
Exploring Electrochemically Mediated ATRP of Styrene


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instrumentation
2.3. Procedures
2.3.1. Typical Procedure for eATRP of Styrene
2.3.2. Preparation of PS-Br Macroinitiator
2.3.3. Procedure for Temporally Controlled eATRP
2.3.4. Chain Extension of PS-Br by eATRP
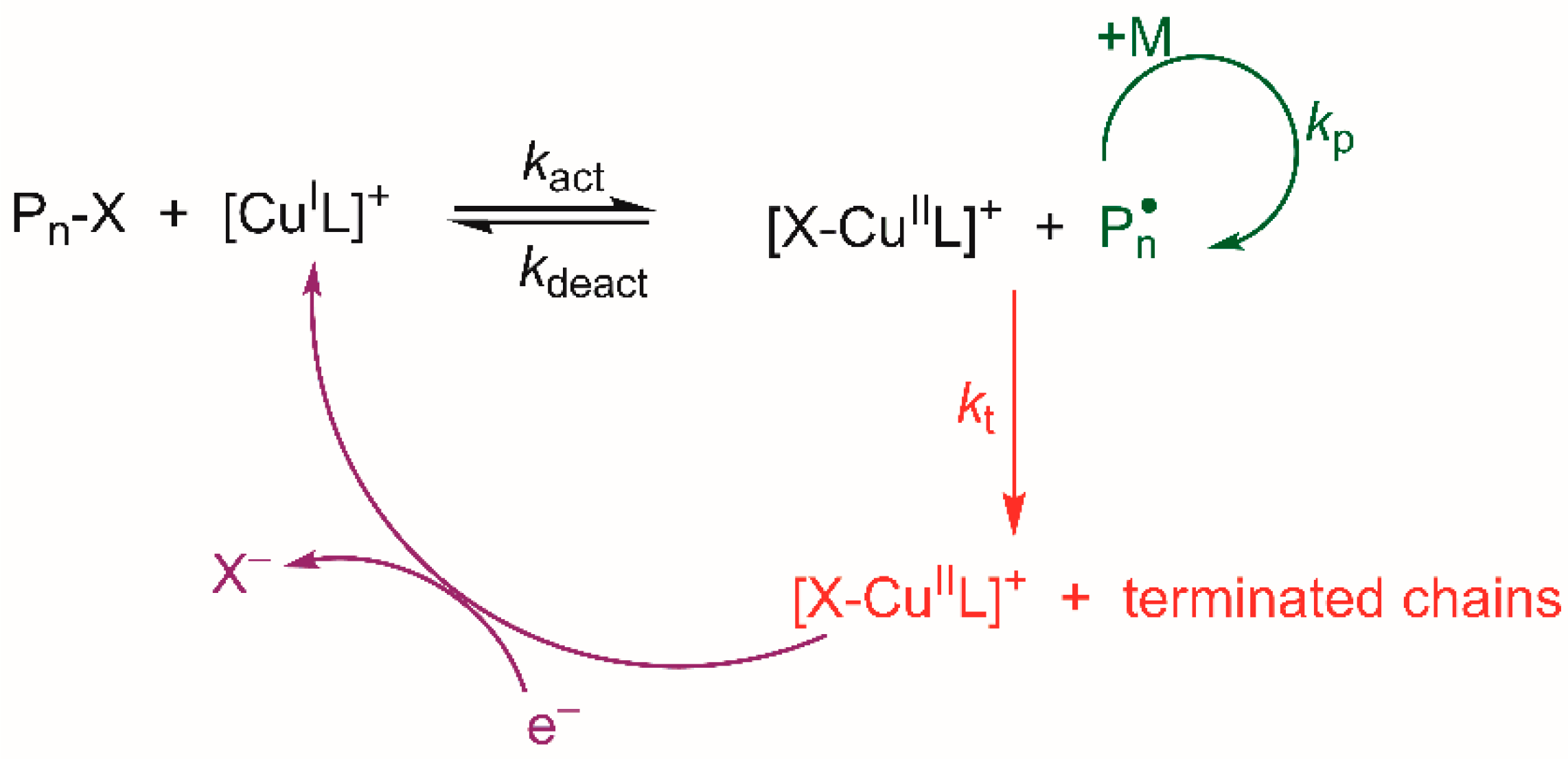
3. Results and Discussion
3.1. Voltammetric Behavior of the Catalyst
3.2. Electrochemically Mediated ATRP of Styrene
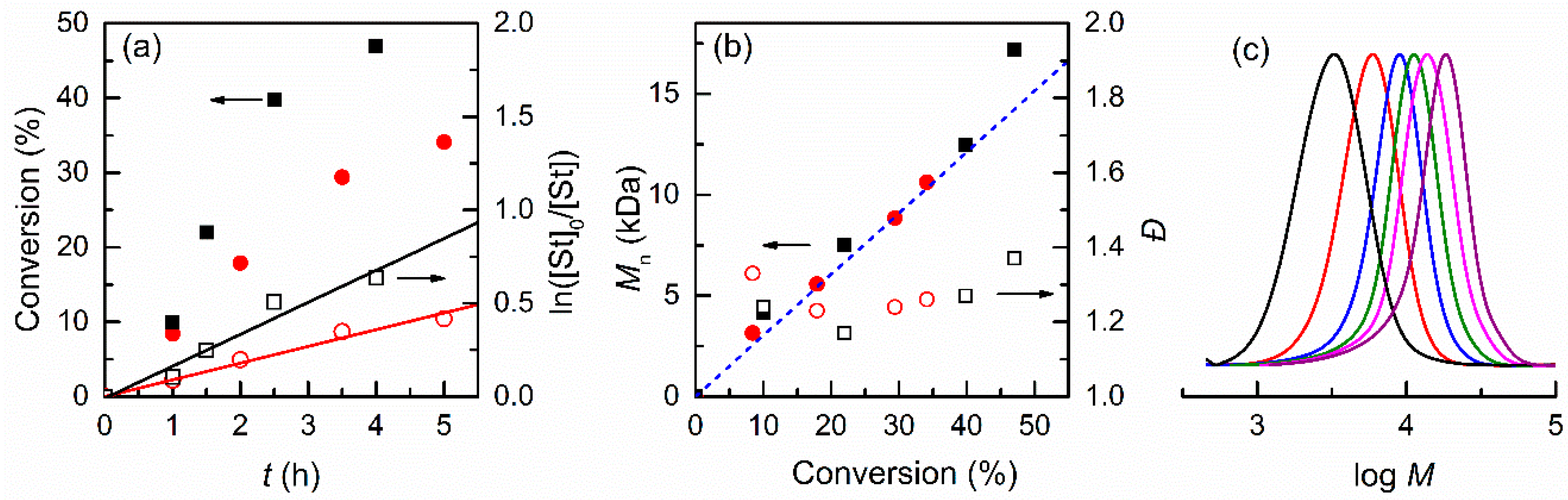
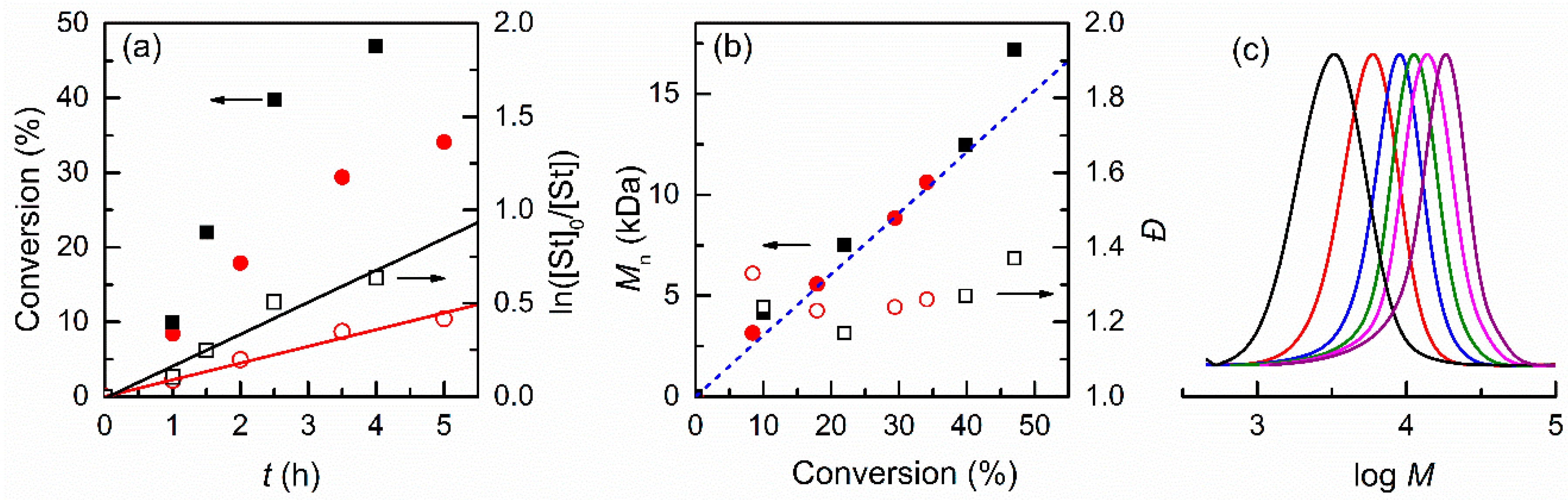
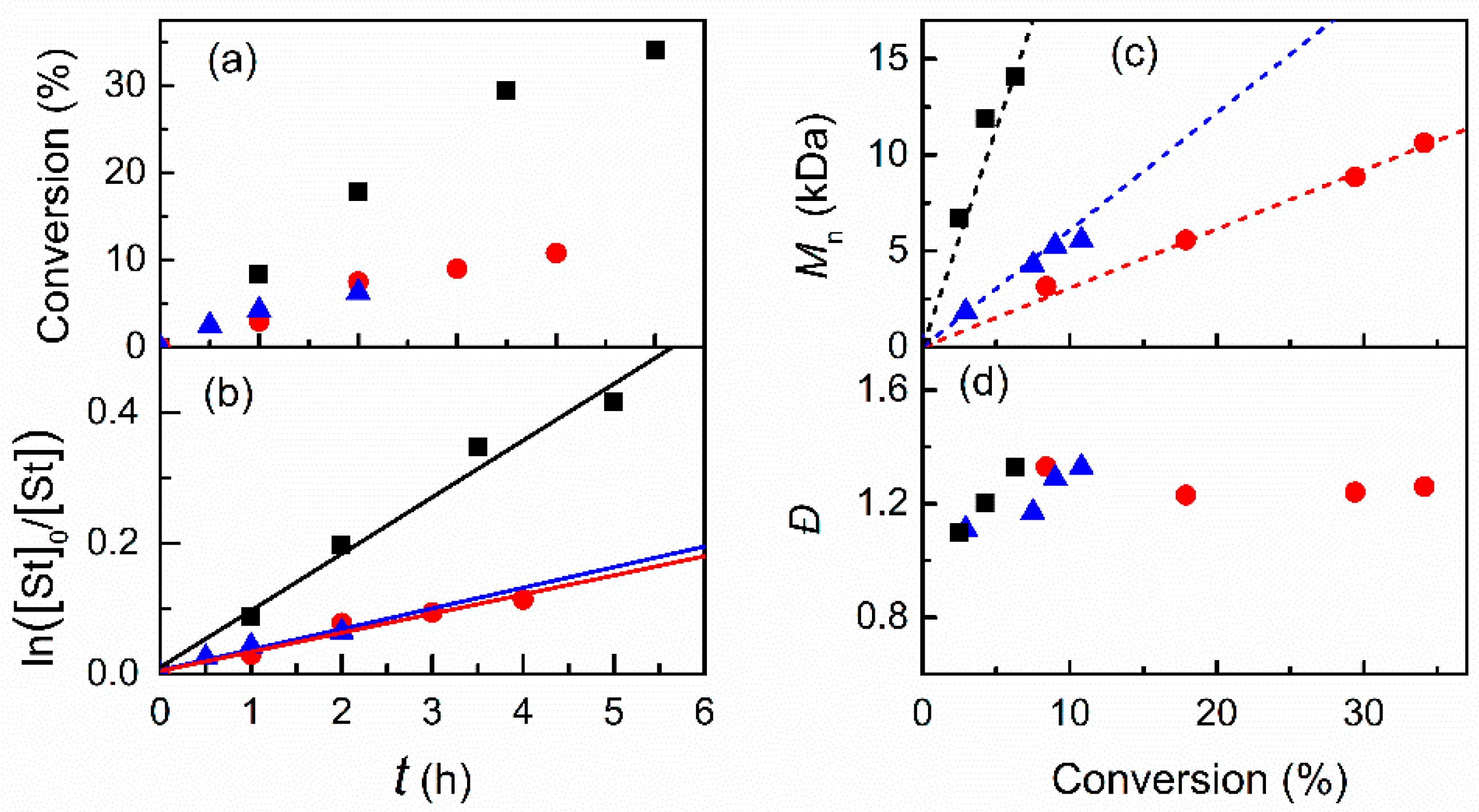
3.2.1. Effect of the Solvent
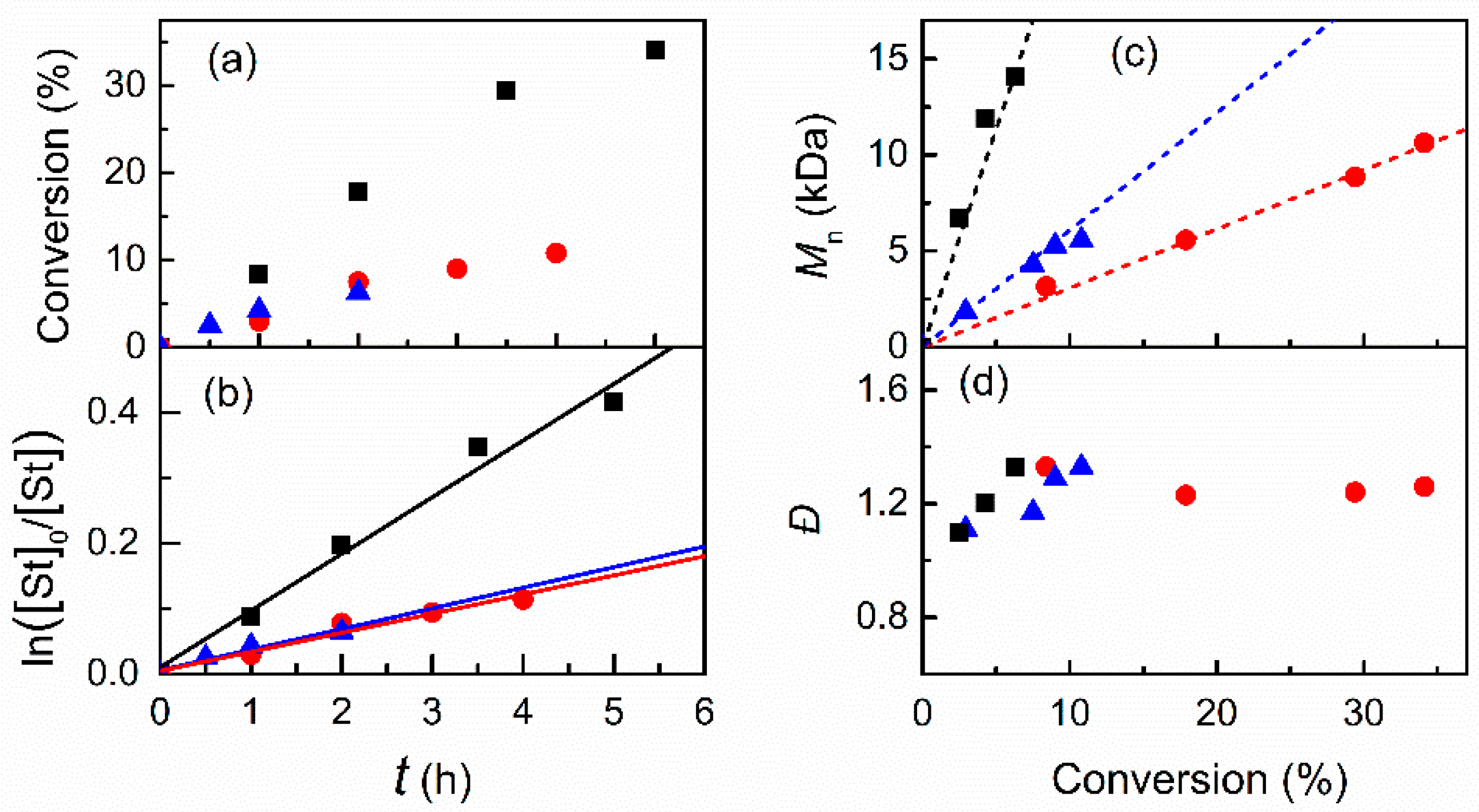
3.2.2. Effect of Initiator and Concentrations of Catalyst and Monomer in DMF
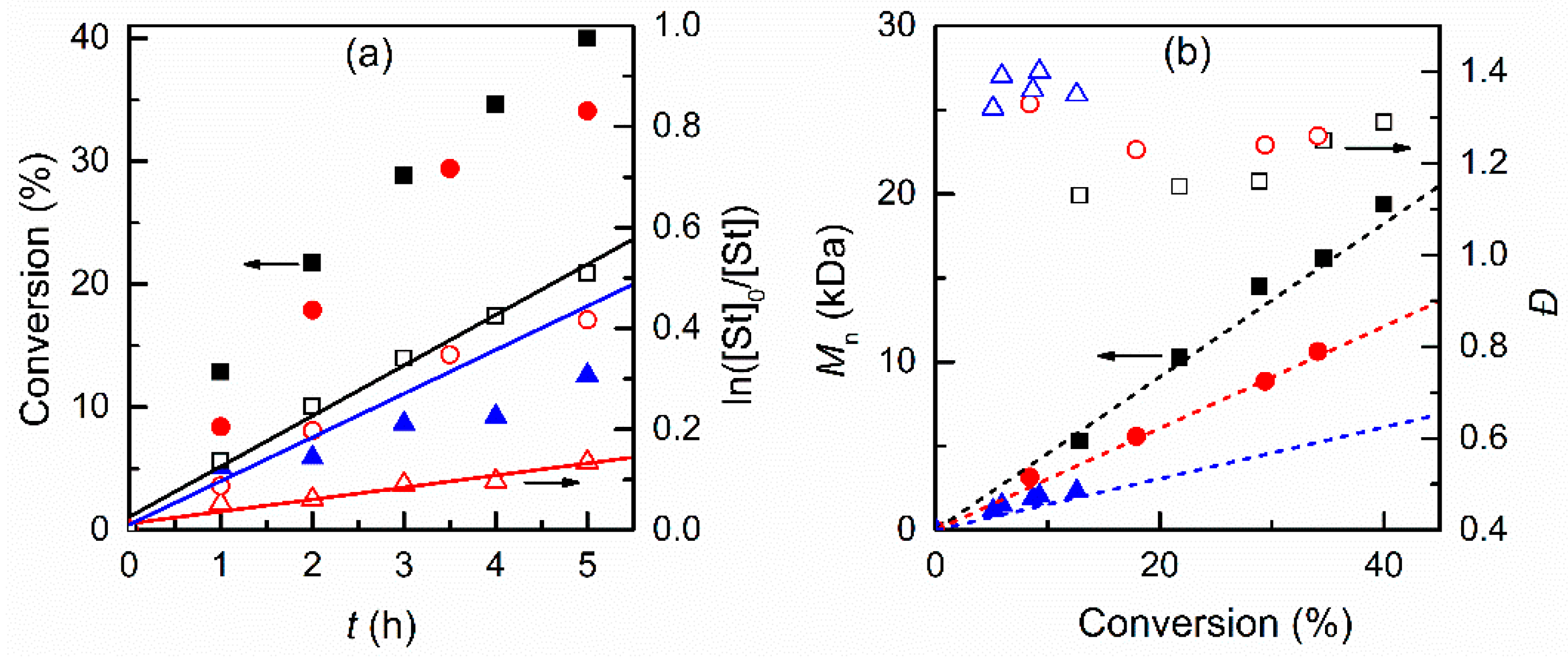
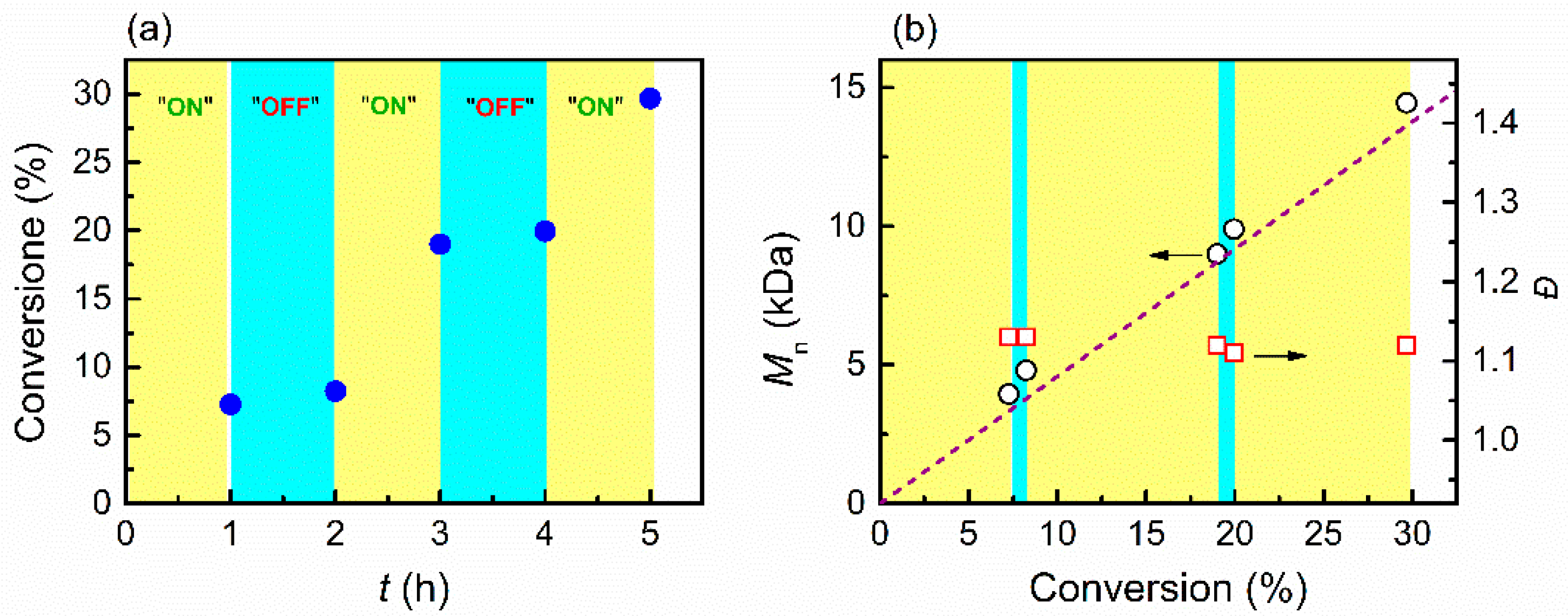
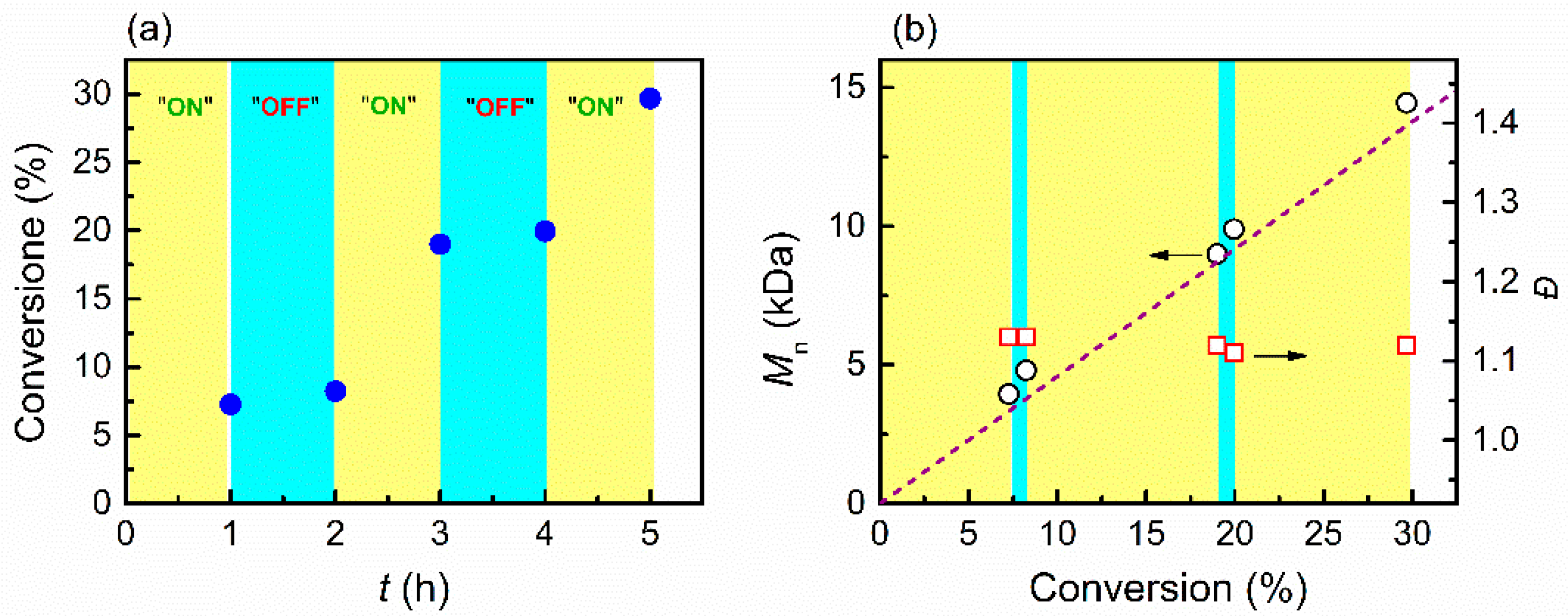
3.3. Temporal Control of Polymerization
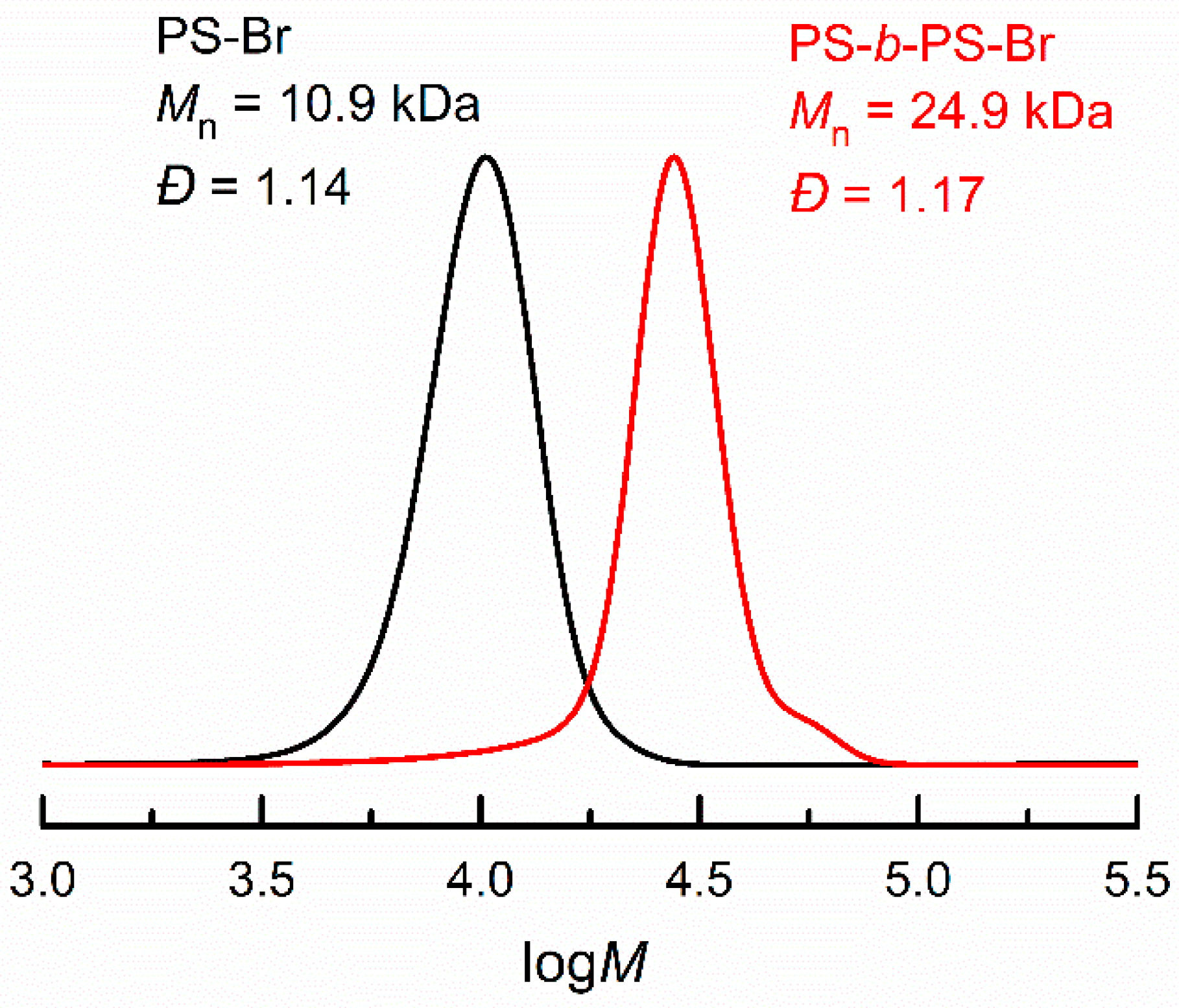
3.4. Electrochemical Chain Extension
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matyjaszewski, K. Atom Transfer Radical Polymerization (ATRP): Current Status and Future Perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Gao, H.; Sumerlin, B.S.; Tsarevsky, N.V. (Eds.) Reversible Deactivation Radical Polymerization: Materials and Applications; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2018; Volume 1285, ISBN 9780841233232. [Google Scholar]
- Matyjaszewski, K.; Jakubowski, W.; Min, K.; Tang, W.; Huang, J.; Braunecker, W.A.; Tsarevsky, N.V. Diminishing Catalyst Concentration in Atom Transfer Radical Polymerization with Reducing Agents. Proc. Natl. Acad. Sci. USA 2006, 103, 15309–15314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konkolewicz, D.; Magenau, A.J.D.; Averick, S.E.; Simakova, A.; He, H.; Matyjaszewski, K. ICAR ATRP with Ppm Cu Catalyst in Water. Macromolecules 2012, 45, 4461–4468. [Google Scholar] [CrossRef]
- Min, K.; Gao, H.; Matyjaszewski, K. Use of Ascorbic Acid as Reducing Agent for Synthesis of Well-Defined Polymers by ARGET ATRP. Macromolecules 2007, 40, 1789–1791. [Google Scholar] [CrossRef]
- Chan, N.; Cunningham, M.F.; Hutchinson, R.A. ARGET ATRP of Methacrylates and Acrylates with Stoichiometric Ratios of Ligand to Copper. Macromol. Chem. Phys. 2008, 209, 1797–1805. [Google Scholar] [CrossRef]
- Simakova, A.; Averick, S.E.; Konkolewicz, D.; Matyjaszewski, K. Aqueous ARGET ATRP. Macromolecules 2012, 45, 6371–6379. [Google Scholar] [CrossRef]
- Mendonça, P.V.; Ribeiro, J.P.M.; Abreu, C.M.R.; Guliashvili, T.; Serra, A.C.; Coelho, J.F.J. Thiourea Dioxide As a Green and Affordable Reducing Agent for the ARGET ATRP of Acrylates, Methacrylates, Styrene, Acrylonitrile, and Vinyl Chloride. ACS Macro Lett. 2019, 8, 315–319. [Google Scholar] [CrossRef]
- Abreu, C.M.R.; Serra, A.C.; Popov, A.V.; Matyjaszewski, K.; Guliashvili, T.; Coelho, J.F.J. Ambient Temperature Rapid SARA ATRP of Acrylates and Methacrylates in Alcohol–Water Solutions Mediated by a Mixed Sulfite/Cu(II)Br2 Catalytic System. Polym. Chem. 2013, 4, 5629. [Google Scholar] [CrossRef]
