
Effects of Geniposide and Geniposidic Acid on Fluoxetine-Induced Muscle Atrophy in C2C12 Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture and Differentiation
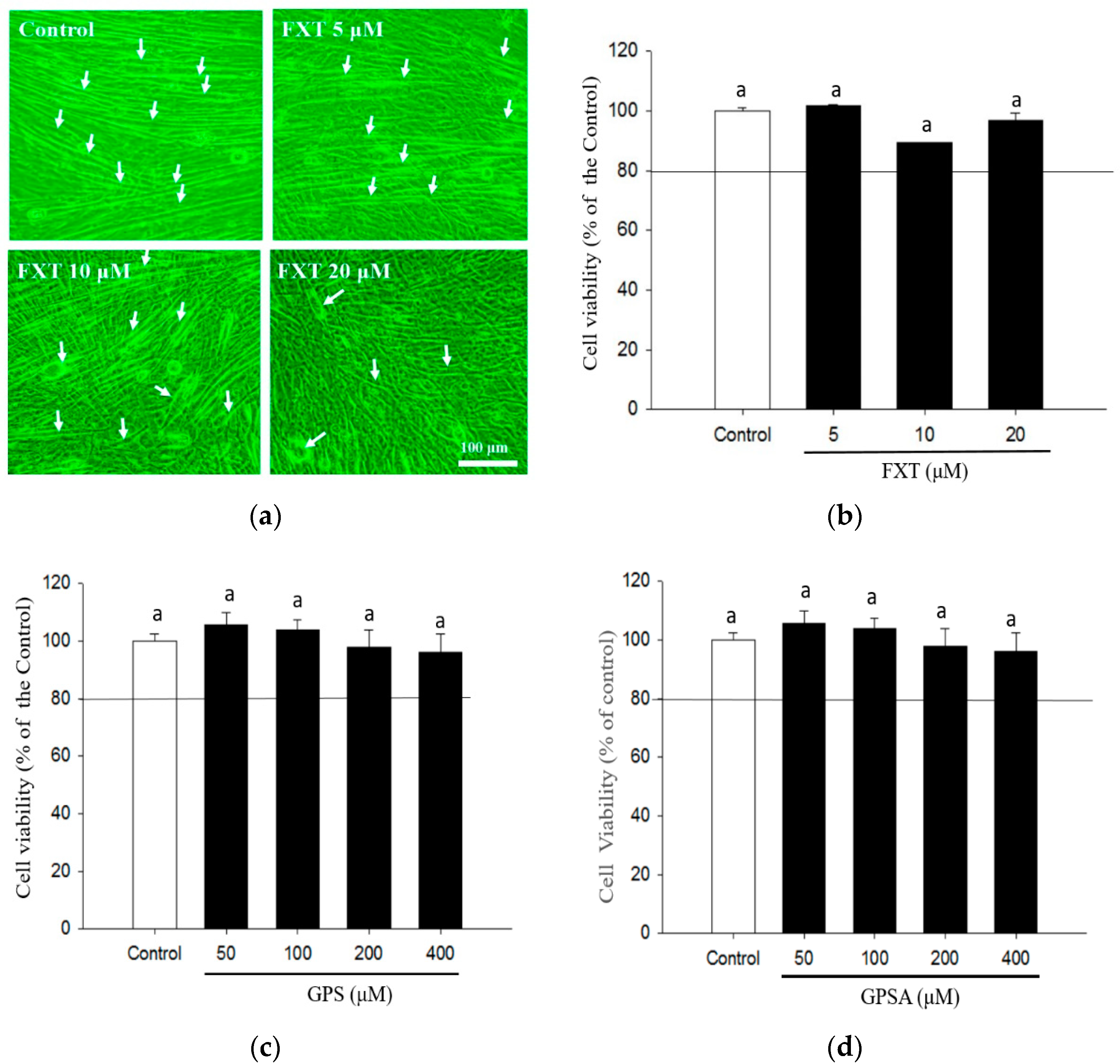
2.3. Cell Viability Assay
2.4. Analysis of Muscle Atrophy-Related Protein Expressions by Western Blot
2.5. Counting of Myotubes by Hematoxylin-Eosin Staining
2.6. Statistical Analysis
3. Results and Discussion
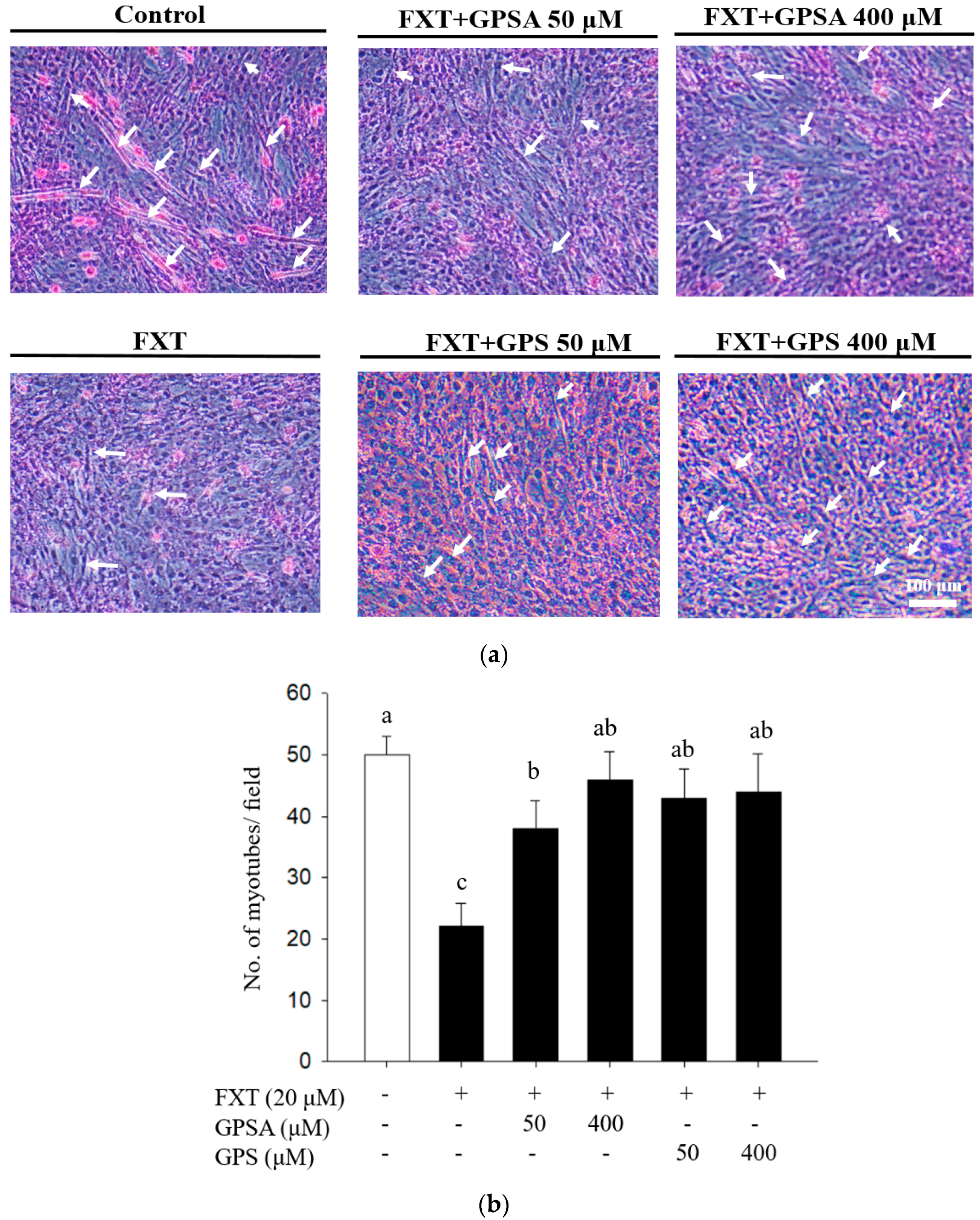
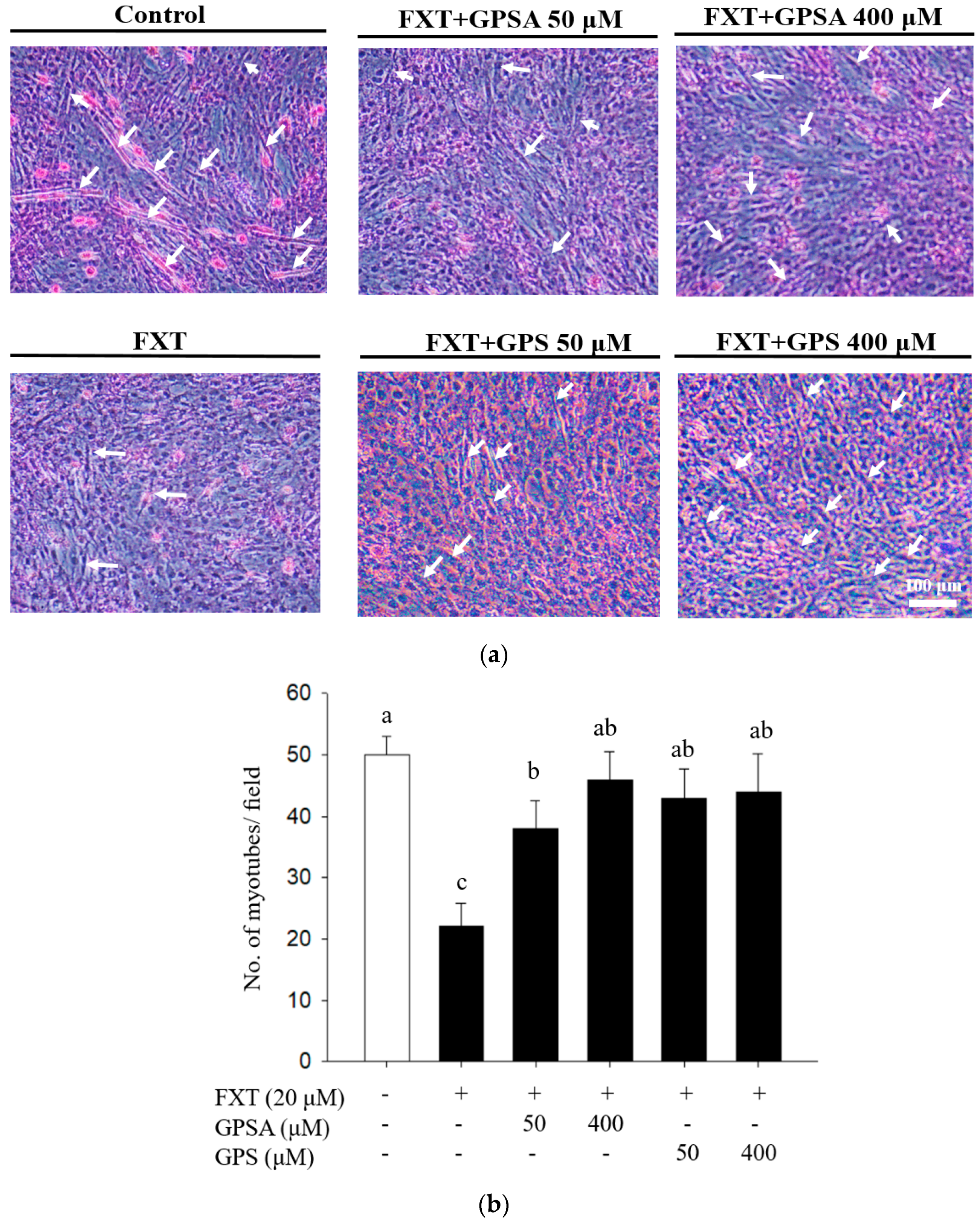
3.1. Effects of GPS and GPSA on FXT-Inhibited Differentiation and Myotube Formation in C2C12 Muscle Cells
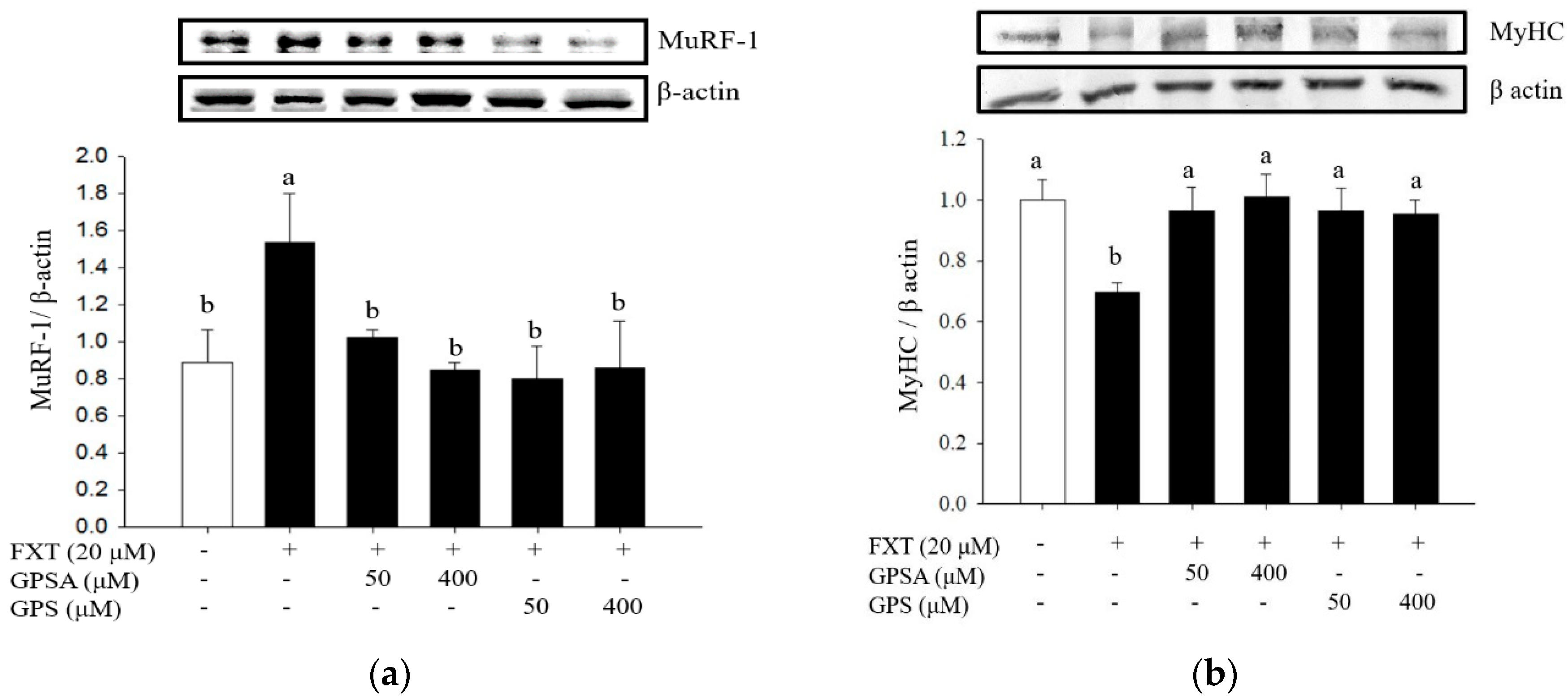
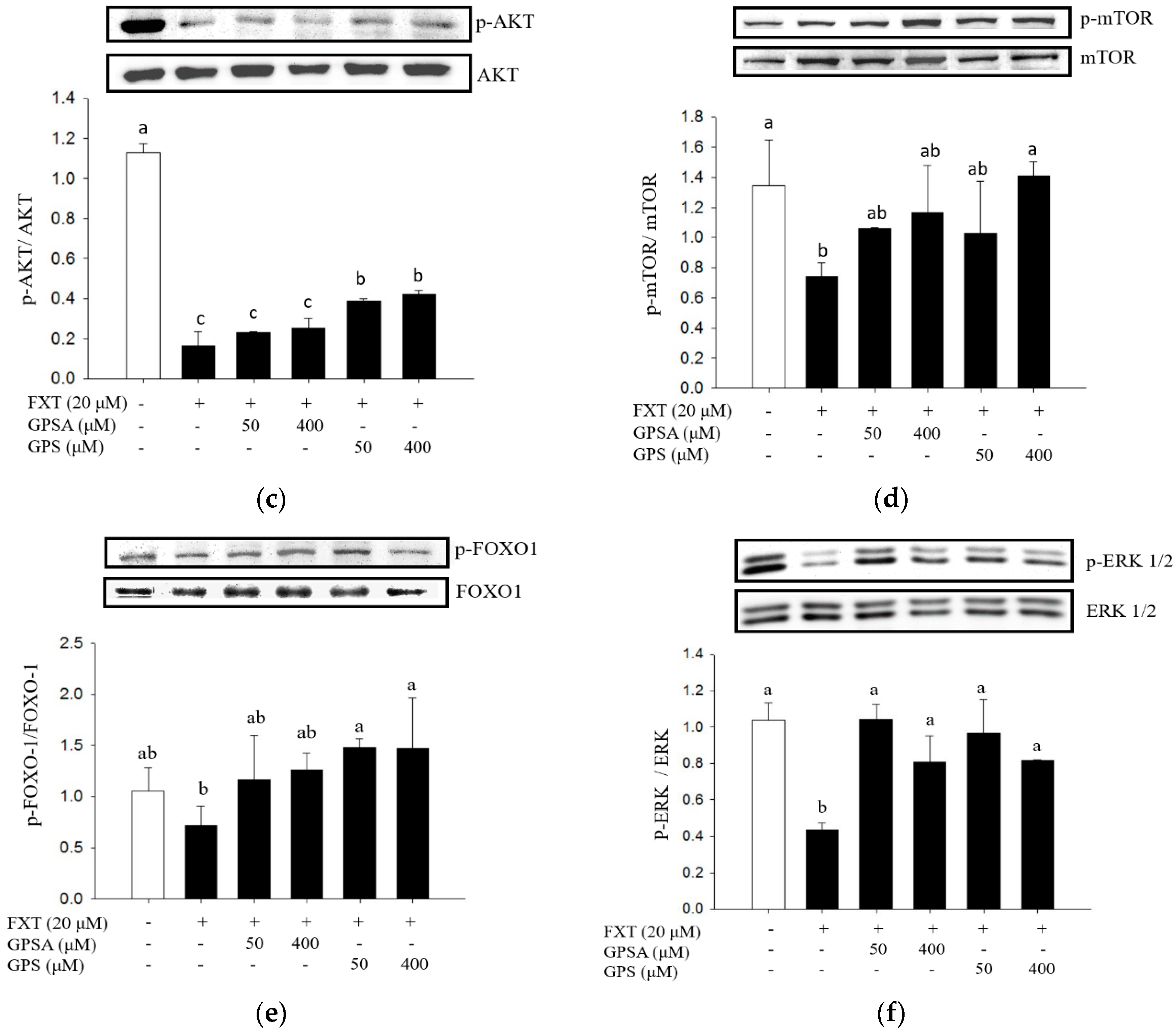
3.2. Effect of GPS or GPSA on the FXT-Induced Muscle Atrophy-Associated Protein Expression and Signaling
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gao, Y.; Arfat, Y.; Wang, H.; Goswami, N. Muscle Atrophy Induced by Mechanical Unloading: Mechanisms and Potential Countermeasures. Front. Physiol. 2018, 9, 235. [Google Scholar] [CrossRef]
- Lu, Y.; Ho, C.S.; Liu, X.; Chua, A.N.; Wang, W.; McIntyre, R.S.; Ho, R.C. Chronic administration of fluoxetine and pro-inflammatory cytokine change in a rat model of depression. PLoS ONE 2017, 12, e0186700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visco, D.B.; Manhães-De-Castro, R.; Chaves, W.F.; Lacerda, D.C.; da Conceição