Non-Alcoholic Fatty Liver Disease: From Pathogenesis to Clinical Impact

 ,
,  , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
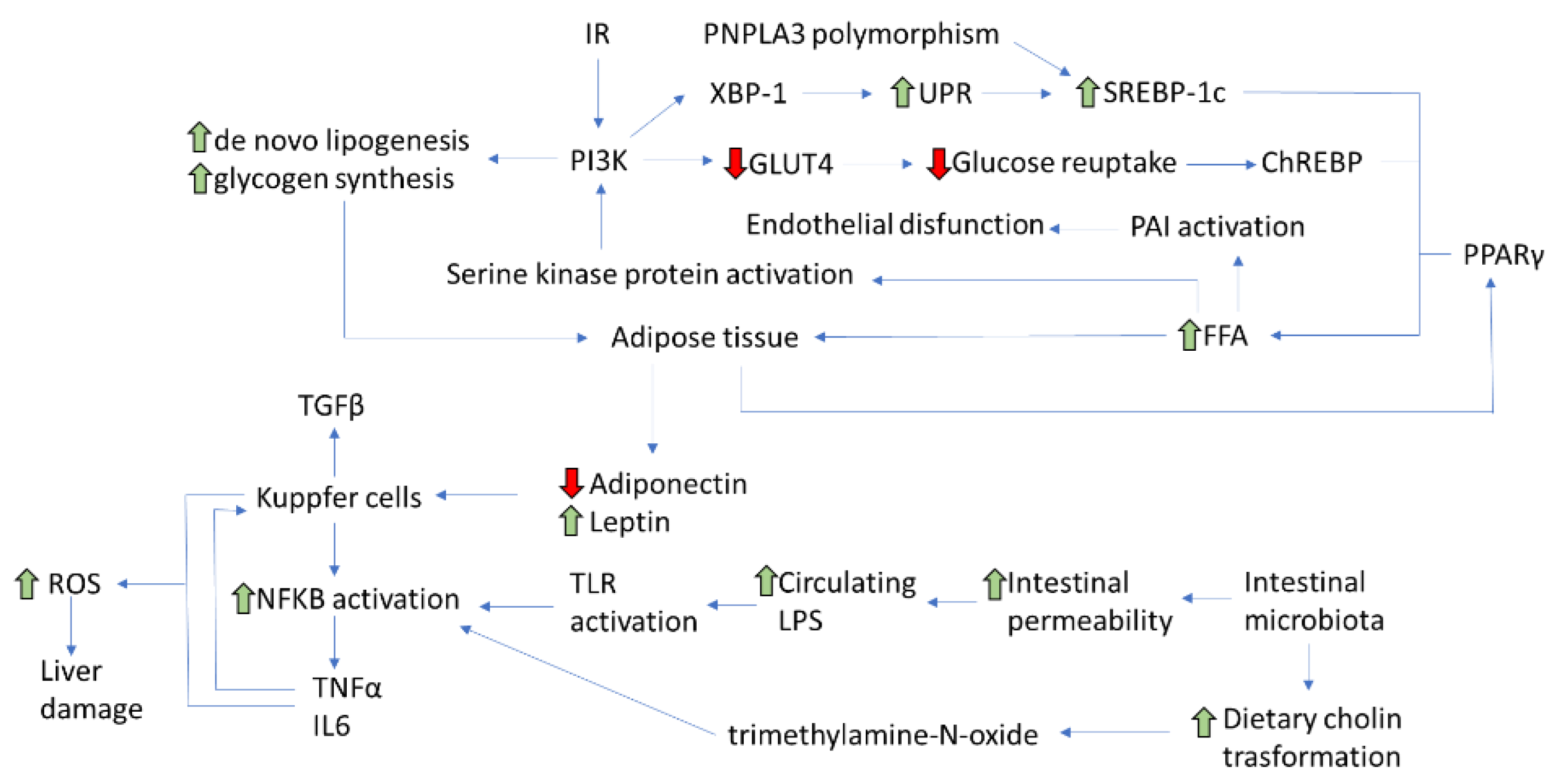
2. Pathophysiological Mechanisms of NAFLD
3. Insulin Resistance
Insulin Resistance and NAFLD Development
4. Role of Visceral Adipose Tissue
5. Role of Diet
6. Role of Intestinal Microbiota
7. Role of Genetic and Epigenetic Factors
8. Role of Low-Degree Chronic Systemic Inflammation
9. Role of Mitochondrial Dysfunction and Endoplasmic Reticulum Stress
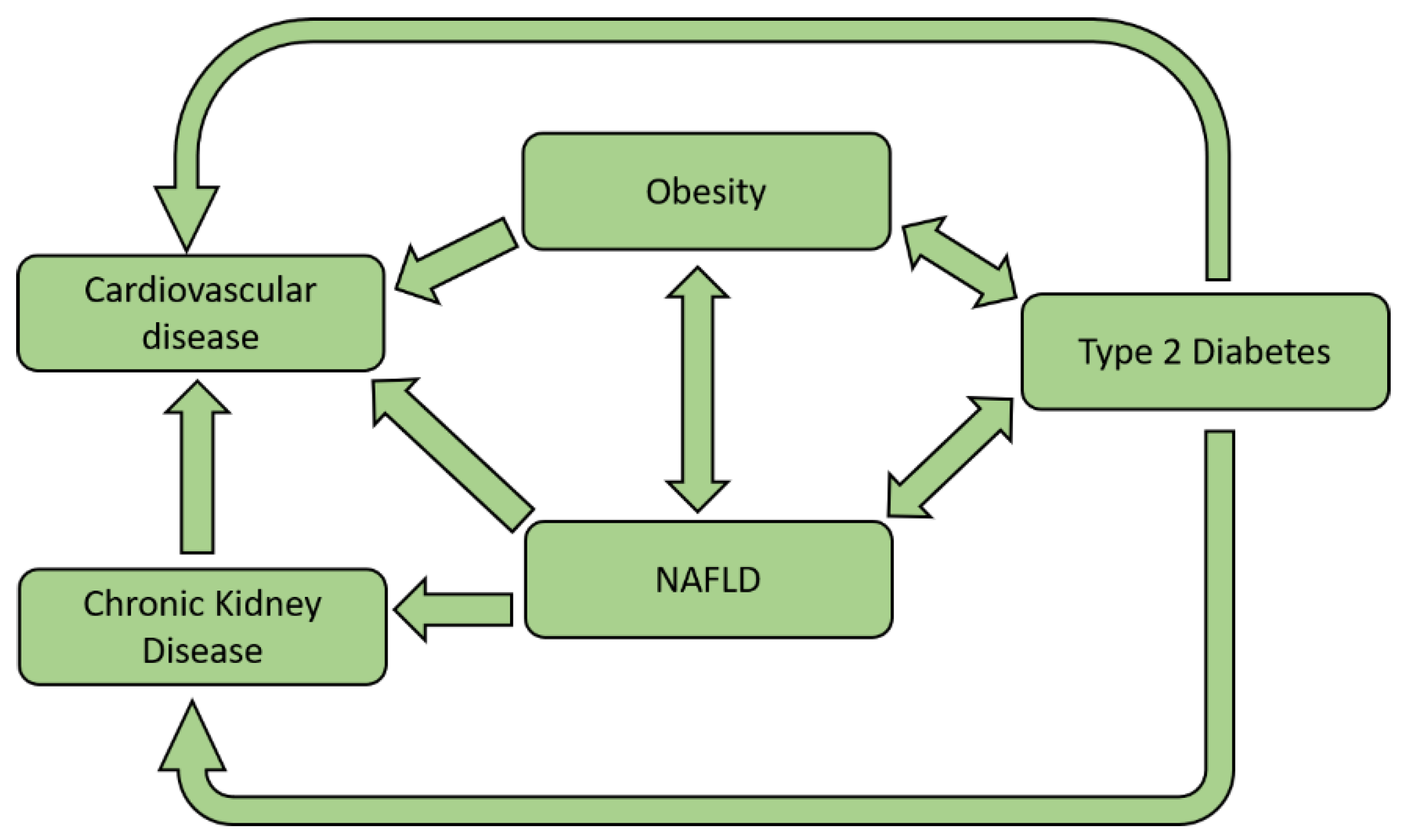
10. Clinical Impact of NAFLD
10.1. NAFLD, CV Disease, and Atherosclerosis
10.2. NAFLD and CKD
11. NAFLD and Type 2 Diabetes
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Makhlouf, H.R. Histology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis in Adults and Children. Clin. Liver Dis. 2016, 20, 293–312. [Google Scholar] [CrossRef] [PubMed]
- Vos, B.; Moreno, C.; Nagy, N.; Féry, F.; Cnop, M.; Vereerstraeten, P.; Devière, J.; Adler, M. Lean non-alcoholic fatty liver disease (Lean-NAFLD): A major cause of cryptogenic liver disease. Acta Gastroenterol. Belg. 2011, 74, 389–394. [Google Scholar]
- Younossi, Z.M.; Stepanova, M.; Negro, F.; Hallaji, S.; Younossi, Y.; Lam, B.; Srishord, M. Nonalcoholic fatty liver disease in lean individuals in the United States. Medicine 2012, 91, 319–327. [Google Scholar] [CrossRef]
- Di Francia, R.; Rinaldi, L.; Troisi, A.; Di Benedetto, F.; Berretta, M. Effect of anti-oxidant agents in patients with hepatocellular diseases. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 3993–3995. [Google Scholar]
- Di Francia, R.; Rinaldi, L.; Cillo, M.; Varriale, E.; Facchini, G.; D’Aniello, C.; Marotta, G.; Berretta, M. Antioxidant diet and genotyping as tools for the prevention of liver disease. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 5155–5163. [Google Scholar]
- Eckel, R.H.; Alberti, K.G.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2010, 375, 181–183. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Maurantonio, M.; Marrazzo, A.; Rinaldi, L.; Adinolfi, L.E. Nonalcoholic fatty liver disease: Evolving paradigms. World J. Gastroenterol. 2017, 23, 6571–6592. [Google Scholar] [CrossRef] [PubMed]
- Acierno, C.; Caturano, A.; Pafundi, P.C.; Nevola, R.; Adinolfi, L.E.; Sasso, F.C. Nonalcoholic fatty liver disease and type 2 diabetes: Pathophysiological mechanisms shared between the two faces of the same coin. Explor. Med. 2020, 1, 287–306. [Google Scholar] [CrossRef]
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr.; et al. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar] [CrossRef] [PubMed]
- Thevis, M.; Thomas, A.; Schänzer, W. Insulin. In Doping in Sports: Biochemical Principles, Effects and Analysis; Springer: Berlin/Heidelberg, Germany, 2010; pp. 209–226. [Google Scholar] [CrossRef]
- Sonksen, P.; Sonksen, J. Insulin: Understanding its action in health and disease. Br. J. Anaesth. 2000, 85, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Posner, B.I. Insulin Signalling: The Inside Story. Can. J. Diabetes 2017, 41, 108–113. [Google Scholar] [CrossRef]
- Goetze, S.; Kim, S.; Xi, X.P.; Graf, K.; Yang, D.C.; Fleck, E.; Meehan, W.P.; Hsueh, W.A.; Law, R.E. Troglitazone inhibits mitogenic signaling by insulin in vascular smooth muscle cells. J. Cardiovasc. Pharmacol. 2000, 35, 749–757. [Google Scholar] [CrossRef]
- Kim, Y.B.; Nikoulina, S.E.; Ciaraldi, T.P.; Henry, R.R.; Kahn, B.B. Normal insulin-dependent activation of Akt/protein kinase B, with diminished activation of phosphoinositide 3-kinase, in muscle in type 2 diabetes. J. Clin. Investig. 1999, 104, 733–741. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin resistance: Review of the underlying molecular mechanisms. J. Cell Physiol. 2019, 234, 8152–8161. [Google Scholar] [CrossRef]
- Romao, I.; Roth, J. Genetic and environmental interactions in obesity and type 2 diabetes. J. Am. Diet. Assoc. 2008, 108, S24–S28. [Google Scholar] [CrossRef]
- Kachur, S.; Lavie, C.J.; de Schutter, A.; Milani, R.V.; Ventura, H.O. Obesity and cardiovascular diseases. Minerva Med. 2017, 108, 212–228. [Google Scholar] [CrossRef]
- Cozzolino, D.; Sessa, G.; Salvatore, T.; Sasso, F.C.; Giugliano, D.; Lefebvre, P.J.; Torella, R. The involvement of the opioid system in human obesity: A study in normal weight relatives of obese people. J. Clin. Endocrinol. Metab. 1996, 81, 713–718. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Montague, C.T.; O’Rahilly, S. The perils of portliness: Causes and consequences of visceral adiposity. Diabetes 2000, 49, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Banerji, M.A.; Lebowitz, J.; Chaiken, R.L.; Gordon, D.; Kral, J.G.; Lebovitz, H.E. Relationship of visceral adipose tissue and glucose disposal is independent of sex in black NIDDM subjects. Am. J. Physiol. 1997, 273, E425–E432. [Google Scholar] [CrossRef] [PubMed]
- Goodpaster, B.H.; Kelley, D.E.; Wing, R.R.; Meier, A.; Thaete, F.L. Effects of weight loss on regional fat distribution and insulin sensitivity in obesity. Diabetes 1999, 48, 839–847. [Google Scholar] [CrossRef]