- Mendes, J.P.; Branco, F.; Abreu, C.M.R.; Mendonça, P.V.; Serra, A.C.; Popov, A.V.; Guliashvili, T.; Coelho, J.F.J. Sulfolane: An Efficient and Universal Solvent for Copper-Mediated Atom Transfer Radical (Co)Polymerization of Acrylates, Methacrylates, Styrene, and Vinyl Chloride. ACS Macro Lett. 2014, 3, 858–861. [Google Scholar] [CrossRef]
- Lorandi, F.; Fantin, M.; Isse, A.A.; Gennaro, A. RDRP in the Presence of Cu0: The Fate of Cu(I) Proves the Inconsistency of SET-LRP Mechanism. Polymer 2015, 72, 238–245. [Google Scholar] [CrossRef]
- Kopeć, M.; Yuan, R.; Gottlieb, E.; Abreu, C.M.R.; Song, Y.; Wang, Z.; Coelho, J.F.J.; Matyjaszewski, K.; Kowalewski, T. Polyacrylonitrile- b -Poly(Butyl Acrylate) Block Copolymers as Precursors to Mesoporous Nitrogen-Doped Carbons: Synthesis and Nanostructure. Macromolecules 2017, 50, 2759–2767. [Google Scholar] [CrossRef]
- Tasdelen, M.A.; Uygun, M.; Yagci, Y. Photoinduced Controlled Radical Polymerization. Macromol. Rapid Commun. 2011, 32, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Konkolewicz, D.; Schröder, K.; Buback, J.; Bernhard, S.; Matyjaszewski, K. Visible Light and Sunlight Photoinduced ATRP with Ppm of Cu Catalyst. ACS Macro Lett. 2012, 1, 1219–1223. [Google Scholar] [CrossRef]
- Anastasaki, A.; Nikolaou, V.; Zhang, Q.; Burns, J.; Samanta, S.R.; Waldron, C.; Haddleton, A.J.; McHale, R.; Fox, D.; Percec, V.; et al. Copper(II)/Tertiary Amine Synergy in Photoinduced Living Radical Polymerization: Accelerated Synthesis of ω-Functional and α,ω-Heterofunctional Poly(Acrylates). J. Am. Chem. Soc. 2014, 136, 1141–1149. [Google Scholar] [CrossRef]
- Treat, N.J.; Sprafke, H.; Kramer, J.W.; Clark, P.G.; Barton, B.E.; Read de Alaniz, J.; Fors, B.P.; Hawker, C.J. Metal-Free Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 2014, 136, 16096–16101. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, G.; Yagci, Y. Photoinduced Metal-Free Atom Transfer Radical Polymerizations: State-of-the-Art, Mechanistic Aspects and Applications. Polym. Chem. 2018, 9, 1757–1762. [Google Scholar] [CrossRef]
- Pan, X.; Fang, C.; Fantin, M.; Malhotra, N.; So, W.Y.; Peteanu, L.A.; Isse, A.A.; Gennaro, A.; Liu, P.; Matyjaszewski, K. Mechanism of Photoinduced Metal-Free Atom Transfer Radical Polymerization: Experimental and Computational Studies. J. Am. Chem. Soc. 2016, 138, 2411–2425. [Google Scholar] [CrossRef] [PubMed]
- Magenau, A.J.D.; Strandwitz, N.C.; Gennaro, A.; Matyjaszewski, K. Electrochemically Mediated Atom Transfer Radical Polymerization. Science 2011, 332, 81–84. [Google Scholar] [CrossRef]
- Magenau, A.J.D.; Bortolamei, N.; Frick, E.; Park, S.; Gennaro, A.; Matyjaszewski, K. Investigation of Electrochemically Mediated Atom Transfer Radical Polymerization. Macromolecules 2013, 46, 4346–4353. [Google Scholar] [CrossRef]
- Lorandi, F.; Fantin, M.; Isse, A.A.; Gennaro, A. Electrochemically Mediated Atom Transfer Radical Polymerization of N-Butyl Acrylate on Non-Platinum Cathodes. Polym. Chem. 2016, 7, 5357–5365. [Google Scholar] [CrossRef]
