lessons for engineering biosynthesis. PLoS Comput. Biol. 2014, 10, e1004016. [Google Scholar] [CrossRef]
- Calvo, A.M.; Wilson, R.A.; Bok, J.W.; Keller, N.P. Relationship between secondary metabolism and fungal development. Microbiol. Mol. Biol. Rev. 2002, 66, 447–459. [Google Scholar]
- Manzoni, M.; Rollini, M. Biosynthesis and biotechnological production of statins by filamentous fungi and application of these cholesterol-lowering drugs. Appl. Microbiol. Biotechnol. 2002, 58, 555–564. [Google Scholar]
- Gluck-Thaler, E.; Haridas, S.; Binder, M.; Grigoriev, I.V.; Crous, P.W.; Spatafora, J.W.; Bushley, K.; Slot, J.C. The architecture of metabolism maximizes biosynthetic diversity in the largest class of fungi. Mol. Biol. Evol. 2020, 37, 2838–2856. [Google Scholar]
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J. Ribosomally synthesized and post-translationally modified peptide natural products: Overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. [Google Scholar]
- Le Govic, Y.; Papon, N.; Le Gal, S.; Bouchara, J.-P.; Vandeputte, P. Non-ribosomal peptide synthetase gene clusters in the human pathogenic fungus Scedosporium apiospermum. Front. Microbiol. 2019, 10, 2062. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.J.; Simpson, T.J. Fungal type I polyketide synthases. Methods Enzymol. 2009, 459, 49–78. [Google Scholar] [PubMed]
- Palmer, J.M.; Keller, N.P. Secondary metabolism in fungi: Does chromosomal location matter? Curr. Opin. Microbiol. 2010, 13, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Tamaru, H. Confining euchromatin/heterochromatin territory: Jumonji crosses the line. Genes Dev. 2010, 24, 1465–1478. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Wang, L.; Zheng, W.; Wang, S. Regulatory roles of histone modifications in filamentous fungal pathogens. J. Fungi 2022, 8, 565. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.N.; Yen, M.-R.; Chiang, C.-Y.; Lin, H.-C.; Chen, P.-Y. Detecting and prioritizing biosynthetic gene clusters for bioactive compounds in bacteria and fungi. Appl. Microbiol. Biotechnol. 2019, 103, 3277–3287. [Google Scholar] [CrossRef]
- Jouhten, P.; Ponomarova, O.; Gonzalez, R.; Patil, K.R. Saccharomyces cerevisiae metabolism in ecological context. FEMS Yeast Res. 2016, 16, fow080. [Google Scholar] [CrossRef]
- Salem-Bango, Z.; Price, T.K.; Chan, J.L.; Chandrasekaran, S.; Garner, O.B.; Yang, S. Fungal whole-genome sequencing for species identification: From test development to clinical utilization. J. Fungi 2023, 9, 183. [Google Scholar]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; Van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Kautsar, S.A.; Blin, K.; Shaw, S.; Navarro-Muñoz, J.C.; Terlouw, B.R.; Van Der Hooft, J.J.; Van Santen, J.A.; Tracanna, V.; Suarez Duran, H.G.; Pascal Andreu, V. MIBiG 2.0: A repository for biosynthetic gene clusters of known function. Nucleic Acids Res. 2020, 48, D454–D458. [Google Scholar] [CrossRef]
- Russell, A.H.; Truman, A.W. Genome mining strategies for ribosomally synthesised and post-translationally modified peptides. Comput. Struct. Biotechnol. J. 2020, 18, 1838–1851. [Google Scholar]
