
Monitoring E. coli Cell Integrity by ATR-FTIR Spectroscopy and Chemometrics: Opportunities and Caveats


Abstract
1. Introduction
2. Materials and Methods
2.1. ATR-FTIR Off-Line Spectra
2.2. Bioreactor Cultivations and In-Line ATR-FTIR Measurements
2.3. Reference Off-Line Measurements
2.4. Programming
2.5. Preprocessing and Validation Split
2.6. Multivariate Regression
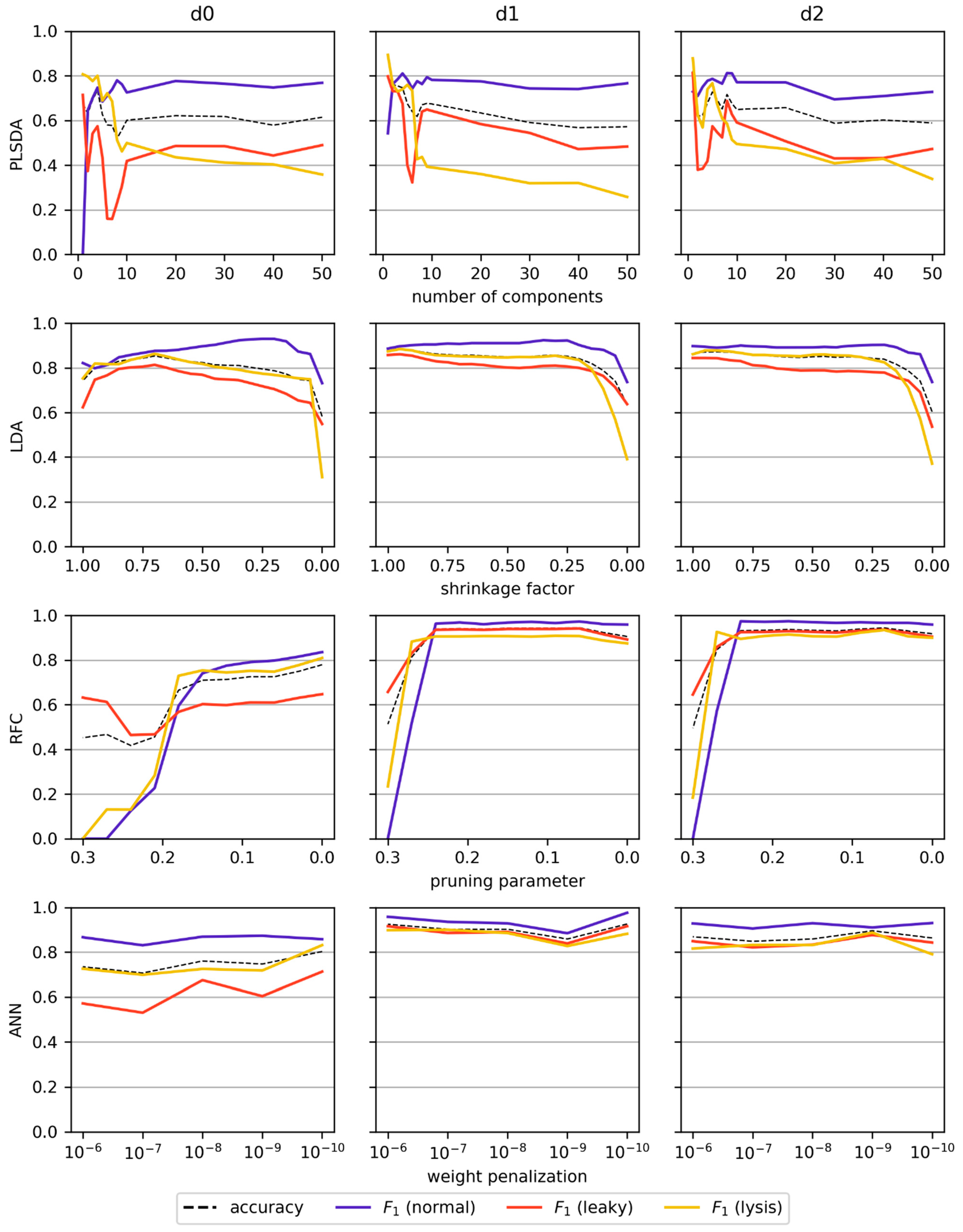
2.7. Classification
3. Results
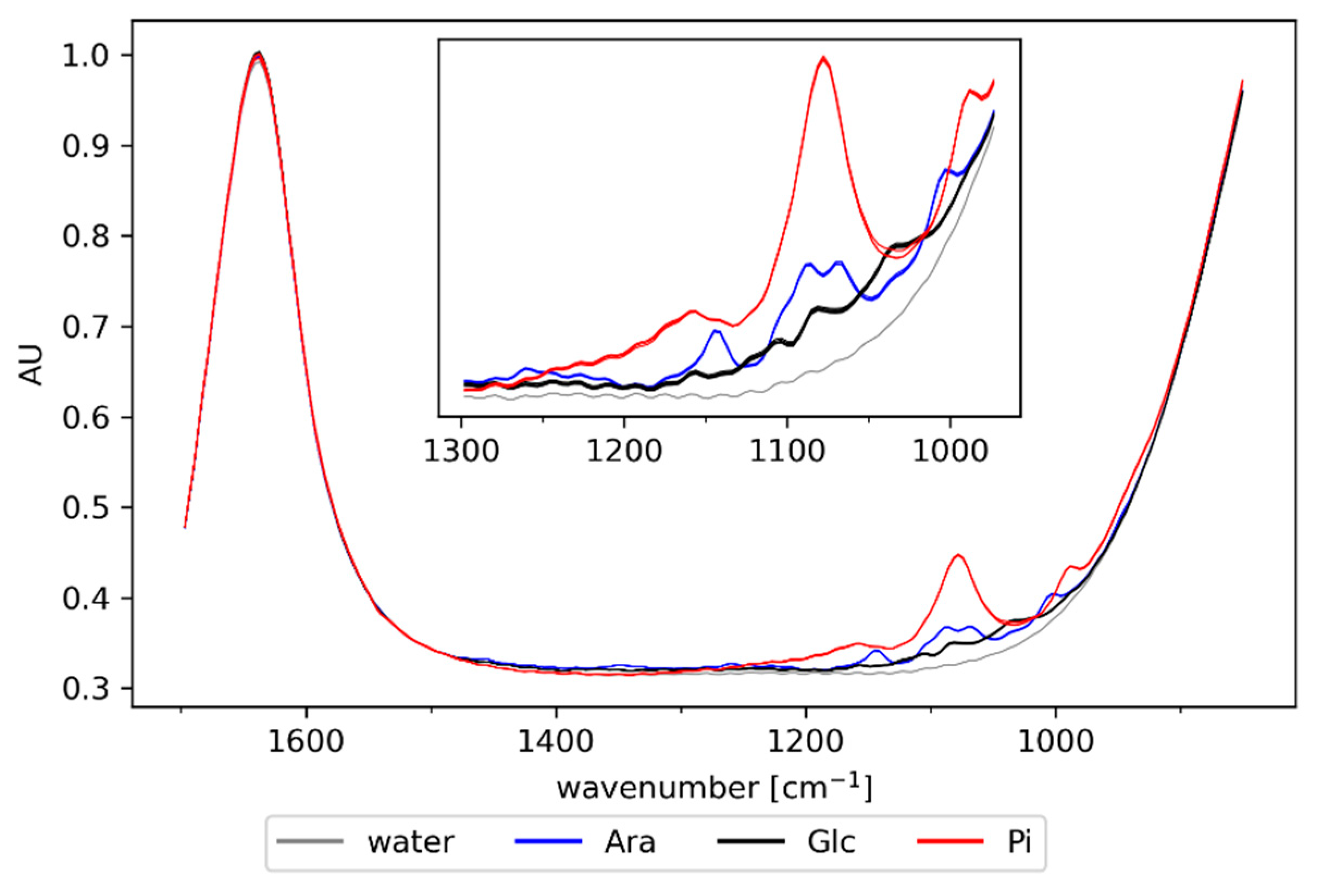
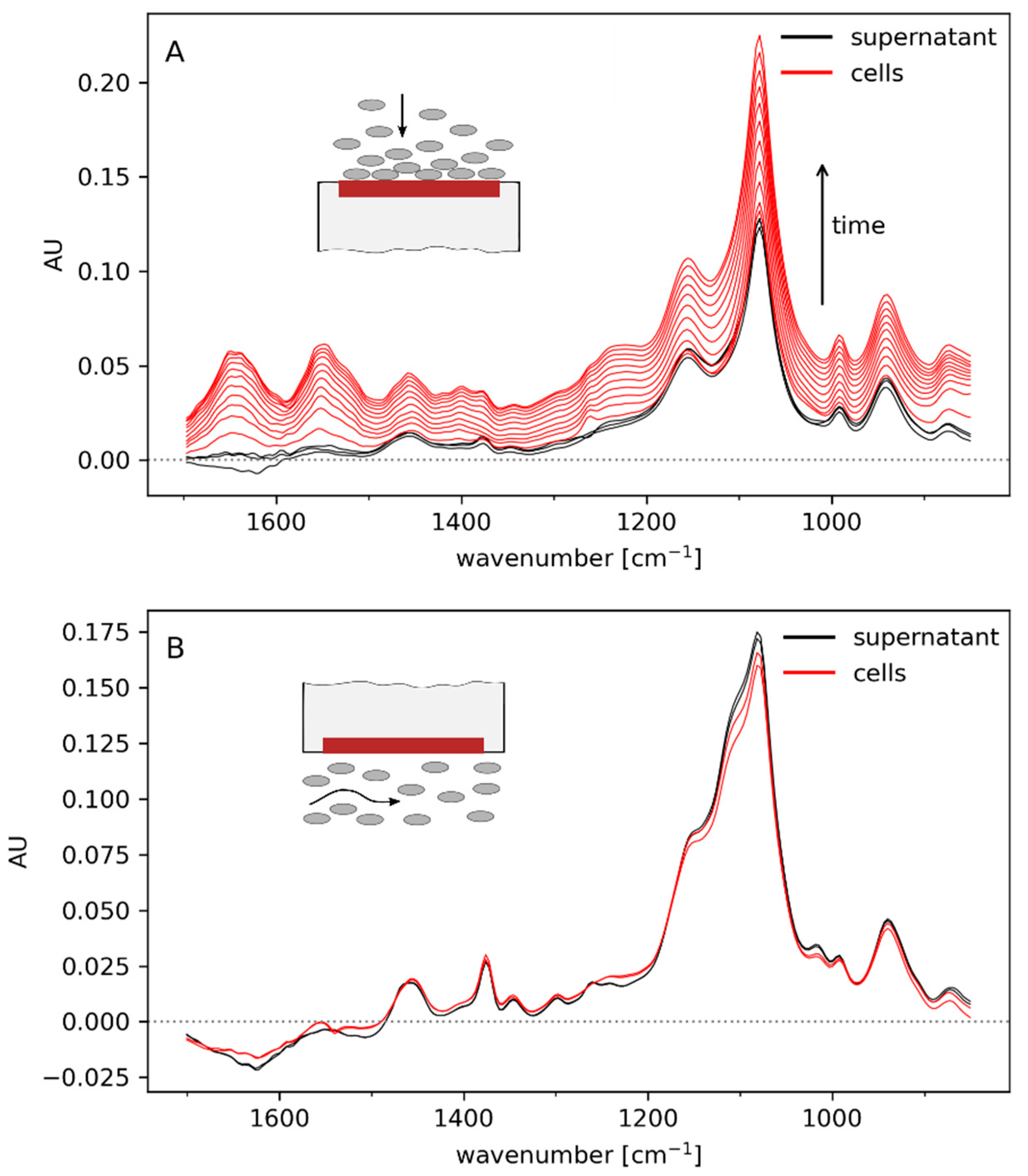
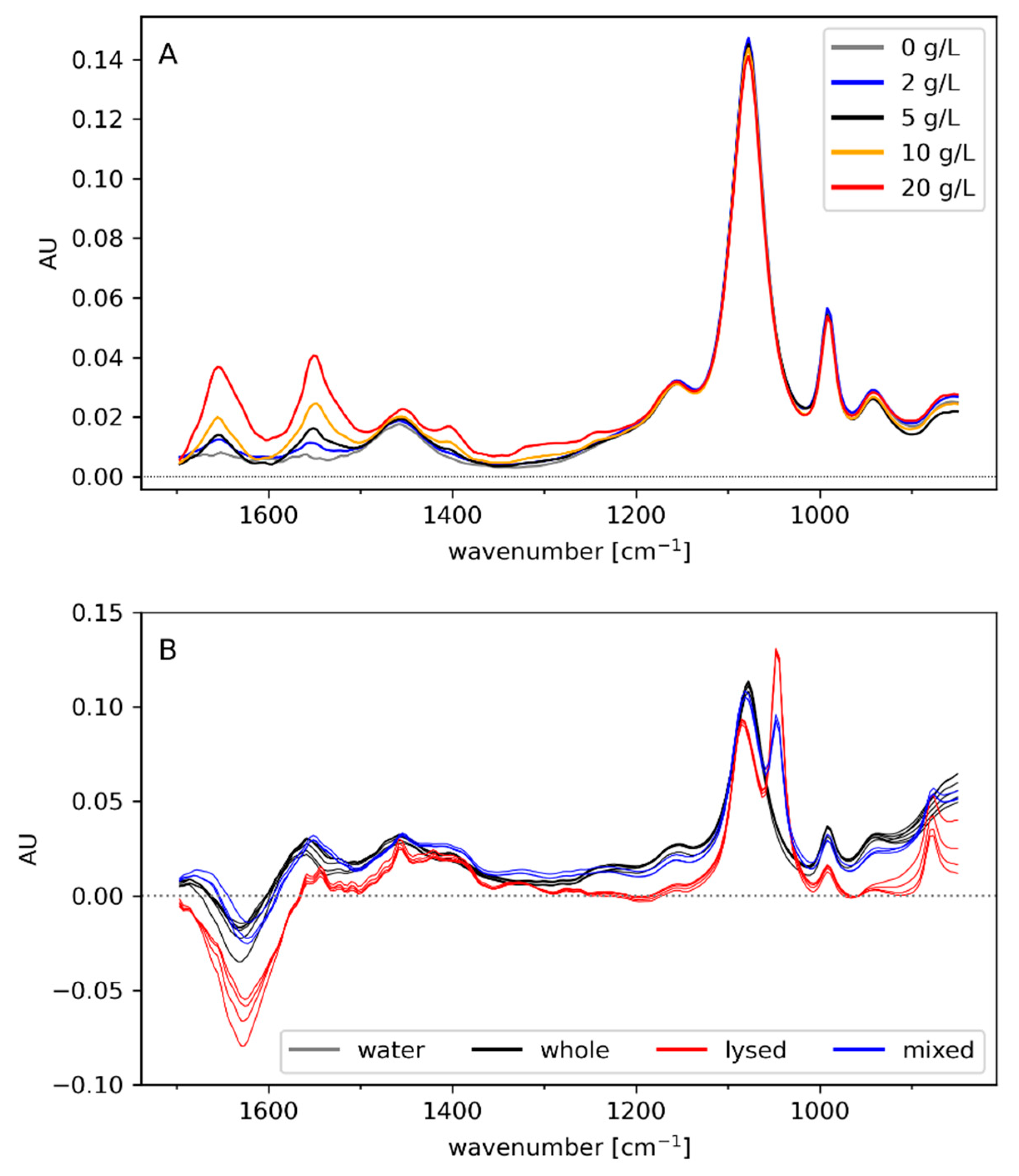
3.1. Off-Line Spectra
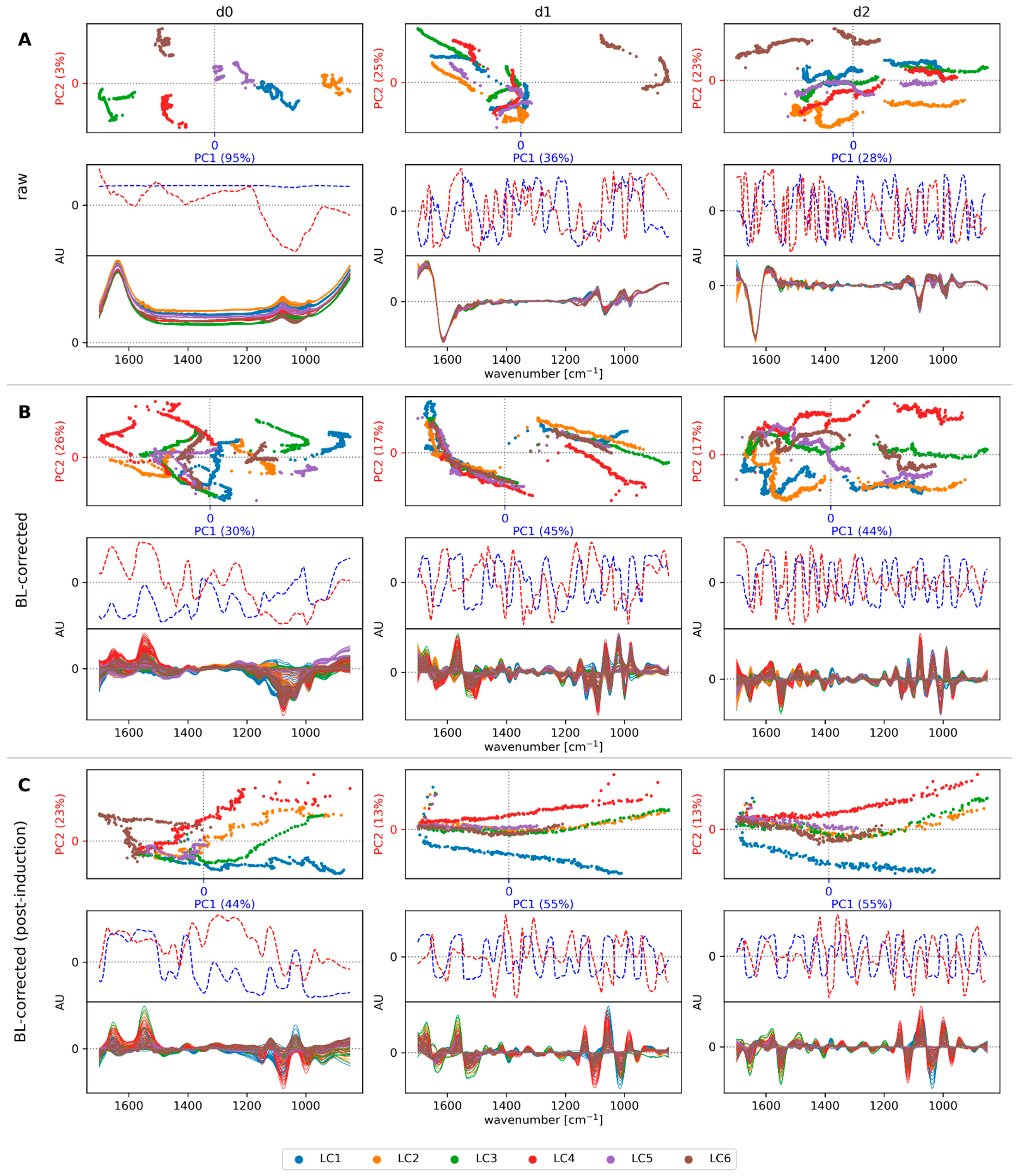
3.2. Preprocessing
3.3. Regression
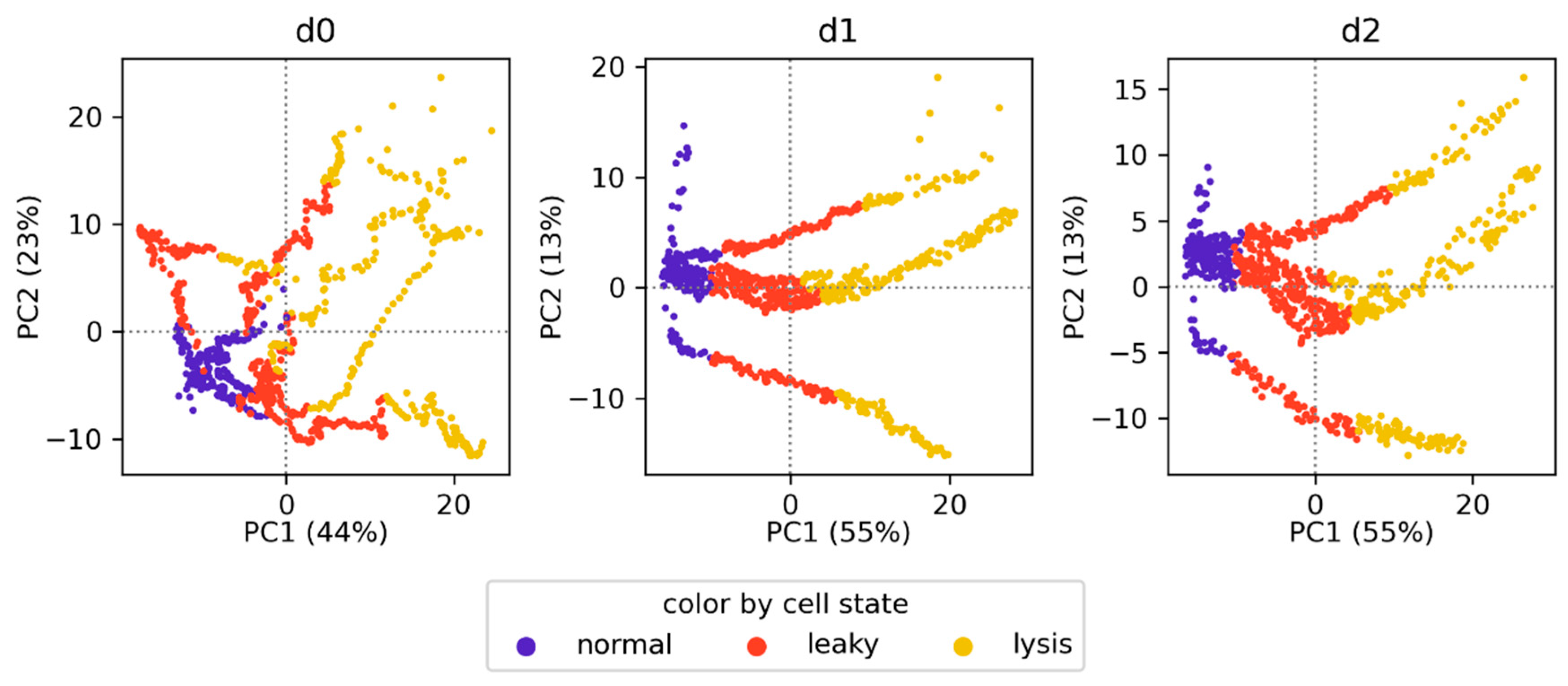
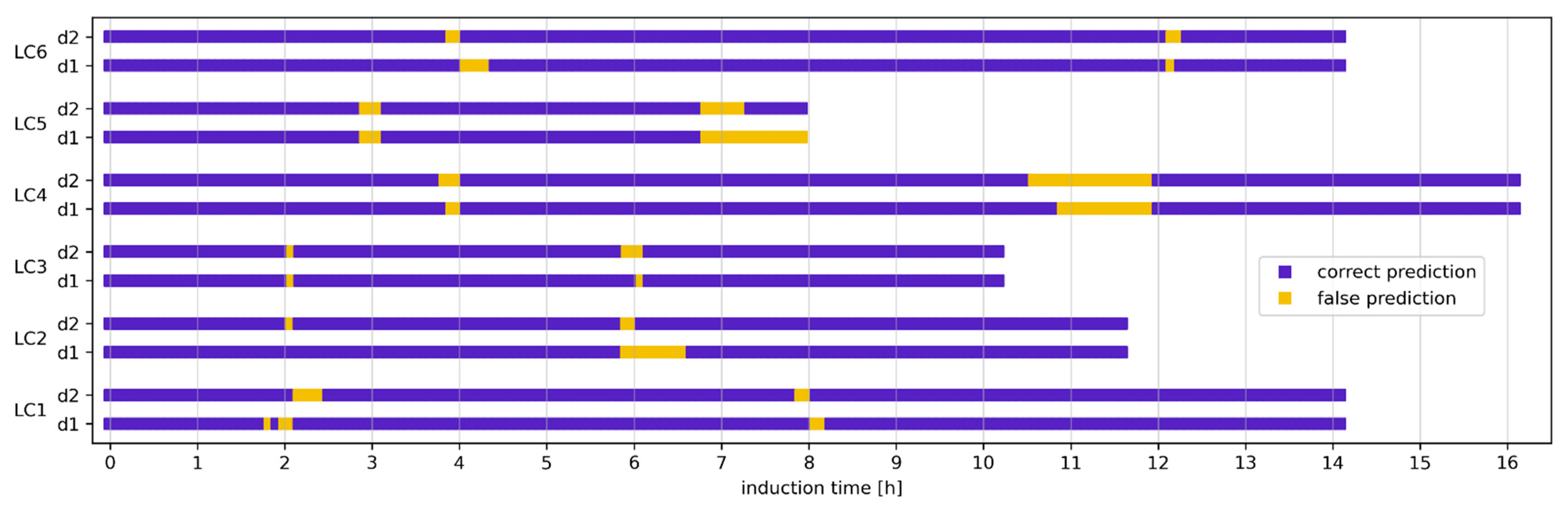
3.4. Classification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kastenhofer, J.; Rajamanickam, V.; Libiseller-Egger, J.; Spadiut, O. Monitoring and control of E. coli cell integrity. J. Biotechnol. 2021, 329, 1–12. [Google Scholar] [CrossRef]
- Landgrebe, D.; Haake, C.; Höpfner, T.; Beutel, S.; Hitzmann, B.; Scheper, T.; Rhiel, M.; Reardon, K.F. On-line infrared spectroscopy for bioprocess monitoring. Appl. Microbiol. Biotechnol. 2010, 88, 11–22. [Google Scholar] [CrossRef]
- Diem, M. Biophysical Applications of Vibrational Spectroscopy. In Modern Vibrational Spectroscopy and Micro-Spectroscopy; Diem, M., Ed.; John Wiley & Sons: West Sussex, UK, 2015; pp. 203–234. [Google Scholar] [CrossRef]