Pereira, S.; Ferraz-Pereira, K.N.; Toscano, A.E. Selective serotonin reuptake inhibitors affect structure, function and metabolism of skeletal muscle: A systematic review. Pharmacol. Res. 2018, 136, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Bonaldo, P.; Sandri, M. Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech. 2013, 6, 25–39. [Google Scholar] [CrossRef] [Green Version]
- McKinnell, I.W.; Rudnicki, M.A. Molecular Mechanisms of Muscle Atrophy. Cell 2004, 119, 907–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandri, M. Autophagy in health and disease. 3. Involvement of autophagy in muscle atrophy. Am. J. Physiol. Physiol. 2010, 298, C1291–C1297. [Google Scholar] [CrossRef] [Green Version]
- Sandri, M. Autophagy in skeletal muscle. FEBS Lett. 2010, 584, 1411–1416. [Google Scholar] [CrossRef]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.-H.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of autophagy and the ubiquitin–proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 2015, 6, 6670. [Google Scholar] [CrossRef] [Green Version]
- Sandri, M.; Barberi, L.; Bijlsma, A.Y.; Blaauw, B.; Dyar, K.; Milan, G.; Mammucari, C.; Meskers, C.; Pallafacchina, G.; Paoli, A.; et al. Signalling pathways regulating muscle mass in ageing skeletal muscle. The role of the IGF1-Akt-mTOR-FoxO pathway. Biogerontology 2013, 14, 303–323. [Google Scholar] [CrossRef]