- Banerji, M.A.; Buckley, M.C.; Chaiken, R.L.; Gordon, D.; Lebovitz, H.E.; Kral, J.G. Liver fat, serum triglycerides and visceral adipose tissue in insulin-sensitive and insulin-resistant black men with NIDDM. Int. J. Obes. Relat. Metab. Disord. 1995, 19, 846–850. [Google Scholar]
- Zac-Varghese, S.; Tan, T.; Bloom, S.R. Hormonal interactions between gut and brain. Discov. Med. 2010, 10, 543–545. [Google Scholar]
- Ahima, R.S.; Lazar, M.A. Adipokines and the peripheral and neural control of energy balance. Mol. Endocrinol. 2008, 22, 1023–1031. [Google Scholar] [CrossRef]
- Park, H.K.; Kwak, M.K.; Kim, H.J.; Ahima, R.S. Linking resistin, inflammation, and cardiometabolic diseases. Korean J. Intern. Med. 2017, 32, 239–247. [Google Scholar] [CrossRef]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef]
- Esposito, K.; Ciotola, M.; Sasso, F.C.; Cozzolino, D.; Saccomanno, F.; Assaloni, R.; Ceriello, A.; Giugliano, D. Effect of a single high-fat meal on endothelial function in patients with the metabolic syndrome: Role of tumor necrosis factor-alpha. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 274–279. [Google Scholar] [CrossRef]
- Dresner, A.; Laurent, D.; Marcucci, M.; Griffin, M.E.; Dufour, S.; Cline, G.W.; Slezak, L.A.; Andersen, D.K.; Hundal, R.S.; Rothman, D.L.; et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J. Clin. Investig. 1999, 103, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Spiegelman, B.M. Tumor necrosis factor alpha: A key component of the obesity-diabetes link. Diabetes 1994, 43, 1271–1278. [Google Scholar] [CrossRef]
- Loskutoff, D.J.; Samad, F. The adipocyte and hemostatic balance in obesity: Studies of PAI-1. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1–6. [Google Scholar] [CrossRef]
- Bugianesi, E.; Moscatiello, S.; Ciaravella, M.F.; Marchesini, G. Insulin resistance in nonalcoholic fatty liver disease. Curr. Pharm Des. 2010, 16, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.J.; Miotto, P.M.; De Nardo, W.; Montgomery, M.K. The Liver as an Endocrine Organ-Linking NAFLD and Insulin Resistance. Endocr. Rev. 2019, 40, 1367–1393. [Google Scholar] [CrossRef] [PubMed]
- Jacome-Sosa, M.M.; Parks, E.J. Fatty acid sources and their fluxes as they contribute to plasma triglyceride concentrations and fatty liver in humans. Curr. Opin. Lipidol. 2014, 25, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog. Lipid Res. 2013, 52, 175–191. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef]
- Jou, J.; Choi, S.S.; Diehl, A.M. Mechanisms of disease progression in nonalcoholic fatty liver disease. Semin. Liver Dis. 2008, 28, 370–379. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- George, J.; Liddle, C. Nonalcoholic fatty liver disease: Pathogenesis and potential for nuclear receptors as therapeutic targets. Mol. Pharm. 2008, 5, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.R.; Tu, H.; Luk, A.; Repa, J.J.; Medina, J.C.; Li, L.; Schwendner, S.; Wang, S.; Thoolen, M.; Mangelsdorf, D.J.; et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000, 14, 2831–2838. [Google Scholar] [CrossRef] [PubMed]
- Schreuder, T.C.; Verwer, B.J.; van Nieuwkerk, C.M.; Mulder, C.J. Nonalcoholic fatty liver disease: An overview of current insights in pathogenesis, diagnosis and treatment. World J. Gastroenterol. 2008, 14, 2474–2486. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Kantartzis, K.; Häring, H.U. Causes and metabolic consequences of Fatty liver. Endocr. Rev. 2008, 29, 939–960. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Girard, J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: Lessons from genetically engineered mice. J. Clin. Investig. 2008, 118, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.F.; Carpentier, A.; Adeli, K.; Giacca, A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr. Rev. 2002, 23, 201–229. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Role of endoplasmic reticulum stress and c-Jun NH2-terminal kinase pathways in inflammation and origin of obesity and diabetes. Diabetes 2005, 54, S73–S78. [Google Scholar] [CrossRef]
- Sabio, G.; Das, M.; Mora, A.; Zhang, Z.; Jun, J.Y.; Ko, H.J.; Barrett, T.; Kim, J.K.; Davis, R.J. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 2008, 322, 1539–1543. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Persico, M.; Capasso, M.; Persico, E.; Svelto, M.; Russo, R.; Spano, D.; Crocè, L.; La Mura, V.; Moschella, F.; Masutti, F.; et al. Suppressor of cytokine signaling 3 (SOCS3) expression and hepatitis C virus-related chronic hepatitis: Insulin resistance and response to antiviral therapy. Hepatology 2007, 46, 1009–1015. [Google Scholar] [CrossRef]
- Torisu, T.; Sato, N.; Yoshiga, D.; Kobayashi, T.; Yoshioka, T.; Mori, H.; Iida, M.; Yoshimura, A. The dual function of hepatic SOCS3 in insulin resistance in vivo. Genes Cells 2007, 12, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Caturano, A.; Galiero, R.; Pafundi, P.C. Metformin for Type 2 Diabetes. JAMA 2019, 322, 1312. [Google Scholar] [CrossRef] [PubMed]