- Fantin, M.; Lorandi, F.; Isse, A.A.; Gennaro, A. Sustainable Electrochemically-Mediated Atom Transfer Radical Polymerization with Inexpensive Non-Platinum Electrodes. Macromol. Rapid Commun. 2016, 37, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Chmielarz, P.; Fantin, M.; Park, S.; Isse, A.A.; Gennaro, A.; Magenau, A.J.D.; Sobkowiak, A.; Matyjaszewski, K. Electrochemically Mediated Atom Transfer Radical Polymerization (eATRP). Prog. Polym. Sci. 2017, 69, 47–78. [Google Scholar] [CrossRef]
- Lorandi, F.; Fantin, M.; Isse, A.A.; Gennaro, A. Electrochemical Triggering and Control of Atom Transfer Radical Polymerization. Curr. Opin. Electrochem. 2018, 8, 1–7. [Google Scholar] [CrossRef]
- Mohapatra, H.; Kleiman, M.; Esser-Kahn, A.P. Mechanically Controlled Radical Polymerization Initiated by Ultrasound. Nat. Chem. 2017, 9, 135–139. [Google Scholar] [CrossRef]
- Wang, Z.; Pan, X.; Li, L.; Fantin, M.; Yan, J.; Wang, Z.; Wang, Z.; Xia, H.; Matyjaszewski, K. Enhancing Mechanically Induced ATRP by Promoting Interfacial Electron Transfer from Piezoelectric Nanoparticles to Cu Catalysts. Macromolecules 2017, 50, 7940–7948. [Google Scholar] [CrossRef]
- Zaborniak, I.; Chmielarz, P. Ultrasound-Mediated Atom Transfer Radical Polymerization (ATRP). Materials 2019, 12, 3600. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Fantin, M.; Yuan, F.; Matyjaszewski, K. Externally Controlled Atom Transfer Radical Polymerization. Chem. Soc. Rev. 2018, 47, 5457–5490. [Google Scholar] [CrossRef]
- Ribelli, T.G.; Lorandi, F.; Fantin, M.; Matyjaszewski, K. Atom Transfer Radical Polymerization: Billion Times More Active Catalysts and New Initiation Systems. Macromol. Rapid Commun. 2019, 40, 1800616. [Google Scholar] [CrossRef]
- De Paoli, P.; Isse, A.A.; Bortolamei, N.; Gennaro, A. New Insights into the Mechanism of Activation of Atom Transfer Radical Polymerization by Cu(I) Complexes. Chem. Commun. 2011, 47, 3580–3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isse, A.A.; Bortolamei, N.; De Paoli, P.; Gennaro, A. On the Mechanism of Activation of Copper-Catalyzed Atom Transfer Radical Polymerization. Electrochim. Acta 2013, 110, 655–662. [Google Scholar] [CrossRef]
- Fantin, M.; Lorandi, F.; Gennaro, A.; Isse, A.; Matyjaszewski, K. Electron Transfer Reactions in Atom Transfer Radical Polymerization. Synthesis 2017, 49, 3311–3322. [Google Scholar] [CrossRef]
- Penczek, S.; Pretula, J.; Lewiński, P. Dormant Polymers and Their Role in Living and Controlled Polymerizations; Influence on Polymer Chemistry, Particularly on the Ring Opening Polymerization. Polymers 2017, 9, 646. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Kwak, Y.; Braunecker, W.; Tsarevsky, N.V.; Coote, M.L.; Matyjaszewski, K. Understanding Atom Transfer Radical Polymerization: Effect of Ligand and Initiator Structures on the Equilibrium Constants. J. Am. Chem. Soc. 2008, 130, 10702–10713. [Google Scholar] [CrossRef] [PubMed]