- Sanchez, A.M.; Candau, R.B.; Bernardi, H. FoxO transcription factors: Their roles in the maintenance of skeletal muscle homeostasis. Cell. Mol. Life Sci. 2013, 71, 1657–1671. [Google Scholar] [CrossRef]
- Kim, S.; Koh, H. Role of FOXO transcription factors in crosstalk between mitochondria and the nucleus. J. Bioenerg. Biomembr. 2017, 16, 18224–18341. [Google Scholar] [CrossRef]
- Gross, D.N.; Van den Heuvel, A.P.J.; Birnbaum, M.J. The role of FoxO in the regulation of metabolism. Oncogene 2008, 27, 2320–2336. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Wong, E. Mitophagy Transcriptome: Mechanistic Insights into Polyphenol-Mediated Mitophagy. Oxid. Med. Cell. Longev. 2017, 2017, 9028435. [Google Scholar] [CrossRef] [PubMed]
- Webb, A.E.; Brunet, A. FOXO transcription factors: Key regulators of cellular quality control. Trends Biochem. Sci. 2014, 39, 159–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhang, F.; Xi, L.; Xia, W.; Wei, Y. Research Progress on Pharmacological Effects and Toxicity of Gardenia jasminoides Ellis and Related Mechanisms. Chin. J. Pharmacovigil. 2021, 18, 94. [Google Scholar]
- Li, N.; Li, L.; Wu, H.; Zhou, H. Antioxidative Property and Molecular Mechanisms Underlying Geniposide-Mediated Therapeutic Effects in Diabetes Mellitus and Cardiovascular Disease. Oxidative Med. Cell. Longev. 