- Sardu, C.; Paolisso, P.; Sacra, C.; Mauro, C.; Minicucci, F.; Portoghese, M.; Rizzo, M.R.; Barbieri, M.; Sasso, F.C.; D’Onofrio, N.; et al. Effects of Metformin Therapy on Coronary Endothelial Dysfunction in Patients with Prediabetes with Stable Angina and Nonobstructive Coronary Artery Stenosis: The CODYCE Multicenter Prospective Study. Diabetes Care 2019, 42, 1946–1955. [Google Scholar] [CrossRef] [PubMed]
- Della Corte, C.M.; Ciaramella, V.; Di Mauro, C.; Castellone, M.D.; Papaccio, F.; Fasano, M.; Sasso, F.C.; Martinelli, E.; Troiani, T.; De Vita, F.; et al. Metformin increases antitumor activity of MEK inhibitors through GLI1 downregulation in LKB1 positive human NSCLC cancer cells. Oncotarget 2016, 7, 4265–4278. [Google Scholar] [CrossRef]
- Tsochatzis, E.A.; Papatheodoridis, G.V.; Archimandritis, A.J. Adipokines in nonalcoholic steatohepatitis: From pathogenesis to implications in diagnosis and therapy. Mediat. Inflamm. 2009, 2009. [Google Scholar] [CrossRef]
- Gregor, M.F.; Hotamisligil, G.S. Thematic review series: Adipocyte Biology. Adipocyte stress: The endoplasmic reticulum and metabolic disease. J. Lipid Res. 2007, 48, 1905–1914. [Google Scholar] [CrossRef]
- Hedjazifar, S.; Khatib Shahidi, R.; Hammarstedt, A.; Bonnet, L.; Church, C.; Boucher, J.; Blüher, M.; Smith, U. The Novel Adipokine Gremlin 1 Antagonizes Insulin Action and Is Increased in Type 2 Diabetes and NAFLD/NASH. Diabetes 2020, 69, 331–341. [Google Scholar] [CrossRef]
- Kakuma, T.; Lee, Y.; Higa, M.; Wang Zw Pan, W.; Shimomura, I.; Unger, R.H. Leptin, troglitazone, and the expression of sterol regulatory element binding proteins in liver and pancreatic islets. Proc. Natl. Acad. Sci. USA 2000, 97, 8536–8541. [Google Scholar] [CrossRef]
- Ikejima, K.; Honda, H.; Yoshikawa, M.; Hirose, M.; Kitamura, T.; Takei, Y.; Sato, N. Leptin augments inflammatory and profibrogenic responses in the murine liver induced by hepatotoxic chemicals. Hepatology 2001, 34, 288–297. [Google Scholar] [CrossRef]
- Wang, J.; Leclercq, I.; Brymora, J.M.; Xu, N.; Ramezani-Moghadam, M.; London, R.M.; Brigstock, D.; George, J. Kupffer cells mediate leptin-induced liver fibrosis. Gastroenterology 2009, 137, 713–723. [Google Scholar] [CrossRef]
- Choi, S.S.; Syn, W.K.; Karaca, G.F.; Omenetti, A.; Moylan, C.A.; Witek, R.P.; Agboola, K.M.; Jung, Y.; Michelotti, G.A.; Diehl, A.M. Leptin promotes the myofibroblastic phenotype in hepatic stellate cells by activating the hedgehog pathway. J. Biol. Chem. 2010, 285, 36551–36560. [Google Scholar] [CrossRef] [PubMed]
- Aleffi, S.; Navari, N.; Delogu, W.; Galastri, S.; Novo, E.; Rombouts, K.; Pinzani, M.; Parola, M.; Marra, F. Mammalian target of rapamycin mediates the angiogenic effects of leptin in human hepatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G210–G219. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Hotamisligil, G.S. Nonalcoholic fatty liver disease: Cytokine-adipokine interplay and regulation of insulin resistance. Gastroenterology 2006, 131, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef]
- Adachi, M.; Brenner, D.A. High molecular weight adiponectin inhibits proliferation of hepatic stellate cells via activation of adenosine monophosphate-activated protein kinase. Hepatology 2008, 47, 677–685. [Google Scholar] [CrossRef]
- Iwabu, M.; Yamauchi, T.; Okada-Iwabu, M.; Sato, K.; Nakagawa, T.; Funata, M.; Yamaguchi, M.; Namiki, S.; Nakayama, R.; Tabata, M.; et al. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature 2010, 464, 1313–1319. [Google Scholar] [CrossRef]
- Tsochatzis, E.; Papatheodoridis, G.V.; Archimandritis, A.J. The evolving role of leptin and adiponectin in chronic liver diseases. Am. J. Gastroenterol. 2006, 101, 2629–2640. [Google Scholar] [CrossRef]
- Kechagias, S.; Ernersson, A.; Dahlqvist, O.; Lundberg, P.; Lindström, T.; Nystrom, F.H.; Fast Food Study Group. Fast-food-based hyper-alimentation can induce rapid and profound elevation of serum alanine aminotransferase in healthy subjects. Gut 2008, 57, 649–654. [Google Scholar] [CrossRef]
- Salvatore, T.; Nevola, R.; Pafundi, P.C.; Monaco, L.; Ricozzi, C.; Imbriani, S.; Rinaldi, L.; Sasso, F.C. Incretin Hormones: The Link between Glycemic Index and Cardiometabolic Diseases. Nutrients 2019, 11, 1878. [Google Scholar] [CrossRef]
- Bergheim, I.; Weber, S.; Vos, M.; Krämer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef]
- Moore, J.B.; Gunn, P.J.; Fielding, B.A. The role of dietary sugars and de novo lipogenesis in non-alcoholic fatty liver disease. Nutrients 2014, 6, 5679–5703. [Google Scholar] [CrossRef] [PubMed]