- Alam, K.; Hao, J.; Zhong, L.; Fan, G.; Ouyang, Q.; Islam, M.; Islam, S.; Sun, H.; Zhang, Y.; Li, R. Complete genome sequencing and in silico genome mining reveal the promising metabolic potential in Streptomyces strain CS-7. Front. Microbiol. 2022, 13, 3751. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Steinberg, K.M.; Larson, D.E.; Wilson, R.K.; Mardis, E.R. The next-generation sequencing revolution and its impact on genomics. Cell 2013, 155, 27–38. [Google Scholar] [PubMed]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of next-generation sequencing technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar]
- Louwen, J.J.; Medema, M.H.; van der Hooft, J.J. Enhanced correlation-based linking of biosynthetic gene clusters to their metabolic products through chemical class matching. Microbiome 2023, 11, 13. [Google Scholar] [CrossRef] [PubMed]
- Van Der Hooft, J.J.; Mohimani, H.; Bauermeister, A.; Dorrestein, P.C.; Duncan, K.R.; Medema, M.H. Linking genomics and metabolomics to chart specialized metabolic diversity. Chem. Soc. Rev. 2020, 49, 3297–3314. [Google Scholar]
- Nevalainen, K.H.; Te’o, V.S.; Bergquist, P.L. Heterologous protein expression in filamentous fungi. Trends Biotechnol. 2005, 23, 468–474. [Google Scholar]
- Qiao, Y.-M.; Yu, R.-L.; Zhu, P. Advances in targeting and heterologous expression of genes involved in the synthesis of fungal secondary metabolites. RSC Adv. 2019, 9, 35124–35134. [Google Scholar] [CrossRef]
- Ning, Y.; Xu, Y.; Jiao, B.; Lu, X. Application of gene knockout and heterologous expression strategy in fungal secondary metabolites biosynthesis. Mar. Drugs 2022, 20, 705. [Google Scholar]
- Murai, K.; Lauterbach, L.; Teramoto, K.; Quan, Z.; Barra, L.; Yamamoto, T.; Nonaka, K.; Shiomi, K.; Nishiyama, M.; Kuzuyama, T. An unusual skeletal rearrangement in the biosynthesis of the sesquiterpene trichobrasilenol from Trichoderma. Angew. Chem. Int. Ed. 2019, 58, 15046–15050. [Google Scholar] [CrossRef]
- Feng, J.; Surup, F.; Hauser, M.; Miller, A.; Wennrich, J.-P.; Stadler, M.; Cox, R.J.; Kuhnert, E. Biosynthesis of oxygenated brasilane terpene glycosides involves a promiscuous N-acetylglucosamine transferase. Chem. Comm. 2020, 56, 12419–12422. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.J.; Tang, M.; Schlecht, U.; Horecka, J.; Fischer, C.R.; Lin, H.-C.; Li, J.; Naughton, B.; Cherry, J.; Miranda, M. HEx: A heterologous expression platform for the discovery of fungal natural products. Sci. Adv. 2018, 4, eaar5459. [Google Scholar] [CrossRef]
- Meng, X.; Fang, Y.; Ding, M.; Zhang, Y.; Jia, K.; Li, Z.; Collemare, J.; Liu, W. Developing fungal heterologous expression platforms to explore and improve the production of natural products from fungal biodiversity. Biotechnol. Adv. 2022, 54, 107866. [Google Scholar]
- Kaneko, A.; Morishita, Y.; Tsukada, K.; Taniguchi, T.; Asai, T. Post-genomic approach based discovery of alkylresorcinols from a cricket-associated fungus, Penicillium soppi. Org. Biomol. Chem. 2019, 17, 5239–5243. [Google Scholar] [CrossRef]
- Song, R.; Zhai, Q.; Sun, L.; Huang, E.; Zhang, Y.; Zhu, Y.; Guo, Q.; Tian, Y.; Zhao, B.; Lu, H. CRISPR/Cas9 genome editing technology in filamentous fungi: Progress and perspective. Appl. Microbiol. Biotechnol. 2019, 103, 6919–6932. [Google Scholar]
- Nødvig, C.S.; Nielsen, J.B.; Kogle, M.E.; Mortensen, U.H. A CRISPR-Cas9 system for genetic engineering of filamentous fungi. PLoS ONE 2015, 10, e0133085. [Google Scholar] [CrossRef]
