N-Tosylcarboxamide in C–H Functionalization: More than a Simple Directing Group



{kind=link}
{kind=link}
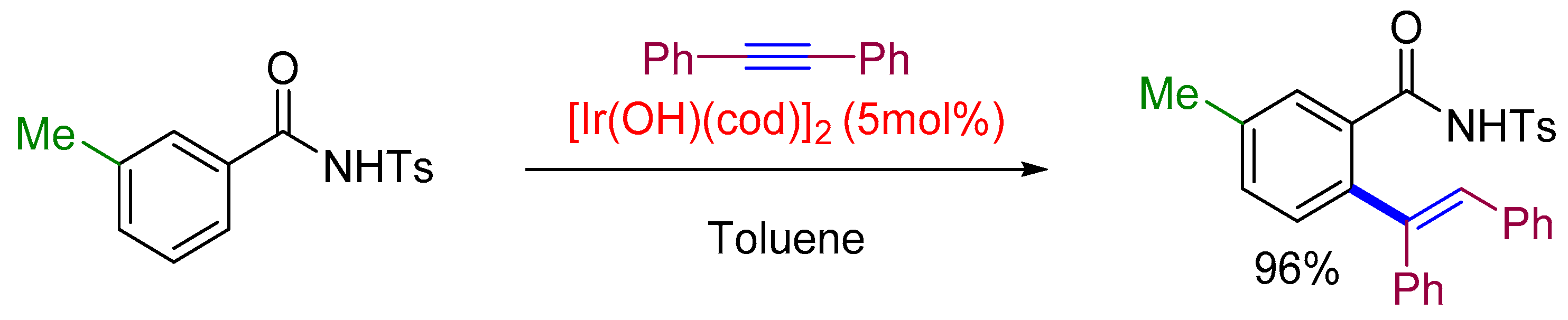{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