- Braunecker, W.A.; Tsarevsky, N.V.; Gennaro, A.; Matyjaszewski, K. Thermodynamic Components of the Atom Transfer Radical Polymerization Equilibrium: Quantifying Solvent Effects. Macromolecules 2009, 42, 6348–6360. [Google Scholar] [CrossRef]
- Buback, M.; Morick, J. Equilibrium Constants and Activation Rate Coefficients for Atom Transfer Radical Polymerizations at Pressures up to 2 500 Bar. Macromol. Chem. Phys. 2010, 211, 2154–2161. [Google Scholar] [CrossRef]
- Wang, Y.; Kwak, Y.; Buback, J.; Buback, M.; Matyjaszewski, K. Determination of ATRP Equilibrium Constants under Polymerization Conditions. ACS Macro Lett. 2012, 1, 1367–1370. [Google Scholar] [CrossRef]
- Lorandi, F.; Fantin, M.; Isse, A.A.; Gennaro, A.; Matyjaszewski, K. New Protocol to Determine the Equilibrium Constant of Atom Transfer Radical Polymerization. Electrochim. Acta 2018, 260, 648–655. [Google Scholar] [CrossRef]
- De Bon, F.; Isse, A.A.; Gennaro, A. Electrochemically Mediated Atom Transfer Radical Polymerization of Methyl Methacrylate: The Importance of Catalytic Halogen Exchange. Chem. Electro. Chem. 2019, 6, 4257–4265. [Google Scholar] [CrossRef]
- Chmielarz, P.; Sobkowiak, A.; Matyjaszewski, K. A Simplified Electrochemically Mediated ATRP Synthesis of PEO-b-PMMA Copolymers. Polymer 2015, 77, 266–271. [Google Scholar] [CrossRef] [Green Version]
- Chmielarz, P.; Yan, J.; Krys, P.; Wang, Y.; Wang, Z.; Bockstaller, M.R.; Matyjaszewski, K. Synthesis of Nanoparticle Copolymer Brushes via Surface-Initiated seATRP. Macromolecules 2017, 50, 4151–4159. [Google Scholar] [CrossRef]
- Wang, J.; Tian, M.; Li, S.; Wang, R.; Du, F.; Xue, Z. Ligand-Free Iron-Based Electrochemically Mediated Atom Transfer Radical Polymerization of Methyl Methacrylate. Polym. Chem. 2018, 9, 4386–4394. [Google Scholar] [CrossRef]
- De Bon, F.; Ribeiro, D.C.M.; Abreu, C.M.R.; Rebelo, R.A.C.; Isse, A.A.; Serra, A.C.; Gennaro, A.; Matyjaszewski, K.; Coelho, J.F.J. Under Pressure: Electrochemically-Mediated Atom Transfer Radical Polymerization of Vinyl Chloride. Polym. Chem. 2020, 11, 6745–6762. [Google Scholar] [CrossRef]
- Guo, J.-K.; Zhou, Y.-N.; Luo, Z.-H. Iron-Based Electrochemically Mediated Atom Transfer Radical Polymerization with Tunable Catalytic Activity. AIChE J. 2018, 64, 961–969. [Google Scholar] [CrossRef]
- De Bon, F.; Isse, A.A.; Gennaro, A. Towards Scale-up of Electrochemically-Mediated Atom Transfer Radical Polymerization: Use of a Stainless-Steel Reactor as Both Cathode and Reaction Vessel. Electrochim. Acta 2019, 304, 505–512. [Google Scholar] [CrossRef]
- Zaborniak, I.; Chmielarz, P.; Martinez, M.R.; Wolski, K.; Wang, Z.; Matyjaszewski, K. Synthesis of High Molecular Weight Poly(n-Butyl Acrylate) Macromolecules via seATRP: From Polymer Stars to Molecular Bottlebrushes. Eur. Polym. J. 2020, 126, 109566. [Google Scholar] [CrossRef]
- Chmielarz, P.; Park, S.; Simakova, A.; Matyjaszewski, K. Electrochemically Mediated ATRP of Acrylamides in Water. Polymer 2015, 60, 302–307. [Google Scholar] [CrossRef]