- Nødvig, C.S.; Hoof, J.B.; Kogle, M.E.; Jarczynska, Z.D.; Lehmbeck, J.; Klitgaard, D.K.; Mortensen, U.H. Efficient oligo nucleotide mediated CRISPR-Cas9 gene editing in Aspergilli. Fungal Genet. Biol. 2018, 115, 78–89. [Google Scholar] [CrossRef]
- Generoso, W.C.; Gottardi, M.; Oreb, M.; Boles, E. Simplified CRISPR-Cas genome editing for Saccharomyces cerevisiae. J. Microbiol. 2016, 127, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Schuster, M.; Kahmann, R. CRISPR-Cas9 genome editing approaches in filamentous fungi and oomycetes. Fungal Genet. Biol. 2019, 130, 43–53. [Google Scholar] [CrossRef]
- Jiang, C.; Lv, G.; Tu, Y.; Cheng, X.; Duan, Y.; Zeng, B.; He, B. Applications of CRISPR/Cas9 in the synthesis of secondary metabolites in filamentous fungi. Front. Microbiol. 2021, 12, 638096. [Google Scholar] [PubMed]
- Roux, I.; Woodcraft, C.; Hu, J.; Wolters, R.; Gilchrist, C.L.; Chooi, Y.-H. CRISPR-mediated activation of biosynthetic gene clusters for bioactive molecule discovery in filamentous fungi. ACS Synth. Biol. 2020, 9, 1843–1854. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhang, J.; Ma, Z.; Zhang, Y.; Bechthold, A.; Yu, X. Double-reporter-guided targeted activation of the oxytetracycline silent gene cluster in Streptomyces rimosus M527. Biotechnol. Bioeng. 2023, 120, 1411–1422. [Google Scholar] [CrossRef] [PubMed]
- El-Hawary, S.S.; Hassan, M.H.; Hudhud, A.O.; Abdelmohsen, U.R.; Mohammed, R. Elicitation for activation of the actinomycete genome’s cryptic secondary metabolite gene clusters. RSC Adv. 2023, 13, 5778–5795. [Google Scholar] [CrossRef]
- Mao, D.; Yoshimura, A.; Wang, R.; Seyedsayamdost, M.R. Reporter-Guided transposon mutant selection for activation of silent gene clusters in Burkholderia thailandensis. ChemBioChem 2020, 21, 1826–1831. [Google Scholar] [CrossRef] [PubMed]
- Mao, D.; Okada, B.K.; Wu, Y.; Xu, F.; Seyedsayamdost, M.R. Recent advances in activating silent biosynthetic gene clusters in bacteria. Curr. Opin. Microbiol. 2018, 45, 156–163. [Google Scholar] [CrossRef]
- Askenazi, M.; Driggers, E.M.; Holtzman, D.A.; Norman, T.C.; Iverson, S.; Zimmer, D.P.; Boers, M.-E.; Blomquist, P.R.; Martinez, E.J.; Monreal, A.W. Integrating transcriptional and metabolite profiles to direct the engineering of lovastatin-producing fungal strains. Nat. Biotechnol. 2003, 21, 150–156. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [PubMed]
- Kandel, E.S.; Lu, T.; Wan, Y.; Agarwal, M.K.; Jackson, M.W.; Stark, G.R. Mutagenesis by reversible promoter insertion to study the activation of NF-κB. Proc. Natl. Acad. Sci. USA 2005, 102, 6425–6430. [Google Scholar] [CrossRef]
- Zheng, W.; Wang, X.; Zhou, H.; Zhang, Y.; Li, A.; Bian, X. Establishment of recombineering genome editing system in Paraburkholderia megapolitana empowers activation of silent biosynthetic gene clusters. Microb. Biotechnol. 2020, 13, 397–405. [Google Scholar] [CrossRef]
- Wei, P.-L.; Fan, J.; Yu, J.; Ma, Z.; Guo, X.; Keller, N.P.; Li, E.; Lou, C.; Yin, W.-B. Quantitative characterization of filamentous fungal promoters on a single-cell resolution to discover cryptic natural products. Sci. China Life Sci. 2022, 66, 848–860. [Google Scholar] [CrossRef]
- Grau, M.F.; Entwistle, R.; Oakley, C.E.; Wang, C.C.; Oakley, B.R. Overexpression of an LaeA-like methyltransferase upregulates secondary metabolite production in Aspergillus nidulans. ACS Chem. Biol. 2019, 14, 1643–1651. [Google Scholar] [CrossRef]