- Spruss, A.; Kanuri, G.; Wagnerberger, S.; Haub, S.; Bischoff, S.C.; Bergheim, I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 2009, 50, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M.; Nonalcoholic Steatohepatitis Clinical Research Network. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Lazo, M.; Horska, A.; Bonekamp, S.; Lipkin, E.W.; Balasubramanyam, A.; Bantle, J.P.; Johnson, R.J.; Diehl, A.M.; Fatty Liver Subgroup of Look AHEAD Research Group; et al. Higher dietary fructose is associated with impaired hepatic adenosine triphosphate homeostasis in obese individuals with type 2 diabetes. Hepatology 2012, 56, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Fox, C.S.; Jacques, P.F.; Speliotes, E.K.; Hoffmann, U.; Smith, C.E.; Saltzman, E.; McKeown, N.M. Sugar-sweetened beverage, diet soda, and fatty liver disease in the Framingham Heart Study cohorts. J. Hepatol. 2015, 63, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Molloy, J.W.; Calcagno, C.J.; Williams, C.D.; Jones, F.J.; Torres, D.M.; Harrison, S.A. Association of coffee and caffeine consumption with fatty liver disease, nonalcoholic steatohepatitis, and degree of hepatic fibrosis. Hepatology 2012, 55, 429–436. [Google Scholar] [CrossRef]
- Gressner, O.A.; Lahme, B.; Rehbein, K.; Siluschek, M.; Weiskirchen, R.; Gressner, A.M. Pharmacological application of caffeine inhibits TGF-beta-stimulated connective tissue growth factor expression in hepatocytes via PPARgamma and SMAD2/3-dependent pathways. J. Hepatol. 2008, 49, 758–767. [Google Scholar] [CrossRef]
- Ryan, M.C.; Itsiopoulos, C.; Thodis, T.; Ward, G.; Trost, N.; Hofferberth, S.; O’Dea, K.; Desmond, P.V.; Johnson, N.A.; Wilson, A.M. The Mediterranean diet improves hepatic steatosis and insulin sensitivity in individuals with non-alcoholic fatty liver disease. J. Hepatol. 2013, 59, 138–143. [Google Scholar] [CrossRef]
- Plaz Torres, M.C.; Aghemo, A.; Lleo, A.; Bodini, G.; Furnari, M.; Marabotto, E.; Miele, L.; Giannini, E.G. Mediterranean Diet and NAFLD: What We Know and Questions That Still Need to Be Answered. Nutrients 2019, 11, 2971. [Google Scholar] [CrossRef]
- Sookoian, S.; Castaño, G.O.; Pirola, C.J. Modest alcohol consumption decreases the risk of non-alcoholic fatty liver disease: A meta-analysis of 43,175 individuals. Gut 2014, 63, 530–532. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Wigg, A.J.; Roberts-Thomson, I.C.; Dymock, R.B.; McCarthy, P.J.; Grose, R.H.; Cummins, A.G. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut 2001, 48, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Spencer, M.D.; Hamp, T.J.; Reid, R.W.; Fischer, L.M.; Zeisel, S.H.; Fodor, A.A. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology 2011, 140, 976–986. [Google Scholar] [CrossRef]
- Tremaroli, V.; Backhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Topping, D.L.; Clifton, P.M. Short-chain fatty acids and human colonic function: Roles of resistant starch and nonstarch polysaccharides. Physiol. Rev. 2001, 81, 1031–1064. [Google Scholar] [CrossRef]
- Noverr, M.C.; Huffnagle, G.B. Does the microbiota regulate immune responses outside the gut? Trends Microbiol. 2004, 12, 562–568. [Google Scholar] [CrossRef]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef]
- Zeisel, S.H.; Wishnok, J.S.; Blusztajn, J.K. Formation of methylamines from ingested choline and lecithin. J. Pharmacol. Exp. Ther. 1983, 225, 320–324. [Google Scholar]
- Kolodziejczyk, A.A.; Zheng, D.; Shibolet, O.; Elinav, E. The role of the microbiome in NAFLD and NASH. EMBO Mol. Med. 2019, 11, e9302. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Day, C.P. The genetics of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Mancina, R.M.; Matikainen, N.; Maglio, C.; Söderlund, S.; Lundbom, N.; Hakkarainen, A.; Rametta, R.; Mozzi, E.; Fargion, S.; Valenti, L.; et al. Paradoxical dissociation between hepatic fat content and de novo lipogenesis due to PNPLA3 sequence variant. J. Clin. Endocrinol. Metab. 2015, 100, E821–E825. [Google Scholar] [CrossRef]
- Zampino, R.; Coppola, N.; Cirillo, G.; Boemio, A.; Minichini, C.; Marrone, A.; Stanzione, M.; Starace, M.; Durante-Mangoni, E.; Sagnelli, E.; et al. Insulin resistance and steatosis in HBV-HCV co-infected patients: Role of PNPLA3 polymorphisms and impact on liver fibrosis progression. World J. Hepatol. 2014, 6, 677–684. [Google Scholar] [CrossRef]
- Xu, R.; Tao, A.; Zhang, S.; Deng, Y.; Chen, G. Association between patatin-like phospholipase domain containing 3 gene (PNPLA3) polymorphisms and nonalcoholic fatty liver disease: A HuGE review and meta-analysis. Sci. Rep. 2015, 5, 9284. [Google Scholar] [CrossRef]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Petta, S.; Maglio, C.; Fracanzani, A.L.; Pipitone, R.; Mozzi, E.; Motta, B.M.; Kaminska, D.; Rametta, R.; Grimaudo, S.; et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015, 61, 506–514. [Google Scholar] [CrossRef]
- Zeybel, M.; Mann, D.A.; Mann, J. Epigenetic modifications as new targets for liver disease therapies. J. Hepatol. 2013, 59, 1349–1353. [Google Scholar] [CrossRef]