2019, 2019, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Liu, K.; Shi, M.; Xie, L.; Deng, M.; Chen, H.; Li, X. Therapeutic potential of catalpol and geniposide in Alzheimer’s and Parkinson’s diseases: A snapshot of their underlying mechanisms. Brain Res. Bull. 2021, 174, 281–295. [Google Scholar] [CrossRef]
- Lautaoja, J.H.; Pekkala, S.; Pasternack, A.; Laitinen, M.; Ritvos, O.; Hulmi, J.J. Differentiation of Murine C2C12 Myoblasts Strongly Reduces the Effects of Myostatin on Intracellular Signaling. Biomolecules 2020, 10, 695. [Google Scholar] [CrossRef]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the MTT Assay. Cold Spring Harb. Protoc. 2018, 2018, 095505. [Google Scholar] [CrossRef]
- Michael, K. Relationship of skeletal muscle atrophy to functional status: A systematic research review. Biol. Res. Nurs. 2000, 2, 117–131. [Google Scholar] [CrossRef]
- Rudrappa, S.S.; Wilkinson, D.J.; Greenhaff, P.L.; Smith, K.; Idris, I.; Atherton, P.J. Human Skeletal Muscle Disuse Atrophy: Effects on Muscle Protein Synthesis, Breakdown, and Insulin Resistance—A Qualitative Review. Front. Physiol. 2016, 7, 361. [Google Scholar] [CrossRef]
- Ryu, Y.; Lee, D.; Jung, S.H.; Lee, K.-J.; Jin, H.; Kim, S.J.; Lee, H.M.; Kim, B.; Won, K.-J. Sabinene Prevents Skeletal Muscle Atrophy by Inhibiting the MAPK-MuRF-1 Pathway in Rats. Int. J. Mol. Sci. 2019, 20, 4955. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Han, J.; Meng, Q.; Xi, Q.; Zhuang, Q.; Jiang, Y.; Han, Y.; Zhang, B.; Fang, J.; Wu, G. Muscle-specific E3 ubiquitin ligases are involved in muscle atrophy of cancer cachexia: An in vitro and in vivo study. Oncol. Rep. 2015, 33, 2261–2268. [Google Scholar] [CrossRef] [Green Version]
- Baehr, L.M.; Furlow, J.D.; Bodine, S.C. Muscle sparing in muscle RING finger 1 null mice: Response to synthetic glucocorticoids. J. Physiol. 2011, 589, 4759–4776. [Google Scholar] [CrossRef] [Green Version]
- Ferraro, E.; Pin, F.; Gorini, S.; Pontecorvo, L.; Ferri, A.; Mollace, V.; Costelli, P.; Rosano, G. Improvement of skeletal muscle performance in ageing by the metabolic modulator Trimetazidine. J. Cachexia Sarcopenia Muscle 2016, 7, 449–457. [Google Scholar] [CrossRef]
- Yadav, A.; Singh, A.; Phogat, J.; Dahuja, A.; Dabur, R. Magnoflorine prevent the skeletal muscle atrophy via Akt/mTOR/FoxO signal pathway and increase slow-MyHC production in streptozotocin-induced diabetic rats. J. Ethnopharmacol. 2021, 267, 113510. [Google Scholar] [CrossRef]
- Wang, M.; Ren, J.; Chen, X.; Liu, J.; Xu, X.; Li, X.; Zhao, D.; Sun, L. 20(S)-ginsenoside Rg3 promotes myoblast differentiation and protects against myotube atrophy via regulation of the Akt/mTOR/FoxO3 pathway. Biochem. Pharmacol. 2020, 180, 114145. [Google Scholar] [CrossRef]