- Shentu, X.; Liu, N.; Tang, G.; Tanaka, Y.; Ochi, K.; Xu, J.; Yu, X. Improved antibiotic production and silent gene activation in Streptomyces diastatochromogenes by ribosome engineering. J. Antibiot. 2016, 69, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhong, C.; Xiao, H. Genetic engineering of filamentous fungi for efficient protein expression and secretion. Front. Bioeng. Biotechnol. 2020, 8, 293. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Duan, Y.; Huang, Y. The application of ribosome engineering to natural product discovery and yield improvement in Streptomyces. Antibiotics 2019, 8, 133. [Google Scholar] [CrossRef]
- Chai, Y.-J.; Cui, C.-B.; Li, C.-W.; Wu, C.-J.; Tian, C.-K.; Hua, W. Activation of the dormant secondary metabolite production by introducing gentamicin-resistance in a marine-derived Penicillium purpurogenum G59. Mar. Drugs 2012, 10, 559–582. [Google Scholar] [CrossRef]
- Wu, C.-J.; Yi, L.; Cui, C.-B.; Li, C.-W.; Wang, N.; Han, X. Activation of the silent secondary metabolite production by introducing neomycin-resistance in a marine-derived Penicillium purpurogenum G59. Mar. Drugs 2015, 13, 2465–2487. [Google Scholar] [CrossRef]
- Dong, Y.; Cui, C.-B.; Li, C.-W.; Hua, W.; Wu, C.-J.; Zhu, T.-J.; Gu, Q.-Q. Activation of dormant secondary metabolite production by introducing neomycin resistance into the deep-sea fungus, Aspergillus versicolor ZBY-3. Mar. Drugs 2014, 12, 4326–4352. [Google Scholar] [CrossRef]
- Perrenoud, A.; Sauer, U. Impact of global transcriptional regulation by ArcA, ArcB, Cra, Crp, Cya, Fnr, and Mlc on glucose catabolism in Escherichia coli. J. Bacteriol. 2005, 187, 3171–3179. [Google Scholar] [CrossRef]
- Glauser, D.A.; Schlegel, W. Mechanisms of transcriptional regulation underlying temporal integration of signals. Nucleic Acids Res. 2006, 34, 5175–5183. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.; Goncharov, T.; Maecker, H.; Zobel, K.; Kömüves, L.G.; Deshayes, K.; Vucic, D. Cellular inhibitors of apoptosis are global regulators of NF-κB and MAPK activation by members of the TNF family of receptors. Sci. Signal. 2012, 5, ra22. [Google Scholar] [CrossRef]
- Peñalva, M.A.; Arst, J.; Herbert, N. Recent advances in the characterization of ambient pH regulation of gene expression in filamentous fungi and yeasts. Annu. Rev. Microbiol. 2004, 58, 425–451. [Google Scholar] [CrossRef]
- Merhej, J.; Richard-Forget, F.; Barreau, C. The pH regulatory factor Pac1 regulates Tri gene expression and trichothecene production in Fusarium graminearum. Fungal Genet. Biol. 2011, 48, 275–284. [Google Scholar] [CrossRef]
- Lyu, H.-N.; Liu, H.-W.; Keller, N.P.; Yin, W.-B. Harnessing diverse transcriptional regulators for natural product discovery in fungi. Nat. Prod. Rep. 2020, 37, 6–16. [Google Scholar] [CrossRef]
- Bok, J.W.; Keller, N.P. LaeA, a regulator of secondary metabolism in Aspergillus spp. Eukaryot. Cell 2004, 3, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Niehaus, E.-M.; Rindermann, L.; Janevska, S.; Münsterkötter, M.; Güldener, U.; Tudzynski, B. Analysis of the global regulator Lae1 uncovers a connection between Lae1 and the histone acetyltransferase HAT1 in Fusarium fujikuroi. Appl. Microbiol. Biotechnol. 2018, 102, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Brakhage, A.A. Regulation of fungal secondary metabolism. Nat. Rev. Microbiol. 2013, 11, 21–32. [Google Scholar] [CrossRef]