- Podrini, C.; Borghesan, M.; Greco, A.; Pazienza, V.; Mazzoccoli, G.; Vinciguerra, M. Redox homeostasis and epigenetics in non-alcoholic fatty liver disease (NAFLD). Curr. Pharm. Des. 2013, 19, 2737–2746. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Zhang, H.W.; Zhou, J.Y.; Liu, Y.; Yang, Y.; Chen, X.L.; Zhu, C.H.; Zheng, R.D.; Ling, W.H.; Zhu, H.L. Betaine attenuates hepatic steatosis by reducing methylation of the MTTP promoter and elevating genomic methylation in mice fed a high-fat diet. J. Nutr. Biochem. 2014, 25, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Pooya, S.; Blaise, S.; Moreno Garcia, M.; Giudicelli, J.; Alberto, J.M.; Guéant-Rodriguez, R.M.; Jeannesson, E.; Gueguen, N.; Bressenot, A.; Nicolas, B.; et al. Methyl donor deficiency impairs fatty acid oxidation through PGC-1α hypomethylation and decreased ER-α, ERR-α, and HNF-4α in the rat liver. J. Hepatol. 2012, 57, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Moschen, A.R.; Wieser, V.; Gerner, R.R.; Bichler, A.; Enrich, B.; Moser, P.; Ebenbichler, C.F.; Kaser, S.; Tilg, H. Adipose tissue and liver expression of SIRT1, 3, and 6 increase after extensive weight loss in morbid obesity. J. Hepatol. 2013, 59, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Panera, N.; Gnani, D.; Crudele, A.; Ceccarelli, S.; Nobili, V.; Alisi, A. MicroRNAs as controlled systems and controllers in non-alcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 15079–15086. [Google Scholar] [CrossRef]
- Cheung, O.; Puri, P.; Eicken, C.; Contos, M.J.; Mirshahi, F.; Maher, J.W.; Kellum, J.M.; Min, H.; Luketic, V.A.; Sanyal, A.J. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology 2008, 48, 1810–1820. [Google Scholar] [CrossRef]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef]
- Moylan, C.A.; Pang, H.; Dellinger, A.; Suzuki, A.; Garrett, M.E.; Guy, C.D.; Murphy, S.K.; Ashley-Koch, A.E.; Choi, S.S.; Michelotti, G.A.; et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014, 59, 471–482. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Wullaert, A.; van Loo, G.; Heyninck, K.; Beyaert, R. Hepatic tumor necrosis factor signaling and nuclear factor-kappaB: Effects on liver homeostasis and beyond. Endocr. Rev. 2007, 28, 365–386. [Google Scholar] [CrossRef]
- Ribeiro, P.S.; Cortez-Pinto, H.; Solá, S.; Castro, R.E.; Ramalho, R.M.; Baptista, A.; Moura, M.C.; Camilo, M.E.; Rodrigues, C.M. Hepatocyte apoptosis, expression of death receptors, and activation of NF-kappaB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am. J. Gastroenterol. 2004, 99, 1708–1717. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Tamiya, G.; Ando, S.; Ohsumi, K.; Chiyo, T.; Mizutani, A.; Kitamura, N.; Toda, K.; Kaneko, T.; Horie, Y.; et al. Tumour necrosis factor alpha signalling through activation of Kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut 2006, 55, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Bastard, J.P.; Jardel, C.; Bruckert, E.; Blondy, P.; Capeau, J.; Laville, M.; Vidal, H.; Hainque, B. Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J. Clin. Endocrinol. Metab. 2000, 85, 3338–3342. [Google Scholar] [CrossRef]
- Crespo, J.; Cayón, A.; Fernández-Gil, P.; Hernández-Guerra, M.; Mayorga, M.; Domínguez-Díez, A.; Fernández-Escalante, J.C.; Pons-Romero, F. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology 2001, 34, 1158–1163. [Google Scholar] [CrossRef]
- Du Plessis, J.; van Pelt, J.; Korf, H.; Mathieu, C.; van der Schueren, B.; Lannoo, M.; Oyen, T.; Topal, B.; Fetter, G.; Nayler, S.; et al. Association of Adipose Tissue Inflammation with Histologic Severity of Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 635–648.e14. [Google Scholar] [CrossRef]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef]
- Sutti, S.; Albano, E. Adaptive immunity: An emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 81–92. [Google Scholar] [CrossRef]
- Pessayre, D.; Fromenty, B. NASH: A mitochondrial disease. J. Hepatol. 2005, 42, 928–940. [Google Scholar] [CrossRef]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; He, W.; Tsai, P.J.; Chen, P.H.; Ye, M.; Guo, J.; Su, Z. Mutual interaction between endoplasmic reticulum and mitochondria in nonalcoholic fatty liver disease. Lipids Health Dis. 2020, 19, 1–19. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 14205–14218. [Google Scholar] [CrossRef]
- Cusi, K. Nonalcoholic fatty liver disease in type 2 diabetes mellitus. Curr. Opin. Endocrinol. Diabetes Obes. 2009, 16, 141–149. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Seki, S.; Kitada, T.; Sakaguchi, H. Clinicopathological significance of oxidative cellular damage in non-alcoholic fatty liver diseases. Hepatol. Res. 2005, 33, 132–134. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Mirshahi, F.; Cheung, O.; Natarajan, R.; Maher, J.W.; Kellum, J.M.; Sanyal, A.J. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef]
- Kapoor, A.; Sanyal, A.J. Endoplasmic reticulum stress and the unfolded protein response. Clin. Liver Dis. 2009, 13, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Zhou, Y.; Lee, J.; Lu, A.; Sun, C.; Chung, J.; Ueki, K.; Ozcan, U. The regulatory subunits of PI3K, p85alpha and p85beta, interact with XBP-1 and increase its nuclear translocation. Nat. Med. 2010, 16, 429–437. [Google Scholar] [CrossRef]