- Biechele, P.; Busse, C.; Solle, D.; Scheper, T.; Reardon, K. Sensor systems for bioprocess monitoring. Eng. Life Sci. 2015, 15, 469–488. [Google Scholar] [CrossRef]
- Schenk, J.; Viscasillas, C.; Marison, I.W.; von Stockar, U. On-line monitoring of nine different batch cultures of E. coli by mid-infrared spectroscopy, using a single spectra library for calibration. J. Biotechnol. 2008, 134, 93–102. [Google Scholar] [CrossRef]
- Koch, C.; Posch, A.E.; Herwig, C.; Lendl, B. Comparison of Fiber Optic and Conduit Attenuated Total Reflection (ATR) Fourier Transform Infrared (FT-IR) Setup for In-Line Fermentation Monitoring. Appl. Spectrosc. 2016, 70, 1965–1973. [Google Scholar] [CrossRef]
- Pontius, K.; Junicke, H.; Gernaey, K.V.; Bevilacqua, M. Monitoring yeast fermentations by nonlinear infrared technology and chemometrics-understanding process correlations and indirect predictions. Appl. Microbiol. Biotechnol. 2020, 104, 5315–5335. [Google Scholar] [CrossRef] [PubMed]
- Pontius, K.; Pratico, G.; Larsen, F.H.; Skov, T.; Arneborg, N.; Lantz, A.E.; Bevilacqua, M. Fast measurement of phosphates and ammonium in fermentation-like media: A feasibility study. New Biotechnol. 2020, 56, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Schalk, R.; Heintz, A.; Braun, F.; Iacono, G.; Rädle, M.; Gretz, N.; Methner, F.-J.; Beuermann, T. Comparison of Raman and Mid-Infrared Spectroscopy for Real-Time Monitoring of Yeast Fermentations: A Proof-of-Concept for Multi-Channel Photometric Sensors. Appl. Sci. 2019, 9, 2472. [Google Scholar] [CrossRef]