- Zheng, R.; Huang, S.; Zhu, J.; Lin, W.; Xu, H.; Zheng, X. Leucine attenuates muscle atrophy and autophagosome formation by activating PI3K/AKT/mTOR signaling pathway in rotator cuff tears. Cell Tissue Res. 2019, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.H.; Jang, E.J.; Kim, Y.W.; Lee, J.-H. Sulforaphane prevents dexamethasone-induced muscle atrophy via regulation of the Akt/Foxo1 axis in C2C12 myotubes. Biomed. Pharmacother. 2017, 95, 1486–1492. [Google Scholar] [CrossRef]
- Kim, J.; Guan, K.-L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef]
- Sartori, R.; Romanello, V.; Sandri, M. Mechanisms of muscle atrophy and hypertrophy: Implications in health and disease. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Menghini, R.; Casagrande, V.; Iuliani, G.; Rizza, S.; Mavilio, M.; Cardellini, M.; Federici, M. Metabolic aspects of cardiovascular diseases: Is FoxO1 a player or a target? Int. J. Biochem. Cell Biol. 2020, 118, 105659. [Google Scholar] [CrossRef]
- Matsuda, R.; Uchitomi, R.; Oyabu, M.; Hatazawa, Y.; Kamei, Y. Metabolomic analysis of C2C12 myoblasts induced by the transcription factor FOXO 1. FEBS Lett. 2019, 593, 1303–1312. [Google Scholar] [CrossRef]
- Penna, F.; Costamagna, D.; Fanzani, A.; Bonelli, G.; Baccino, F.M.; Costelli, P. Muscle Wasting and Impaired Myogenesis in Tumor Bearing Mice Are Prevented by ERK Inhibition. PLoS ONE 2010, 5, e13604. [Google Scholar] [CrossRef]