- Winnay, J.N.; Boucher, J.; Mori, M.A.; Ueki, K.; Kahn, C.R. A regulatory subunit of phosphoinositide 3-kinase increases the nuclear accumulation of X-box-binding protein-1 to modulate the unfolded protein response. Nat. Med. 2010, 16, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Marfella, R.; D’Amico, M.; Di Filippo, C.; Siniscalchi, M.; Sasso, F.C.; Ferraraccio, F.; Rossi, F.; Paolisso, G. The possible role of the ubiquitin proteasome system in the development of atherosclerosis in diabetes. Cardiovasc. Diabetol. 2007, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Day, C.P.; Bonora, E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N. Engl. J. Med. 2010, 363, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Byrne, C.D.; Lonardo, A.; Zoppini, G.; Barbui, C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: A meta-analysis. J. Hepatol. 2016, 65, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Hagström, H.; Nasr, P.; Fredrikson, M.; Stål, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Motamed, N.; Rabiee, B.; Poustchi, H.; Dehestani, B.; Hemasi, G.R.; Khonsari, M.R.; Maadi, M.; Saeedian, F.S.; Zamani, F. Non-alcoholic fatty liver disease (NAFLD) and 10-year risk of cardiovascular diseases. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 31–38. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Kozakova, M.; Højlund, K.; Flyvbjerg, A.; Favuzzi, A.; Mitrakou, A.; Balkau, B.; RISC Investigators. Fatty liver is associated with insulin resistance, risk of coronary heart disease, and early atherosclerosis in a large European population. Hepatology 2009, 49, 1537–1544. [Google Scholar] [CrossRef]
- Targher, G.; Bertolini, L.; Padovani, R.; Poli, F.; Scala, L.; Tessari, R.; Zenari, L.; Falezza, G. Increased prevalence of cardiovascular disease in Type 2 diabetic patients with non-alcoholic fatty liver disease. Diabet. Med. 2006, 23, 403–409. [Google Scholar] [CrossRef]
- Mellinger, J.L.; Pencina, K.M.; Massaro, J.M.; Hoffmann, U.; Seshadri, S.; Fox, C.S.; O’Donnell, C.J.; Speliotes, E.K. Hepatic steatosis and cardiovascular disease outcomes: An analysis of the Framingham Heart Study. J. Hepatol. 2015, 63, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Wong, G.L.; Yeung, J.C.; Fung, C.Y.; Chan, J.K.; Chang, Z.H.; Kwan, C.T.; Lam, H.W.; Limquiaco, J.; Chim, A.M.; et al. Long-term clinical outcomes after fatty liver screening in patients undergoing coronary angiogram: A prospective cohort study. Hepatology 2016, 63, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Assy, N.; Djibre, A.; Farah, R.; Grosovski, M.; Marmor, A. Presence of coronary plaques in patients with nonalcoholic fatty liver disease. Radiology 2010, 254, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Bertolini, L.; Padovani, R.; Zenari, L.; Zoppini, G.; Falezza, G. Relation of nonalcoholic hepatic steatosis to early carotid atherosclerosis in healthy men: Role of visceral fat accumulation. Diabetes Care 2004, 27, 2498–2500. [Google Scholar] [CrossRef] [PubMed]
- Fracanzani, A.L.; Burdick, L.; Raselli, S.; Pedotti, P.; Grigore, L.; Santorelli, G.; Valenti, L.; Maraschi, A.; Catapano, A.; Fargion, S. Carotid artery intima-media thickness in nonalcoholic fatty liver disease. Am. J. Med. 2008, 121, 72–78. [Google Scholar] [CrossRef]
- Villanova, N.; Moscatiello, S.; Ramilli, S.; Bugianesi, E.; Magalotti, D.; Vanni, E.; Zoli, M.; Marchesini, G. Endothelial dysfunction and cardiovascular risk profile in nonalcoholic fatty liver disease. Hepatology 2005, 42, 473–480. [Google Scholar] [CrossRef]
- Sinn, D.H.; Kang, D.; Chang, Y.; Ryu, S.; Gu, S.; Kim, H.; Seong, D.; Cho, S.J.; Yi, B.K.; Park, H.D.; et al. Non-alcoholic fatty liver disease and progression of coronary artery calcium score: A retrospective cohort study. Gut 2017, 66, 323–329. [Google Scholar] [CrossRef]
- Sinn, D.H.; Cho, S.J.; Gu, S.; Seong, D.; Kang, D.; Kim, H.; Yi, B.K.; Paik, S.W.; Guallar, E.; Cho, J.; et al. Persistent Nonalcoholic Fatty Liver Disease Increases Risk for Carotid Atherosclerosis. Gastroenterology 2016, 151, 481–488.e1. [Google Scholar] [CrossRef]
- Howard, G.; O’Leary, D.H.; Zaccaro, D.; Haffner, S.; Rewers, M.; Hamman, R.; Selby, J.V.; Saad, M.F.; Savage, P.; Bergman, R. Insulin sensitivity and atherosclerosis. The Insulin Resistance Atherosclerosis Study (IRAS) Investigators. Circulation 1996, 93, 1809–1817. [Google Scholar] [CrossRef]
- Kozakova, M.; Natali, A.; Dekker, J.; Beck-Nielsen, H.; Laakso, M.; Nilsson, P.; Balkau, B.; Ferrannini, E.; RISC Investigators. Insulin sensitivity and carotid intima-media thickness: Relationship between insulin sensitivity and cardiovascular risk study. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1409–1417. [Google Scholar] [CrossRef]
- Sasso, F.C.; Pafundi, P.C.; Marfella, R.; Calabrò, P.; Piscione, F.; Furbatto, F.; Esposito, G.; Galiero, R.; Gragnano, F.; Rinaldi, L.; et al. Adiponectin and insulin resistance are related to restenosis and overall new PCI in subjects with normal glucose tolerance: The prospective AIRE Study. Cardiovasc. Diabetol. 2019, 18, 24. [Google Scholar] [CrossRef] [PubMed]
- Zavaroni, I.; Bonini, L.; Gasparini, P.; Barilli, A.L.; Zuccarelli, A.; Dall’Aglio, E.; Delsignore, R.; Reaven, G.M. Hyperinsulinemia in a normal population as a predictor of non-insulin-dependent diabetes mellitus, hypertension, and coronary heart disease: The Barilla factory revisited. Metabolism 1999, 48, 989–994. [Google Scholar] [CrossRef]