- Fantin, M.; Isse, A.A.; Gennaro, A.; Matyjaszewski, K. Understanding the Fundamentals of Aqueous ATRP and Defining Conditions for Better Control. Macromolecules 2015, 48, 6862–6875. [Google Scholar] [CrossRef]
- Strover, L.T.; Malmström, J.; Stubbing, L.A.; Brimble, M.A.; Travas-Sejdic, J. Electrochemically-Controlled Grafting of Hydrophilic Brushes from Conducting Polymer Substrates. Electrochim. Acta 2016, 188, 57–70. [Google Scholar] [CrossRef]
- Sun, Y.; Lathwal, S.; Wang, Y.; Fu, L.; Olszewski, M.; Fantin, M.; Enciso, A.E.; Szczepaniak, G.; Das, S.; Matyjaszewski, K. Preparation of Well-Defined Polymers and DNA–Polymer Bioconjugates via Small-Volume EATRP in the Presence of Air. ACS Macro Lett. 2019, 8, 603–609. [Google Scholar] [CrossRef]
- Michieletto, A.; Lorandi, F.; De Bon, F.; Isse, A.A.; Gennaro, A. Biocompatible Polymers via Aqueous Electrochemically Mediated Atom Transfer Radical Polymerization. J. Polym. Sci. 2020, 58, 114–123. [Google Scholar] [CrossRef]
- Lorandi, F.; Fantin, M.; Wang, Y.; Isse, A.A.; Gennaro, A.; Matyjaszewski, K. Atom Transfer Radical Polymerization of Acrylic and Methacrylic Acids: Preparation of Acidic Polymers with Various Architectures. ACS Macro Lett. 2020, 9, 693–699. [Google Scholar] [CrossRef]
- Zaborniak, I.; Chmielarz, P.; Matyjaszewski, K. Synthesis of Riboflavin-Based Macromolecules through Low Ppm ATRP in Aqueous Media. Macromol. Chem. Phys. 2020, 221, 1900496. [Google Scholar] [CrossRef]
- De Bon, F.; Marenzi, S.; Isse, A.A.; Durante, C.; Gennaro, A. Electrochemically Mediated Aqueous Atom Transfer Radical Polymerization of N,N-Dimethylacrylamide. ChemElectroChem 2020, 7, 1378–1388. [Google Scholar] [CrossRef]
- Fantin, M.; Park, S.; Wang, Y.; Matyjaszewski, K. Electrochemical Atom Transfer Radical Polymerization in Miniemulsion with a Dual Catalytic System. Macromolecules 2016, 49, 8838–8847. [Google Scholar] [CrossRef]
- Fantin, M.; Chmielarz, P.; Wang, Y.; Lorandi, F.; Isse, A.A.; Gennaro, A.; Matyjaszewski, K. Harnessing the Interaction between Surfactant and Hydrophilic Catalyst To Control eATRP in Miniemulsion. Macromolecules 2017, 50, 3726–3732. [Google Scholar] [CrossRef] [PubMed]
- Zaborniak, I.; Chmielarz, P. Miniemulsion Switchable Electrolysis under Constant Current Conditions. Polym. Adv. Technol. 2020, 31, 2806–2815. [Google Scholar] [CrossRef]
- De Bon, F.; Fantin, M.; Isse, A.A.; Gennaro, A. Electrochemically Mediated ATRP in Ionic Liquids: Controlled Polymerization of Methyl Acrylate in [BMIm][OTf]. Polym. Chem. 2018, 9, 646–655. [Google Scholar] [CrossRef]
- Guo, J.K.; Zhou, Y.N.; Luo, Z.H. Electrochemically Mediated ATRP Process Intensified by Ionic Liquid: A “Flash” Polymerization of Methyl Acrylate. Chem. Eng. J. 2019, 372, 163–170. [Google Scholar] [CrossRef]
- Wang, J.-S.; Matyjaszewski, K. Controlled/”Living” Radical Polymerization. Halogen Atom Transfer Radical Polymerization Promoted by a Cu(I)/Cu(II) Redox Process. Macromolecules 1995, 28, 7901–7910. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Patten, T.E.; Xia, J. Controlled/“Living” Radical Polymerization. Kinetics of the Homogeneous Atom Transfer Radical Polymerization of Styrene. J. Am. Chem. Soc. 1997, 119, 674–680. [Google Scholar] [CrossRef]