- Swiderski, K.; Brock, C.J.; Trieu, J.; Chee, A.; Thakur, S.S.; Baum, D.M.; Gregorevic, P.; Murphy, K.T.; Lynch, G.S. Phosphorylation of ERK and dystrophin S3059 protects against inflammation-associated C2C12 myotube atrophy. Am. J. Physiol. Physiol. 2021, 320, C956–C965. [Google Scholar] [CrossRef]
- Karimi-Khouzani, O.; Heidarian, E.; Amini, S.A. Anti-inflammatory and ameliorative effects of gallic acid on fluoxetine-induced oxidative stress and liver damage in rats. Pharmacol. Rep. 2017, 69, 830–835. [Google Scholar] [CrossRef]
- Inkielewicz-Stępniak, I. Impact of fluoxetine on liver damage in rats. Pharmacol. Rep. 2011, 63, 441–447. [Google Scholar] [CrossRef]
- Feng, X.-M.; Xiong, J.; Qin, H.; Liu, W.; Chen, R.-N.; Shang, W.; Ning, R.; Hu, G.; Yang, J. Fluoxetine Induces Hepatic Lipid Accumulation Via Both Promotion of the SREBP1c-Related Lipogenesis and Reduction of Lipolysis in Primary Mouse Hepatocytes. CNS Neurosci. Ther. 2012, 18, 974–980. [Google Scholar] [CrossRef] [PubMed]
- Pieczenik, S.R.; Neustadt, J. Mitochondrial dysfunction and molecular pathways of disease. Exp. Mol. Pathol. 2007, 83, 84–92. [Google Scholar] [CrossRef]
- Chung, L.-H.; Liu, S.-T.; Huang, S.-M.; Salter, D.M.; Lee, H.-S.; Hsu, Y.-J. High phosphate induces skeletal muscle atrophy and suppresses myogenic differentiation by increasing oxidative stress and activating Nrf2 signaling. Aging 2020, 12, 21446–21468. [Google Scholar] [CrossRef]