- Eddy, D.; Schlessinger, L.; Kahn, R.; Peskin, B.; Schiebinger, R. Relationship of insulin resistance and related metabolic variables to coronary artery disease: A mathematical analysis. Diabetes Care 2009, 32, 361–366. [Google Scholar] [CrossRef]
- Hanley, A.J.; Williams, K.; Stern, M.P.; Haffner, S.M. Homeostasis model assessment of insulin resistance in relation to the incidence of cardiovascular disease: The San Antonio Heart Study. Diabetes Care 2002, 25, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Gast, K.B.; Tjeerdema, N.; Stijnen, T.; Smit, J.W.; Dekkers, O.M. Insulin resistance and risk of incident cardiovascular events in adults without diabetes: Meta-analysis. PLoS ONE 2012, 7, e52036. [Google Scholar] [CrossRef]
- VanWagner, L.B.; Wilcox, J.E.; Colangelo, L.A.; Lloyd-Jones, D.M.; Carr, J.J.; Lima, J.A.; Lewis, C.E.; Rinella, M.E.; Shah, S.J. Association of nonalcoholic fatty liver disease with subclinical myocardial remodeling and dysfunction: A population-based study. Hepatology 2015, 62, 773–783. [Google Scholar] [CrossRef]
- Valbusa, F.; Agnoletti, D.; Scala, L.; Grillo, C.; Arduini, P.; Bonapace, S.; Calabria, S.; Scaturro, G.; Mantovani, A.; Zoppini, G.; et al. Non-alcoholic fatty liver disease and increased risk of all-cause mortality in elderly patients admitted for acute heart failure. Int. J. Cardiol. 2018, 265, 162–168. [Google Scholar] [CrossRef]
- Yoshihisa, A.; Sato, Y.; Yokokawa, T.; Sato, T.; Suzuki, S.; Oikawa, M.; Kobayashi, A.; Yamaki, T.; Kunii, H.; Nakazato, K.; et al. Liver fibrosis score predicts mortality in heart failure patients with preserved ejection fraction. ESC Heart Fail. 2018, 5, 262–270. [Google Scholar] [CrossRef]
- Wijarnpreecha, K.; Boonpheng, B.; Thongprayoon, C.; Jaruvongvanich, V.; Ungprasert, P. The association between non-alcoholic fatty liver disease and atrial fibrillation: A meta-analysis. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 525–532. [Google Scholar] [CrossRef]
- Kim, S.U.; Song, D.; Heo, J.H.; Yoo, J.; Kim, B.K.; Park, J.Y.; Kim, D.Y.; Ahn, S.H.; Kim, K.J.; Han, K.H.; et al. Liver fibrosis assessed with transient elastography is an independent risk factor for ischemic stroke. Atherosclerosis 2017, 260, 156–162. [Google Scholar] [CrossRef]
- Tziomalos, K.; Giampatzis, V.; Bouziana, S.D.; Spanou, M.; Papadopoulou, M.; Pavlidis, A.; Kostaki, S.; Bozikas, A.; Savopoulos, C.; Hatzitolios, A.I. Association between nonalcoholic fatty liver disease and acute ischemic stroke severity and outcome. World J. Hepatol. 2013, 5, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Ryu, S.; Sung, E.; Woo, H.Y.; Oh, E.; Cha, K.; Jung, E.; Kim, W.S. Nonalcoholic fatty liver disease predicts chronic kidney disease in nonhypertensive and nondiabetic Korean men. Metabolism 2008, 57, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Tabibian, J.H.; Ekstedt, M.; Kechagias, S.; Hamaguchi, M.; Hultcrantz, R.; Hagström, H.; Yoon, S.K.; Charatcharoenwitthaya, P.; et al. Association of non-alcoholic fatty liver disease with chronic kidney disease: A systematic review and meta-analysis. PLoS Med. 2014, 11, e1001680. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Chonchol, M.; Bertolini, L.; Rodella, S.; Zenari, L.; Lippi, G.; Franchini, M.; Zoppini, G.; Muggeo, M. Increased risk of CKD among type 2 diabetics with nonalcoholic fatty liver disease. J. Am. Soc. Nephrol. 2008, 19, 1564–1570. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Testa, F.; Zacchia, M.; Crispo, A.A.; Di Iorio, V.; Capolongo, G.; Rinaldi, L.; D’Antonio, M.; Fioretti, T.; Salvatore, F.; et al. Genetic characterization of Italian patients with Bardet-Biedl syndrome and correlation to ocular, renal and audio-vestibular phenotype: Identification of eleven novel pathogenic sequence variants. BMC Med. Genet. 2017, 18, 10. [Google Scholar] [CrossRef] [PubMed]
- Valente, G.; Rinaldi, L.; Sgambato, M.; Piai, G. Conversion from twice-daily to once-daily tacrolimus in stable liver transplant patients: Effectiveness in a real-world setting. Transplant. Proc. 2013, 45, 1273–1275. [Google Scholar] [CrossRef] [PubMed]
- Giordano, M.; Ciarambino, T.; Castellino, P.; Malatino, L.; Cataliotti, A.; Rinaldi, L.; Paolisso, G.; Adinolfi, L.E. Seasonal variations of hyponatremia in the emergency department: Age-related changes. Am. J. Emerg. Med. 2017, 35, 749–752. [Google Scholar] [CrossRef]
- Bril, F.; Cusi, K. Management of Nonalcoholic Fatty Liver Disease in Patients with Type 2 Diabetes: A Call to Action. Diabetes Care 2017, 40, 419–430. [Google Scholar] [CrossRef]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef]
- Dai, W.; Ye, L.; Liu, A.; Wen, S.W.; Deng, J.; Wu, X.; Lai, Z. Prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus: A meta-analysis. Medicine 2017, 96, e8179. [Google Scholar] [CrossRef]
- Portillo, P.; Yavuz, S.; Bril, F.; Cusi, K. Role of Insulin Resistance and Diabetes in the Pathogenesis and Treatment of Nonalcoholic Fatty Liver Disease. Curr. Hepatol. Rep. 2014, 13, 159–170. [Google Scholar] [CrossRef]