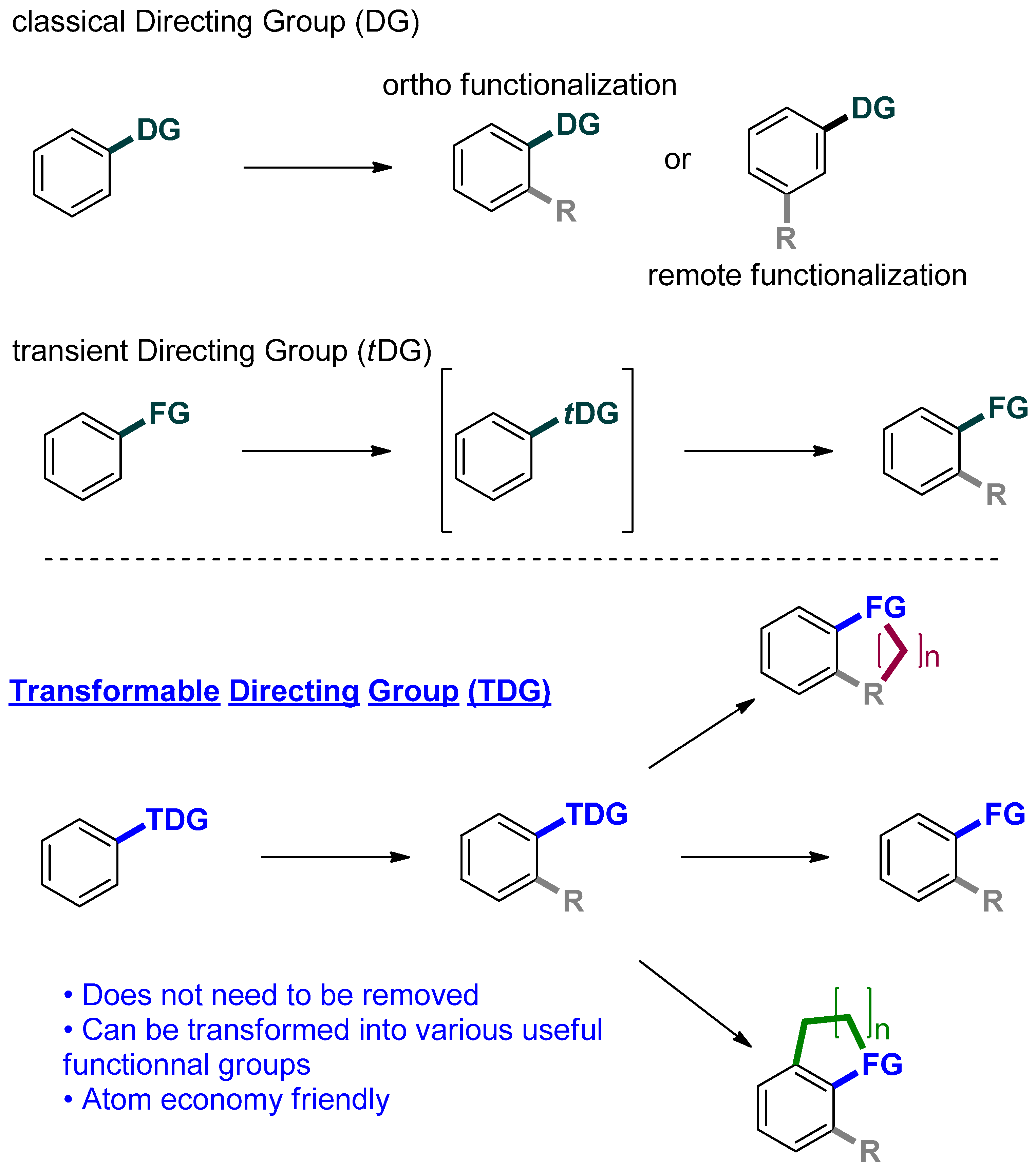
1. Introduction
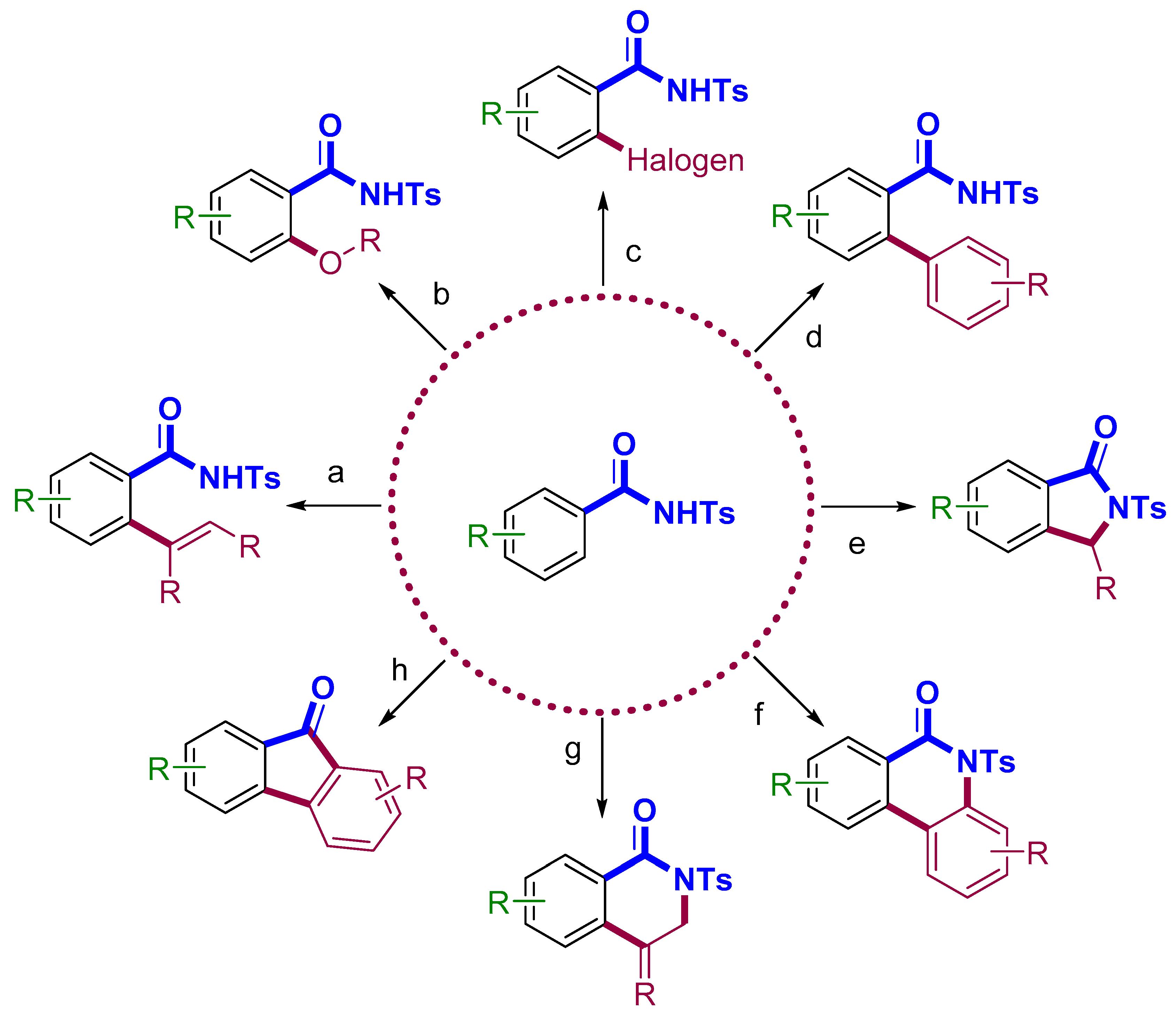
2. C–H Functionalization