- Strauss, J.; Reyes-Dominguez, Y. Regulation of secondary metabolism by chromatin structure and epigenetic codes. Fungal Genet. Biol. 2011, 48, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Wang, M.; Li, L.; Si, J.; Song, B.; Zhou, C.; Yu, M.; Wang, X.; Zhang, Y.; Ding, G. Overexpression of the global regulator LaeA in Chaetomium globosum leads to the biosynthesis of chaetoglobosin Z. J. Nat. Prod. 2016, 79, 2487–2494. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Han, H.; Zhang, X.; Ma, C.; Sun, C.; Che, Q.; Gu, Q.; Zhu, T.; Zhang, G.; Li, D. Discovery of two new sorbicillinoids by overexpression of the global regulator LaeA in a marine-derived fungus Penicillium dipodomyis YJ-11. Mar. Drugs 2019, 17, 446. [Google Scholar] [CrossRef]
- Bok, J.W.; Hoffmeister, D.; Maggio-Hall, L.A.; Murillo, R.; Glasner, J.D.; Keller, N.P. Genomic mining for Aspergillus natural products. Chem. Biol. 2006, 13, 31–37. [Google Scholar] [CrossRef]
- Hong, E.J.; Kim, N.K.; Lee, D.; Kim, W.G.; Lee, I. Overexpression of the laeA gene leads to increased production of cyclopiazonic acid in Aspergillus fumisynnematus. Fungal Biol. 2015, 119, 973–983. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, X.; Zhang, K.; Zhang, X.; Zhu, T.; Che, Q.; Zhang, G.; Li, D. Overexpression of global regulator PbrlaeA leads to the discovery of new polyketide in fungus Penicillium brocae HDN-12-143. Front. Chem. 2020, 8, 270. [Google Scholar] [CrossRef]
- Ding, Z.; Wang, X.; Kong, F.-D.; Huang, H.-M.; Zhao, Y.-N.; Liu, M.; Wang, Z.-P.; Han, J. Overexpression of global regulator Talae1 leads to the discovery of new antifungal polyketides from endophytic fungus Trichoderma afroharzianum. Front. Microbiol. 2020, 11, 622785. [Google Scholar] [CrossRef]
- Bode, H.B.; Bethe, B.; Höfs, R.; Zeeck, A. Big effects from small changes: Possible ways to explore nature’s chemical diversity. ChemBioChem 2002, 3, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Bai, X.; Chen, J.; Zhang, H.; Wang, H. Exploring structural diversity of microbe secondary metabolites using OSMAC strategy: A literature review. Front. Microbiol. 2019, 10, 294. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.; Jackson, S.A.; Patry, S.; Dobson, A.D. Extending the “one strain many compounds”(OSMAC) principle to marine microorganisms. Mar. Drugs 2018, 16, 244. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Seok, S.; Ryoo, R.; Choi, S.U.; Kim, K.H. Macrocyclic trichothecene mycotoxins from a deadly poisonous mushroom, Podostroma cornu-damae. J. Nat. Prod. 2018, 82, 122–128. [Google Scholar] [CrossRef]
- Staropoli, A.; Iacomino, G.; De Cicco, P.; Woo, S.L.; Di Costanzo, L.; Vinale, F. Induced secondary metabolites of the beneficial fungus Trichoderma harzianum M10 through OSMAC approach. Chem. Biol. Technol. Agric. 2023, 10, 28. [Google Scholar] [CrossRef]
- Hewage, R.T.; Aree, T.; Mahidol, C.; Ruchirawat, S.; Kittakoop, P. One strain-many compounds (OSMAC) method for production of polyketides, azaphilones, and an isochromanone using the endophytic fungus Dothideomycete sp. Phytochemistry 2014, 108, 87–94. [Google Scholar] [CrossRef]