- Capito, F.; Skudas, R.; Kolmar, H.; Hunzinger, C. Mid-infrared spectroscopy-based antibody aggregate quantification in cell culture fluids. Biotechnol. J. 2013, 8, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Capito, F.; Skudas, R.; Kolmar, H.; Hunzinger, C. At-line mid infrared spectroscopy for monitoring downstream processing unit operations. Process Biochem. 2015, 50, 997–1005. [Google Scholar] [CrossRef]
- Naumann, D. Vibrational Spectroscopy in Microbiology and Medical Diagnostics. In Biomedical Vibrational Spectroscopy; Lasch, P., Kneipp, J., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2008; pp. 1–8. [Google Scholar] [CrossRef]
- Chalmers, J.M.; Griffiths, P.R. Vibrational Spectroscopy: Sampling Techniques and Fiber-Optic Probes. In Handbook of Vibrational Spectroscopy; Chalmers, J.M., Griffiths, P.R., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar] [CrossRef]
- Griffiths, P.R. Introduction to the Theory and Instrumentation for Vibrational Spectroscopy. In Handbook of Vibrational Spectroscopy; Chalmers, J.M., Griffiths, P.R., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar] [CrossRef]
- Koch, C.; Brandstetter, M.; Lendl, B.; Radel, S. Ultrasonic manipulation of yeast cells in suspension for absorption spectroscopy with an immersible mid-infrared fiberoptic probe. Ultrasound Med. Biol. 2013, 39, 1094–1101. [Google Scholar] [CrossRef]