- Angot, S.; Murthy, K.S.; Taton, D.; Gnanou, Y. Atom Transfer Radical Polymerization of Styrene Using a Novel Octafunctional Initiator: Synthesis of Well-Defined Polystyrene Stars. Macromolecules 1998, 31, 7218–7225. [Google Scholar] [CrossRef]
- Plichta, A.; Li, W.; Matyjaszewski, K. ICAR ATRP of Styrene and Methyl Methacrylate with Ru(Cp*)Cl(PPh 3) 2. Macromolecules 2009, 42, 2330–2332. [Google Scholar] [CrossRef]
- Zhang, L.; Miao, J.; Cheng, Z.; Zhu, X. Iron-Mediated ICAR ATRP of Styrene and Methyl Methacrylate in the Absence of Thermal Radical Initiator. Macromol. Rapid Commun. 2010, 31, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Mukumoto, K.; Wang, Y.; Matyjaszewski, K. Iron-Based ICAR ATRP of Styrene with Ppm Amounts of Fe III Br 3 and 1,1′-Azobis(Cyclohexanecarbonitrile). ACS Macro Lett. 2012, 1, 599–602. [Google Scholar] [CrossRef]
- Jakubowski, W.; Matyjaszewski, K. Activator Generated by Electron Transfer for Atom Transfer Radical Polymerization. Macromolecules 2005, 38, 4139–4146. [Google Scholar] [CrossRef]
- Jakubowski, W.; Min, K.; Matyjaszewski, K. Activators Regenerated by Electron Transfer for Atom Transfer Radical Polymerization of Styrene. Macromolecules 2006, 39, 39–45. [Google Scholar] [CrossRef]
- Jakubowski, W.; Kirci-Denizli, B.; Gil, R.R.; Matyjaszewski, K. Polystyrene with Improved Chain-End Functionality and Higher Molecular Weight by ARGET ATRP. Macromol. Chem. Phys. 2008, 209, 32–39. [Google Scholar] [CrossRef]
- Braidi, N.; Buffagni, M.; Ghelfi, F.; Imperato, M.; Menabue, A.; Parenti, F.; Gennaro, A.; Isse, A.A.; Bedogni, E.; Bonifaci, L.; et al. Copper-Catalysed “Activators Regenerated by Electron Transfer” “Atom Transfer Radical Polymerisation” of Styrene from a Bifunctional Initiator in Ethyl Acetate/Ethanol, Using Ascorbic Acid/Sodium Carbonate as Reducing System. Macromol. Res. 2020, 28, 751–761. [Google Scholar] [CrossRef]
- Braidi, N.; Buffagni, M.; Buzzoni, V.; Ghelfi, F.; Parenti, F.; Focarete, M.L.; Gualandi, C.; Bedogni, E.; Bonifaci, L.; Cavalca, G.; et al. Unusual Cross-Linked Polystyrene by Copper-Catalyzed ARGET ATRP Using a Bifunctional Initiator and No Cross-Linking Agent. Macromol. Res. 2021, 29, 280–288. [Google Scholar] [CrossRef]
- Hsiao, C.-Y.; Han, H.-A.; Lee, G.-H.; Peng, C.-H. AGET and SARA ATRP of Styrene and Methyl Methacrylate Mediated by Pyridyl-Imine Based Copper Complexes. Eur. Polym. J. 2014, 51, 12–20. [Google Scholar] [CrossRef]
- Mendes, J.P.; Góis, J.R.; Costa, J.R.C.; Maximiano, P.; Serra, A.C.; Guliashvili, T.; Coelho, J.F.J. Ambient Temperature SARAATRP for Meth(Acrylates), Styrene, and Vinyl Chloride Using Sulfolane/1-Butyl-3-Methylimidazolium Hexafluorophosphate-Based Mixtures. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 1322–1328. [Google Scholar] [CrossRef]
- Chmielarz, P.; Krol, P. PSt-b-PU-b-PSt Copolymers Using Tetraphenylethane-Urethane Macroinitiator through SARA ATRP. Express Polym. Lett. 2016, 10, 302–310. [Google Scholar] [CrossRef]