- Kang, J.S.; Han, M.H.; Kim, G.-Y.; Kim, C.M.; Kim, B.W.; Hwang, H.J.; Choi, Y.H. Nrf2-Mediated HO-1 Induction Contributes to Antioxidant Capacity of a Schisandrae Fructus Ethanol Extract in C2C12 Myoblasts. Nutrients 2014, 6, 5667–5678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitman, S.A.; Long, M.; Wondrak, G.T.; Zheng, H.; Zhang, D.D. Nrf2 modulates contractile and metabolic properties of skeletal muscle in streptozotocin-induced diabetic atrophy. Exp. Cell Res. 2013, 319, 2673–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, S.-M.; Lin, S.-Y.; Chen, M.-K.; Peng, C.-C.; Hsieh, C.-L. Effects of Geniposide and Geniposidic Acid on Fluoxetine-Induced Muscle Atrophy in C2C12 Cells. Processes 2021, 9, 1649. https://doi.org/10.3390/pr9091649
Huang S-M, Lin S-Y, Chen M-K, Peng C-C, Hsieh C-L. Effects of Geniposide and Geniposidic Acid on Fluoxetine-Induced Muscle Atrophy in C2C12 Cells. Processes. 2021; 9(9):1649. https://doi.org/10.3390/pr9091649
Chicago/Turabian StyleHuang, Shang-Ming, Shuan-Ying Lin, Ming-Kai Chen, Chiung-Chi Peng, and Chiu-Lan Hsieh. 2021. "Effects of Geniposide and Geniposidic Acid on Fluoxetine-Induced Muscle Atrophy in C2C12 Cells" Processes 9, no. 9: 1649. https://doi.org/10.3390/pr9091649
APA StyleHuang, S.-M., Lin, S.-Y., Chen, M.-K., Peng, C.-C., & Hsieh, C.-L. (2021). Effects of Geniposide and Geniposidic Acid on Fluoxetine-Induced Muscle Atrophy in C2C12 Cells. Processes, 9(9), 1649. https://doi.org/10.3390/pr9091649