- Koch, C.; Brandstetter, M.; Wechselberger, P.; Lorantfy, B.; Plata, M.R.; Radel, S.; Herwig, C.; Lendl, B. Ultrasound-enhanced attenuated total reflection mid-infrared spectroscopy in-line probe: Acquisition of cell spectra in a bioreactor. Anal. Chem. 2015, 87, 2314–2320. [Google Scholar] [CrossRef]
- Rolinger, L.; Rudt, M.; Hubbuch, J. A critical review of recent trends, and a future perspective of optical spectroscopy as PAT in biopharmaceutical downstream processing. Anal. Bioanal. Chem. 2020, 412, 2047–2064. [Google Scholar] [CrossRef]
- Barchi, A.C.; Ito, S.; Escaramboni, B.; Neto, P.d.O.; Herculano, R.D.; Romeiro Miranda, M.C.; Passalia, F.J.; Rocha, J.C.; Fernández Núñez, E.G. Artificial intelligence approach based on near-infrared spectral data for monitoring of solid-state fermentation. Process Biochem. 2016, 51, 1338–1347. [Google Scholar] [CrossRef]
- Jiang, H.; Mei, C.; Chen, Q. Rapid identification of fermentation stages of bioethanol solid-state fermentation (SSF) using FT-NIR spectroscopy: Comparisons of linear and non-linear algorithms for multiple classification issues. Anal. Methods 2017, 9, 5769–5776. [Google Scholar] [CrossRef]
- Pais, D.A.M.; Portela, R.M.C.; Carrondo, M.J.T.; Isidro, I.A.; Alves, P.M. Enabling PAT in insect cell bioprocesses: In situ monitoring of recombinant adeno-associated virus production by fluorescence spectroscopy. Biotechnol. Bioeng. 2019, 116, 2803–2814. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Xu, W.; Ding, Y.; Chen, Q. Quantitative analysis of yeast fermentation process using Raman spectroscopy: Comparison of CARS and VCPA for variable selection. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 228, 117781. [Google Scholar] [CrossRef]