- Xue, M.; Hou, X.; Fu, J.; Zhang, J.; Wang, J.; Zhao, Z.; Xu, D.; Lai, D.; Zhou, L. Recent advances in search of bioactive secondary metabolites from fungi triggered by chemical epigenetic modifiers. J. Fungi 2023, 9, 172. [Google Scholar] [CrossRef]
- Bind, S.; Bind, S.; Sharma, A.; Chaturvedi, P. Epigenetic modification: A key tool for secondary metabolite production in microorganisms. Front. Microbiol. 2022, 13, 230. [Google Scholar] [CrossRef]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [Google Scholar] [CrossRef]
- Anjum, K.; Xuewei, Y. Epigenetic strategies to discover novel fungal secondary metabolites. J. ISSN 2022, 2766, 2276. [Google Scholar] [CrossRef]
- Lei, H.; Zhang, D.; Ding, N.; Chen, S.; Song, C.; Luo, Y.; Fu, X.; Bi, X.; Niu, H. New cytotoxic natural products from the marine sponge-derived fungus Pestalotiopsis sp. by epigenetic modification. RSC Adv. 2020, 10, 37982–37988. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, N.; Shi, Z.; Yang, Z.; Hu, X. Histone deacetylase inhibitor suberoyl bis-hydroxamic acid suppresses cell proliferation and induces apoptosis in breast cancer cells. Mol. Med. Rep. 2015, 11, 2908–2912. [Google Scholar] [CrossRef]
- Pinto, A.A.; Barúa, J.E.; Almeida, M.O.; Viaud, M.; Zorrilla, D.; Collado, I.G.; Macías-Sánchez, A.J.; Durán-Patrón, R. Structural and biosynthetic studies of botrycinereic acid, a new cryptic metabolite from the fungus Botrytis cinerea. Bioorg. Chem. 2022, 127, 105979. [Google Scholar] [CrossRef]
- Wang, Z.; He, X.; Niu, C.; Zhou, J.; Zhang, Z. Induction of funitatin A, a new polyketide from the Yellow River wetland-derived fungus Talaromyces funiculosus. Phytochem. Lett. 2022, 47, 42–45. [Google Scholar] [CrossRef]
- Abdelhakim, I.A.; Bin Mahmud, F.; Motoyama, T.; Futamura, Y.; Takahashi, S.; Osada, H. Dihydrolucilactaene, a potent antimalarial compound from Fusarium sp. RK97-94. J. Nat. Prod. 2021, 85, 63–69. [Google Scholar] [CrossRef]
- Xu, F.; Nazari, B.; Moon, K.; Bushin, L.B.; Seyedsayamdost, M.R. Discovery of a cryptic antifungal compound from Streptomyces albus J1074 using high-throughput elicitor screens. J. Am. Chem. Soc. 2017, 139, 9203–9212. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Wu, Y.; Zhang, C.; Davis, K.M.; Moon, K.; Bushin, L.B.; Seyedsayamdost, M.R. A genetics-free method for high-throughput discovery of cryptic microbial metabolites. Nat. Chem. Biol. 2019, 15, 161–168. [Google Scholar] [CrossRef]
- Moon, K.; Xu, F.; Zhang, C.; Seyedsayamdost, M.R. Bioactivity-HiTES unveils cryptic antibiotics encoded in actinomycete bacteria. ACS Chem. Biol. 2019, 14, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Seyedsayamdost, M.R. Induction of diverse cryptic fungal metabolites by steroids and channel blockers. Angew. Chem. Int. Ed. 2022, 61, e202204519. [Google Scholar] [CrossRef]
- Qiu, S.; Cai, Y.; Yao, H.; Lin, C.; Xie, Y.; Tang, S.; Zhang, A. Small molecule metabolites: Discovery of biomarkers and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 132. [Google Scholar] [CrossRef]
- Soares, J.X.; Santos, Á.; Fernandes, C.; Pinto, M.M. Liquid chromatography on the different methods for the determination of lipophilicity: An essential analytical tool in medicinal chemistry. Chemosensors 2022, 10, 340. [Google Scholar] [CrossRef]