Using N-Tosylcarboxamide as Directing Group
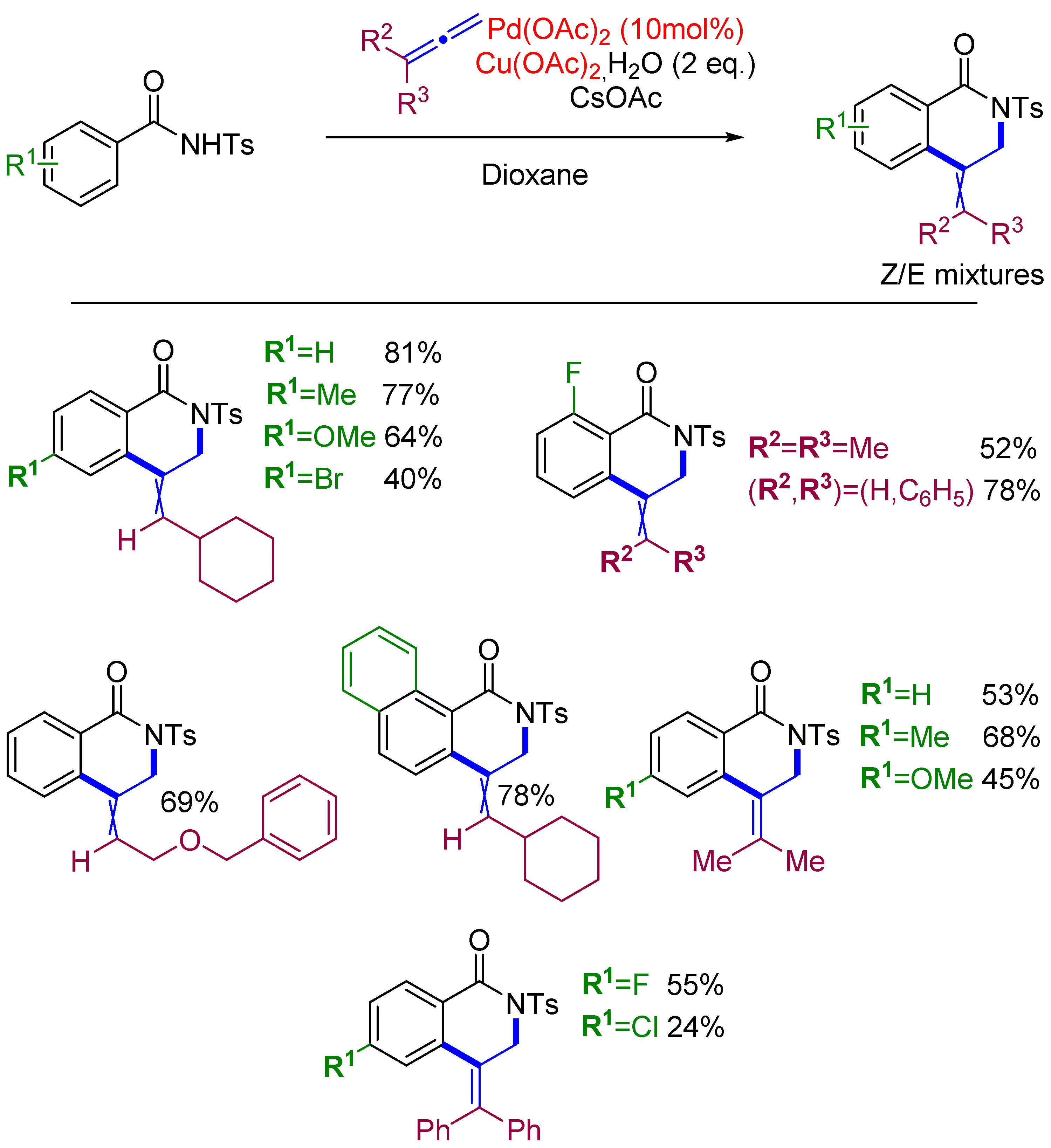
2.1. Alkenylation