- Sampaio, P.N.; Calado, C.R.C. Classification of recombinant Saccharomyces cerevisiae cells using PLS-DA modelling based on MIR spectroscopy. In Proceedings of the 6th IEEE Portuguese Meeting on Bioengineering, Lisbon, Portugal, 22–23 February 2019. [Google Scholar] [CrossRef]
- Meza Ramirez, C.A.; Greenop, M.; Ashton, L.; Rehman, I. Applications of machine learning in spectroscopy. Appl. Spectrosc. Rev. In press. [CrossRef]
- Peris-Díaz, M.D.; Krężel, A. A guide to good practice in chemometric methods for vibrational spectroscopy, electrochemistry, and hyphenated mass spectrometry. TrAC Trends Anal. Chem. 2021, 135. [Google Scholar] [CrossRef]
- Brereton, R.G. Consequences of sample size, variable selection, and model validation and optimisation, for predicting classification ability from analytical data. TrAC Trends Anal. Chem. 2006, 25, 1103–1111. [Google Scholar] [CrossRef]
- Miller, C.E. Chemometrics in Process Analytical Technology (PAT). In Process Analytical Technology; Bakeev, K.A., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2010; pp. 353–438. [Google Scholar] [CrossRef]
- Xiaobo, Z.; Jiewen, Z.; Povey, M.J.; Holmes, M.; Hanpin, M. Variables selection methods in near-infrared spectroscopy. Anal. Chim. Acta 2010, 667, 14–32. [Google Scholar] [CrossRef]