- Non-Alcoholic Fatty Liver Disease Study Group; Lonardo, A.; Bellentani, S.; Byrne, C.D.; Caldwell, S.H.; Cortez-Pinto, H.; Grieco, A.; Machado, M.V.; Miele, L.; Targher, G. Epidemiological modifiers of non-alcoholic fatty liver disease: Focus on high-risk groups. Dig. Liver Dis. 2015, 47, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Inchiostro, S.; Bertoli, G.; Zanette, G.; Donadon, V. Evidence of higher insulin resistance in NIDDM patients with ischaemic heart disease. Diabetologia 1994, 37, 597–603. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Torella, D.; Ellison, G.M.; Torella, M.; Vicinanza, C.; Aquila, I.; Iaconetti, C.; Scalise, M.; Marino, F.; Henning, B.J.; Lewis, F.C.; et al. Carbonic anhydrase activation is associated with worsened pathological remodeling in human ischemic diabetic cardiomyopathy. J. Am. Heart Assoc. 2014, 3, e000434. [Google Scholar] [CrossRef] [PubMed]
- Marfella, R.; Sasso, F.C.; Siniscalchi, M.; Paolisso, P.; Rizzo, M.R.; Ferraro, F.; Stabile, E.; Sorropago, G.; Calabrò, P.; Carbonara, O.; et al. Peri-procedural tight glycemic control during early percutaneous coronary intervention is associated with a lower rate of in-stent restenosis in patients with acute ST-elevation myocardial infarction. J. Clin. Endocrinol. Metab. 2012, 97, 2862–2871. [Google Scholar] [CrossRef]
- Sasso, F.C.; Rinaldi, L.; Lascar, N.; Marrone, A.; Pafundi, P.C.; Adinolfi, L.E.; Marfella, R. Role of Tight Glycemic Control during Acute Coronary Syndrome on CV Outcome in Type 2 Diabetes. J. Diabetes Res. 2018, 4, 3106056. [Google Scholar] [CrossRef]
- Zhou, Y.Y.; Zhou, X.D.; Wu, S.J.; Hu, X.Q.; Tang, B.; Poucke, S.V.; Pan, X.-Y.; Wu, W.-J.; Gu, X.-M.; Zheng, M.H.; et al. Synergistic increase in cardiovascular risk in diabetes mellitus with nonalcoholic fatty liver disease: A meta-analysis. Eur. J. Gastroenterol. Hepatol. 2018, 30, 631–636. [Google Scholar] [CrossRef]
- Paik, J.; Golabi, P.; Younoszai, Z.; Mishra, A.; Trimble, G.; Younossi, Z.M. Chronic kidney disease is independently associated with increased mortality in patients with nonalcoholic fatty liver disease. Liver Int. 2019, 39, 342–352. [Google Scholar] [CrossRef]
- Sasso, F.C.; Chiodini, P.; Carbonara, O.; De Nicola, L.; Conte, G.; Salvatore, T.; Nasti, R.; Marfella, R.; Gallo, C.; Nephropathy in Type 2 Diabetes Study Group; et al. High cardiovascular risk in patients with Type 2 diabetic nephropathy: The predictive role of albuminuria and glomerular filtration rate. The NID-2 Prospective Cohort Study. Nephrol. Dial. Transplant. 2012, 27, 2269–2274. [Google Scholar] [CrossRef]
- Minutolo, R.; Gabbai, F.B.; Provenzano, M.; Chiodini, P.; Borrelli, S.; Garofalo, C.; Sasso, F.C.; Santoro, D.; Bellizzi, V.; Conte, G.; et al. Cardiorenal prognosis by residual proteinuria level in diabetic chronic kidney disease: Pooled analysis of four cohort studies. Nephrol. Dial. Transplant. 2018, 33, 1942–1949. [Google Scholar] [CrossRef]
- Minutolo, R.; Sasso, F.C.; Chiodini, P.; Cianciaruso, B.; Carbonara, O.; Zamboli, P.; Tirino, G.; Pota, A.; Torella, R.; Conte, G.; et al. Management of cardiovascular risk factors in advanced type 2 diabetic nephropathy: A comparative analysis in nephrology, diabetology and primary care settings. J. Hypertens. 2006, 24, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Önnerhag, K.; Dreja, K.; Nilsson, P.M.; Lindgren, S. Increased mortality in non-alcoholic fatty liver disease with chronic kidney disease is explained by metabolic comorbidities. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Sasso, F.C.; Salvatore, T.; Tranchino, G.; Cozzolino, D.; Caruso, A.A.; Persico, M.; Gentile, S.; Torella, D.; Torella, R. Cochlear dysfunction in type 2 diabetes: A complication independent of neuropathy and acute hyperglycemia. Metabolism 1999, 48, 1346–1350. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caturano, A.; Acierno, C.; Nevola, R.; Pafundi, P.C.; Galiero, R.; Rinaldi, L.; Salvatore, T.; Adinolfi, L.E.; Sasso, F.C. Non-Alcoholic Fatty Liver Disease: From Pathogenesis to Clinical Impact. Processes 2021, 9, 135. https://doi.org/10.3390/pr9010135
Caturano A, Acierno C, Nevola R, Pafundi PC, Galiero R, Rinaldi L, Salvatore T, Adinolfi LE, Sasso FC. Non-Alcoholic Fatty Liver Disease: From Pathogenesis to Clinical Impact. Processes. 2021; 9(1):135. https://doi.org/10.3390/pr9010135
Chicago/Turabian StyleCaturano, Alfredo, Carlo Acierno, Riccardo Nevola, Pia Clara Pafundi, Raffaele Galiero, Luca Rinaldi, Teresa Salvatore, Luigi Elio Adinolfi, and Ferdinando Carlo Sasso. 2021. "Non-Alcoholic Fatty Liver Disease: From Pathogenesis to Clinical Impact" Processes 9, no. 1: 135. https://doi.org/10.3390/pr9010135
APA StyleCaturano, A., Acierno, C., Nevola, R., Pafundi, P. C., Galiero, R., Rinaldi, L., Salvatore, T., Adinolfi, L. E., & Sasso, F. C. (2021). Non-Alcoholic Fatty Liver Disease: From Pathogenesis to Clinical Impact. Processes, 9(1), 135. https://doi.org/10.3390/pr9010135