- Whitfield, R.; Anastasaki, A.; Nikolaou, V.; Jones, G.R.; Engelis, N.G.; Discekici, E.H.; Fleischmann, C.; Willenbacher, J.; Hawker, C.J.; Haddleton, D.M. Universal Conditions for the Controlled Polymerization of Acrylates, Methacrylates, and Styrene via Cu(0)-RDRP. J. Am. Chem. Soc. 2017, 139, 1003–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhang, L.; Cheng, Z.; Zhu, X. Metal-Free Photoinduced Electron Transfer–Atom Transfer Radical Polymerization (PET–ATRP) via a Visible Light Organic Photocatalyst. Polym. Chem. 2016, 7, 689–700. [Google Scholar] [CrossRef]
- Kütahya, C.; Schmitz, C.; Strehmel, V.; Yagci, Y.; Strehmel, B. Near-Infrared Sensitized Photoinduced Atom-Transfer Radical Polymerization (ATRP) with a Copper(II) Catalyst Concentration in the Ppm Range. Angew. Chemie Int. Ed. 2018, 57, 7898–7902. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Wu, Z.; Lin, C.; Han, H.; Chen, Y.; Chou, P.; Fu, X.; Peng, C. Polystyrene with Persistently Enhanced Fluorescence: Photo-Induced Atom Transfer Radical Polymerization Using a Pyrene-Based Initiator. Chem. Photo. Chem. 2019, 3, 1153–1161. [Google Scholar] [CrossRef]
- Bonometti, V.; Labbé, E.; Buriez, O.; Mussini, P.; Amatore, C. Exploring the First Steps of an Electrochemically-Triggered Controlled Polymerization Sequence: Activation of Alkyl- and Benzyl Halide Initiators by an Electrogenerated FeIISalen Complex. J. Electroanal. Chem. 2009, 633, 99–105. [Google Scholar] [CrossRef]
- Buback, M.; Gilbert, R.G.; Hutchinson, R.A.; Klumperman, B.; Kuchta, F.-D.; Manders, B.G.; O’Driscoll, K.F.; Russell, G.T.; Schweer, J. Critically Evaluated Rate Coefficients for Free-Radical Polymerization, 1. Propagation Rate Coefficient for Styrene. Macromol. Chem. Phys. 1995, 196, 3267–3280. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, R.S. Theory and Application of Cyclic Voltammetry for Measurement of Electrode Reaction Kinetics. Anal. Chem. 1965, 37, 1351–1355. [Google Scholar] [CrossRef]
- Fawcett, W.R.; Opallo, M. The Kinetics of Heterogeneous Electron Transfer Reaction in Polar Solvents. Angew. Chem. Int. Ed. Engl. 1994, 33, 2131–2143. [Google Scholar] [CrossRef]
- Pavan, P.; Lorandi, F.; De Bon, F.; Gennaro, A.; Isse, A.A. Enhancement of the Rate of Atom Transfer Radical Polymerization in Organic Solvents by Addition of Water: An Electrochemical Study. Chem. Electro. Chem. 2021, 8, 2450–2458. [Google Scholar] [CrossRef]
- Bortolamei, N.; Isse, A.A.; Di Marco, V.B.; Gennaro, A.; Matyjaszewski, K. Thermodynamic Properties of Copper Complexes Used as Catalysts in Atom Transfer Radical Polymerization. Macromolecules 2010, 43, 9257–9267. [Google Scholar] [CrossRef]
- Zerk, T.J.; Bernhardt, P.V. Organo-Copper(II) Complexes as Products of Radical Atom Transfer. Inorg. Chem. 2017, 56, 5784–5792. [Google Scholar] [CrossRef] [PubMed]