- Engel, J.; Gerretzen, J.; Szymańska, E.; Jansen, J.J.; Downey, G.; Blanchet, L.; Buydens, L.M.C. Breaking with trends in pre-processing? TrAC Trends Anal. Chem. 2013, 50, 96–106. [Google Scholar] [CrossRef]
- Westad, F.; Marini, F. Validation of chemometric models—A tutorial. Anal. Chim. Acta 2015, 893, 14–24. [Google Scholar] [CrossRef]
- Stargardt, P.; Feuchtenhofer, L.; Cserjan-Puschmann, M.; Striedner, G.; Mairhofer, J. Bacteriophage inspired growth-decoupled recombinant protein production in E. coli. ACS Synth. Biol. 2020, 9, 1336–1348. [Google Scholar] [CrossRef]
- Mairhofer, J.; Striedner, G.; Grabherr, R.; Wilde, M. Uncoupling Growth and Protein Production. WO 2016174195A1, 29 April 2016. [Google Scholar]
- Kastenhofer, J.; Rettenbacher, L.; Feuchtenhofer, L.; Mairhofer, J.; Spadiut, O. Inhibition of E. coli host RNA polymerase allows efficient extracellular recombinant protein production by enhancing outer membrane leakiness. Biotechnol. J. 2020, e2000274. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar] [CrossRef]
- Abadi, M.; Barham, P.; Chen, J.; Chen, Z.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Irving, G.; Isard, M.; et al. TensorFlow: A system for large-scale machine learning. In Proceedings of the 12th USENIX Symposium on Operating Systems Design and Implementation, Svannah, GA, USA, 2–4 November 2016; pp. 265–283. [Google Scholar]
- Chollet, F. Keras. Available online: https://keras.io (accessed on 1 February 2021).