- Xiao, J.F.; Zhou, B.; Ressom, H.W. Metabolite identification and quantitation in LC-MS/MS-based metabolomics. TrAC Trends Anal. Chem. 2012, 32, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Marco-Ramell, A.; Palau-Rodriguez, M.; Alay, A.; Tulipani, S.; Urpi-Sarda, M.; Sanchez-Pla, A.; Andres-Lacueva, C. Evaluation and comparison of bioinformatic tools for the enrichment analysis of metabolomics data. BMC Bioinform. 2018, 19, 1–11. [Google Scholar] [CrossRef]
- Zhang, Q.-W.; Lin, L.-G.; Ye, W.-C. Techniques for extraction and isolation of natural products: A comprehensive review. Chin. Med. 2018, 13, 20. [Google Scholar] [CrossRef]
- Li, X.-C.; Ferreira, D.; Ding, Y. Determination of absolute configuration of natural products: Theoretical calculation of electronic circular dichroism as a tool. Curr. Org. Chem. 2010, 14, 1678–1697. [Google Scholar] [CrossRef]
- Fleming, A.M.; Alshykhly, O.; Orendt, A.M.; Burrows, C.J. Computational studies of electronic circular dichroism spectra predict absolute configuration assignments for the guanine oxidation product 5-carboxamido-5-formamido-2-iminohydantoin. Tetrahedron Lett. 2015, 56, 3191–3196. [Google Scholar] [CrossRef]
- Koenis, M.A.; Chibueze, C.; Jinks, M.A.; Nicu, V.P.; Visscher, L.; Goldup, S.; Buma, W.J. Vibrational circular dichroism spectroscopy for probing the expression of chirality in mechanically planar chiral rotaxanes. Chem. Sci. 2020, 11, 8469–8475. [Google Scholar] [CrossRef]
- Felippe, L.G.; Batista, J.M., Jr.; Baldoqui, D.C.; Nascimento, I.R.; Kato, M.J.; He, Y.; Nafie, L.A.; Furlan, M. VCD to determine absolute configuration of natural product molecules: Secolignans from Peperomia blanda. Org. Biomol. Chem. 2012, 10, 4208–4214. [Google Scholar] [CrossRef]
- Grassin, C.; Santoro, E.; Merten, C. 7-Azaindole breaks carboxylic acid dimers and simplifies VCD spectra analyses of natural products. Chem. Comm. 2022, 58, 11527–11530. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.W.; Wang, J.W. How cryo-electron microscopy and X-ray crystallography complement each other. Protein Sci. 2017, 26, 32–39. [Google Scholar] [CrossRef]
- Guss, J.M.; King, G.F. Macromolecular structure determination: Comparison of crystallography and NMR. Encycl. Life Sci. 2002, 11, 1–5. [Google Scholar]
- Danelius, E.; Halaby, S.; van der Donk, W.A.; Gonen, T. MicroED in natural product and small molecule research. Nat. Prod. Rep. 2021, 38, 423–431. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hur, J.Y.; Jeong, E.; Kim, Y.C.; Lee, S.R. Strategies for Natural Product Discovery by Unlocking Cryptic Biosynthetic Gene Clusters in Fungi. Separations 2023, 10, 333. https://doi.org/10.3390/separations10060333
Hur JY, Jeong E, Kim YC, Lee SR. Strategies for Natural Product Discovery by Unlocking Cryptic Biosynthetic Gene Clusters in Fungi. Separations. 2023; 10(6):333. https://doi.org/10.3390/separations10060333
Chicago/Turabian StyleHur, Ji Yun, Eunju Jeong, Young Chan Kim, and Seoung Rak Lee. 2023. "Strategies for Natural Product Discovery by Unlocking Cryptic Biosynthetic Gene Clusters in Fungi" Separations 10, no. 6: 333. https://doi.org/10.3390/separations10060333
APA StyleHur, J. Y., Jeong, E., Kim, Y. C., & Lee, S. R. (2023). Strategies for Natural Product Discovery by Unlocking Cryptic Biosynthetic Gene Clusters in Fungi. Separations, 10(6), 333. https://doi.org/10.3390/separations10060333