- Fantin, M.; Lorandi, F.; Ribelli, T.G.; Szczepaniak, G.; Enciso, A.E.; Fliedel, C.; Thevenin, L.; Isse, A.A.; Poli, R.; Matyjaszewski, K. Impact of Organometallic Intermediates on Copper-Catalyzed Atom Transfer Radical Polymerization. Macromolecules 2019, 52, 4079–4090. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent 1 | Cu(II) | E° 2 (V) | 102 × k° (cm s−1) | βII/βI | KBrII/KBrI |
|---|---|---|---|---|---|
| DMF | Cu(OTf)2 | −0.486 | |||
| DMF | [CuTPMA]2+ | −0.621 | 2.8 | 1.89 × 102 | |
| DMF | [BrCuTPMA]+ | −0.708 | 4.4 | 29.9 | |
| DMSO | Cu(OTf)2 | −0.418 | |||
| DMSO | [CuTPMA]2+ | −0.615 | 1.6 | 2.14 × 103 | |
| DMSO | [BrCuTPMA]+ | −0.687 | 2.3 | 16.5 | |
| CH3CN | Cu(OTf)2 | 0.611 | |||
| CH3CN | [CuTPMA]2+ | −0.408 | 7.5 | 1.67 × 1017 | |
| CH3CN | [BrCuTPMA]+ | −0.658 | 10 | 1.69 × 104 | |
| DMF/St | Cu(OTf)2 | −0.266 | |||
| DMF/St | [CuTPMA]2+ | −0.505 | 0.8 | 1.10 × 104 | |
| DMF/St | [BrCuTPMA]+ | −0.710 | 3.0 | 2.92 × 103 | |
| DMSO/St | Cu(OTf)2 | −0.346 | |||
| DMSO/St | [CuTPMA]2+ | −0.514 | 0.7 | 6.92 × 102 | |
| DMSO/St | [BrCuTPMA]+ | −0.693 | 2.4 | 1.05 × 103 | |
| CH3CN/St | Cu(OTf)2 | - | - | ||
| CH3CN/St | [CuTPMA]2+ | −0.384 | 1.6 | ||
| CH3CN/St | [BrCuTPMA]+ | −0.683 | 7.4 | 1.13 × 105 |
| Entry | Solvent | t (h) | Q (C) | Conversion (%) | 102 × kpapp 2 (h−1) | Mn,th3 (kDa) | Mn4 (kDa) | Ð |
|---|---|---|---|---|---|---|---|---|
| 1 | DMF | 5 | 5.82 | 34 | 8.7 | 10.5 | 10.6 | 1.26 |
| 2 | DMSO | 2 | 0.99 | 15 | - 5 | 4.7 | 4.8 | 1.22 |
| 3 | CH3CN | 4 | 3.39 | 47 | 17.1 | 14.4 | 17.2 | 1.37 |
| Entry | St (vol%) | [CuII] (mM) | [EBiB] (mM) | DP | t (h) | Q (C) | Conversion (%) | 102 × kpapp 2 (h−1) | Mn,th3 (kDa) | Mn4 (kDa) | Ð |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 50 | 1.0 | 15 | 290 | 5 | 5.8 | 34 | 8.7 | 10.5 | 10.7 | 1.26 |
| 2 | 50 | 1.0 | 7.5 | 580 | 4 | 7.7 | 11 | 2.9 | 6.7 | 5.6 | 1.33 |
| 3 | 50 | 1.0 | 2 | 2175 | 2 | 2.5 | 6.3 | 3.1 | 14.5 | 14.1 | 1.33 |
| 4 | 50 | 0.5 | 2 | 2175 | 2 | 2.6 | 7.7 | 3.0 | 17.6 | 19.1 | 1.32 |
| 5 | 50 | 0.5 | 2 | 2175 | 4 | 3.5 | 11.4 | 3.0 | 26.0 | 26.6 | 1.46 |
| 6 | 50 | 0.2 | 2 | 2175 | 2 | 1.4 | 8.8 | 3.4 | 20.2 | 24.3 | 1.32 |
| 7 | 50 | 0.2 | 2 | 2175 | 4 | 1.9 | 12.5 | 3.4 | 28.5 | 31.9 | 1.52 |
| 8 | 25 | 1.0 | 15 | 145 | 5 | 3.1 | 13 | 2.4 | 2.1 | 2.3 | 1.35 |
| 9 | 75 | 1.0 | 15 | 435 | 5 | 10.1 | 40 | 10.0 | 18.3 | 19.4 | 1.29 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Bon, F.; Carlan, G.M.; Tognella, E.; Isse, A.A. Exploring Electrochemically Mediated ATRP of Styrene. Processes 2021, 9, 1327. https://doi.org/10.3390/pr9081327
De Bon F, Carlan GM, Tognella E, Isse AA. Exploring Electrochemically Mediated ATRP of Styrene. Processes. 2021; 9(8):1327. https://doi.org/10.3390/pr9081327
Chicago/Turabian StyleDe Bon, Francesco, Gian Marco Carlan, Enrico Tognella, and Abdirisak Ahmed Isse. 2021. "Exploring Electrochemically Mediated ATRP of Styrene" Processes 9, no. 8: 1327. https://doi.org/10.3390/pr9081327
APA StyleDe Bon, F., Carlan, G. M., Tognella, E., & Isse, A. A. (2021). Exploring Electrochemically Mediated ATRP of Styrene. Processes, 9(8), 1327. https://doi.org/10.3390/pr9081327