- Goutte, C.; Gaussier, E. A Probabilistic Interpretation of Precision, Recall and F-Score, with Implication for Evaluation. In Proceedings of the 27th European Conference on IR Research, Santiago de Compostela, Spain, 21–23 March 2005; pp. 345–359. [Google Scholar]
- Louppe, G. Understanding Random Forests: From Theory to Practice. Ph.D. Thesis, University of Liege, Liege, Belgium, 2014. [Google Scholar]
- Barth, A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta 2007, 1767, 1073–1101. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.J.; Hussain, S.R.; Lovergne, L.; Untereiner, V.; Hughes, C.; Lukaszewski, R.A.; Thiefin, G.; Sockalingum, G.D. Developing and understanding biofluid vibrational spectroscopy: A critical review. Chem. Soc. Rev. 2016, 45, 1803–1818. [Google Scholar] [CrossRef]
- Quintelas, C.; Ferreira, E.C.; Lopes, J.A.; Sousa, C. An Overview of the Evolution of Infrared Spectroscopy Applied to Bacterial Typing. Biotechnol. J. 2018, 13. [Google Scholar] [CrossRef]
- Parikh, S.J.; Chorover, J. Infrared spectroscopy studies of cation effects on lipopolysaccharides in aqueous solution. Colloids Surf. B. Biointerfaces 2007, 55, 241–250. [Google Scholar] [CrossRef]
- Jarute, G.; Kainz, A.; Schroll, G.; Baena, J.R.; Lendl, B. On-line determination of the intracellular poly(b-hydroxybutyric acid) content in transformed E. coli and glucose during PHB production using stopped-flow attenuated total reflection FT-IR spectrometry. Anal. Chem. 2004, 76, 6353–6358. [Google Scholar] [CrossRef]
- Kosa, G.; Shapaval, V.; Kohler, A.; Zimmermann, B. FTIR spectroscopy as a unified method for simultaneous analysis of intra- and extracellular metabolites in high-throughput screening of microbial bioprocesses. Microb. Cell Factories 2017, 16, 1–11. [Google Scholar] [CrossRef]
- Wieland, K.; Tauber, S.; Gasser, C.; Rettenbacher, L.A.; Lux, L.; Radel, S.; Lendl, B. In-Line Ultrasound-Enhanced Raman Spectroscopy Allows for Highly Sensitive Analysis with Improved Selectivity in Suspensions. Anal. Chem. 2019, 91, 14231–14238. [Google Scholar] [CrossRef]
- Rafferty, C.; Johnson, K.; O’Mahony, J.; Burgoyne, B.; Rea, R.; Balss, K.M. Analysis of chemometric models applied to Raman spectroscopy for monitoring key metabolites of cell culture. Biotechnol. Prog. 2020, 36, e2977. [Google Scholar] [CrossRef]
- Wasalathanthri, D.P.; Rehmann, M.S.; Song, Y.; Gu, Y.; Mi, L.; Shao, C.; Chemmalil, L.; Lee, J.; Ghose, S.; Borys, M.C.; et al. Technology outlook for real-time quality attribute and process parameter monitoring in biopharmaceutical development—A review. Biotechnol. Bioeng. 2020, 117, 3182–3198. [Google Scholar] [CrossRef]
- Kastenhofer, J.; Spadiut, O. Culture medium density as a simple monitoring tool for cell integrity of E. coli. J. Biotechnol. X 2020, 6, 100017. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
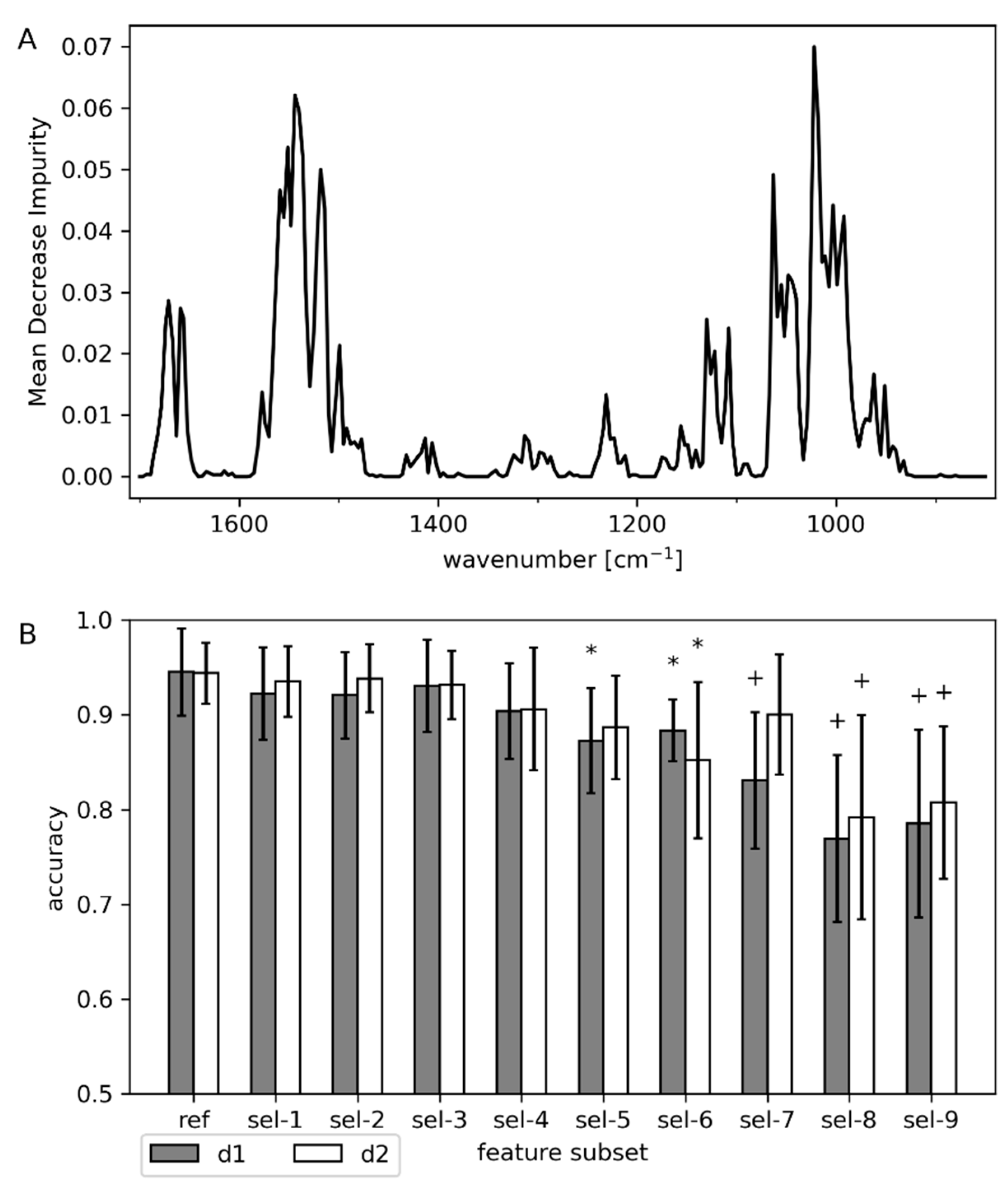
| Subset Name | Retained Features (Wavenumbers (cm−1)) | Comment |
|---|---|---|
| ref | 850–1700 | reference; whole fingerprint region |
| sel-1 | 1505–1700 | retain only amide I and II region |
| sel-2 | 1080–1700 | remove part of carbohydrate, phosphate, and lysed cells fingerprint |
| sel-3 | 850–1505 | remove amide I and II region |
| sel-4 | 1080–1505 | sel-2 and sel-3 combined |
| sel-5 | 1190–1450 | remove amide I and II region, full carbohydrate and phosphate fingerprint |
| sel-6 | 1190–1360 | sel-5 and remove putative amino acid band |
| sel-7 | 1255–1450 | based on MDI only |
| sel-8 | 1255–1360 | retain only putative amide III region |
| sel-9 | 1360–1450 | retain only putative amino acid band |
| Baseline-Corrected | Baseline-Corrected (Post-Induction) | ||||||
|---|---|---|---|---|---|---|---|
| d0 | d1 | d2 | d0 | d1 | d2 | ||
| PLSR | Components | 6 | 2 | 5 | 6 | 2 | 2 |
| NRMSE | |||||||
| AP | 0.13 | 0.11 | 0.10 | 0.18 | 0.24 | 0.22 | |
| SpA | 0.28 | 0.35 | 0.23 | 0.22 | 0.27 | 0.26 | |
| DNA | 0.14 | 0.13 | 0.15 | 0.21 | 0.20 | 0.18 | |
| RFR | Pruning | 0 | 0 | 0 | 0 | 0 | 0 |
| NRMSE | |||||||
| AP | 0.18 | 0.12 | 0.11 | 0.17 | 0.15 | 0.14 | |
| SpA | 0.28 | 0.21 | 0.22 | 0.32 | 0.25 | 0.21 | |
| DNA | 0.16 | 0.12 | 0.12 | 0.21 | 0.15 | 0.15 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kastenhofer, J.; Libiseller-Egger, J.; Rajamanickam, V.; Spadiut, O. Monitoring E. coli Cell Integrity by ATR-FTIR Spectroscopy and Chemometrics: Opportunities and Caveats. Processes 2021, 9, 422. https://doi.org/10.3390/pr9030422
Kastenhofer J, Libiseller-Egger J, Rajamanickam V, Spadiut O. Monitoring E. coli Cell Integrity by ATR-FTIR Spectroscopy and Chemometrics: Opportunities and Caveats. Processes. 2021; 9(3):422. https://doi.org/10.3390/pr9030422
Chicago/Turabian StyleKastenhofer, Jens, Julian Libiseller-Egger, Vignesh Rajamanickam, and Oliver Spadiut. 2021. "Monitoring E. coli Cell Integrity by ATR-FTIR Spectroscopy and Chemometrics: Opportunities and Caveats" Processes 9, no. 3: 422. https://doi.org/10.3390/pr9030422
APA StyleKastenhofer, J., Libiseller-Egger, J., Rajamanickam, V., & Spadiut, O. (2021). Monitoring E. coli Cell Integrity by ATR-FTIR Spectroscopy and Chemometrics: Opportunities and Caveats. Processes, 9(3), 422. https://doi.org/10.3390/pr9030422