2.2. Alkoxylation
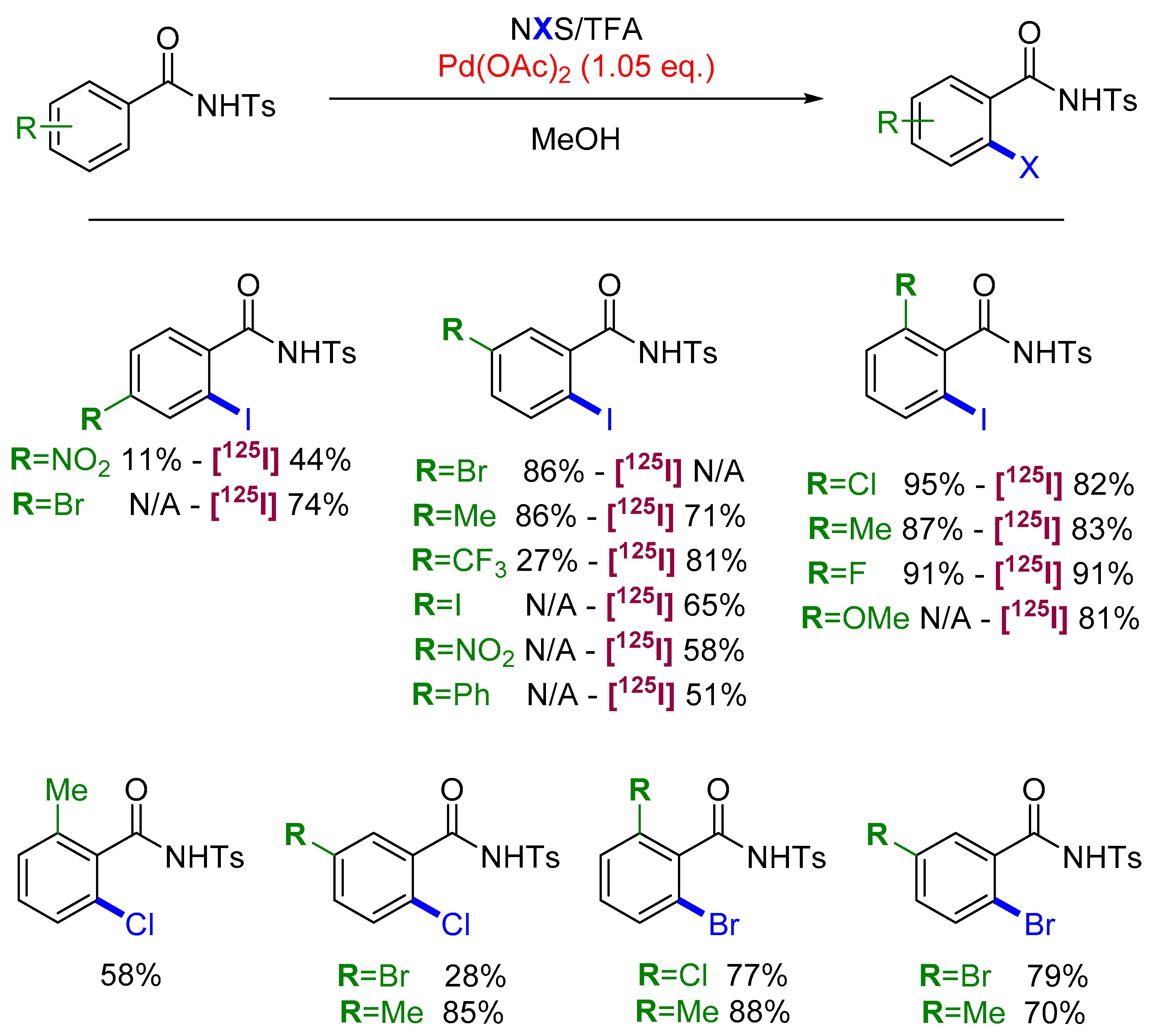
2.3. Halogenation
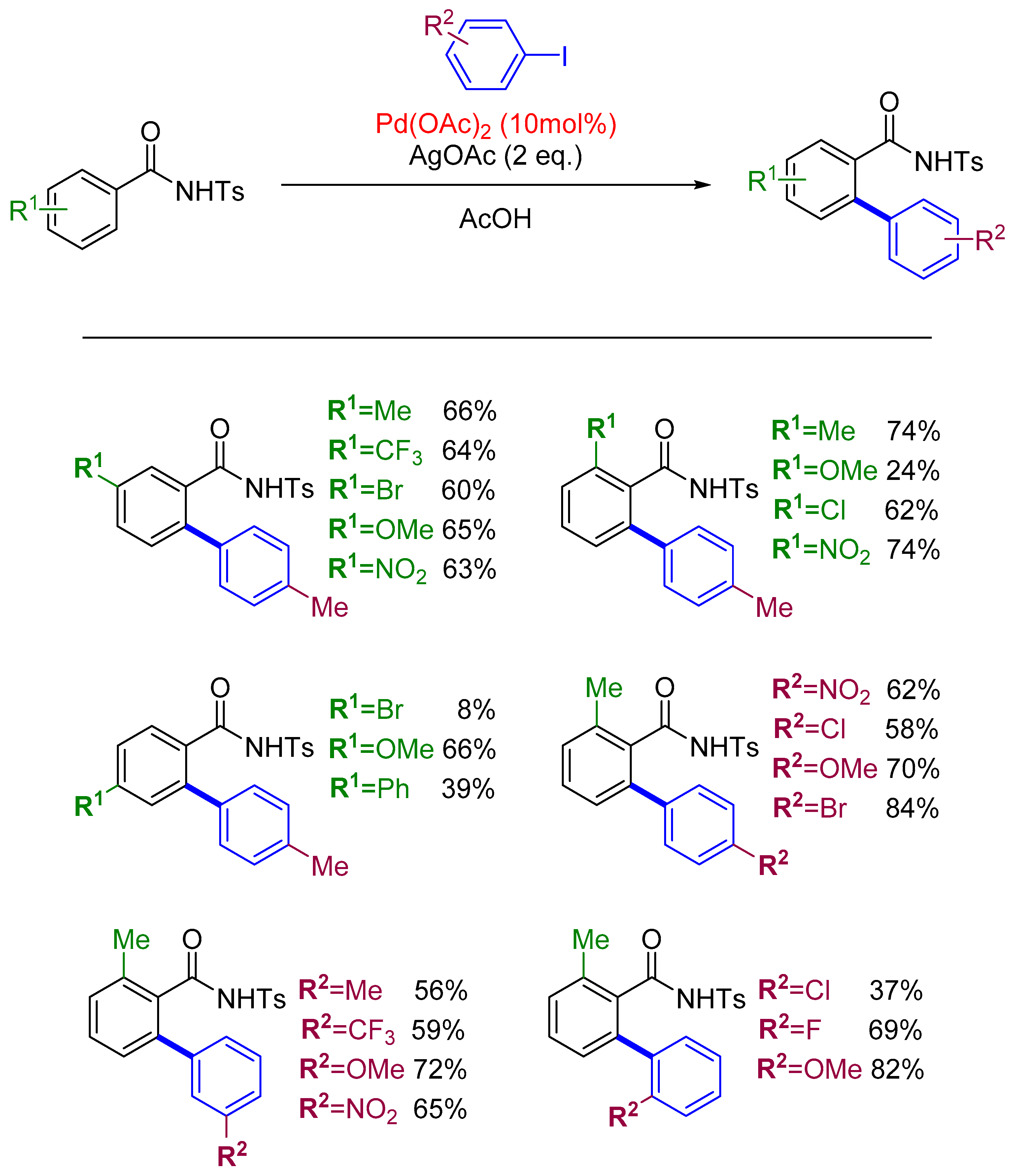
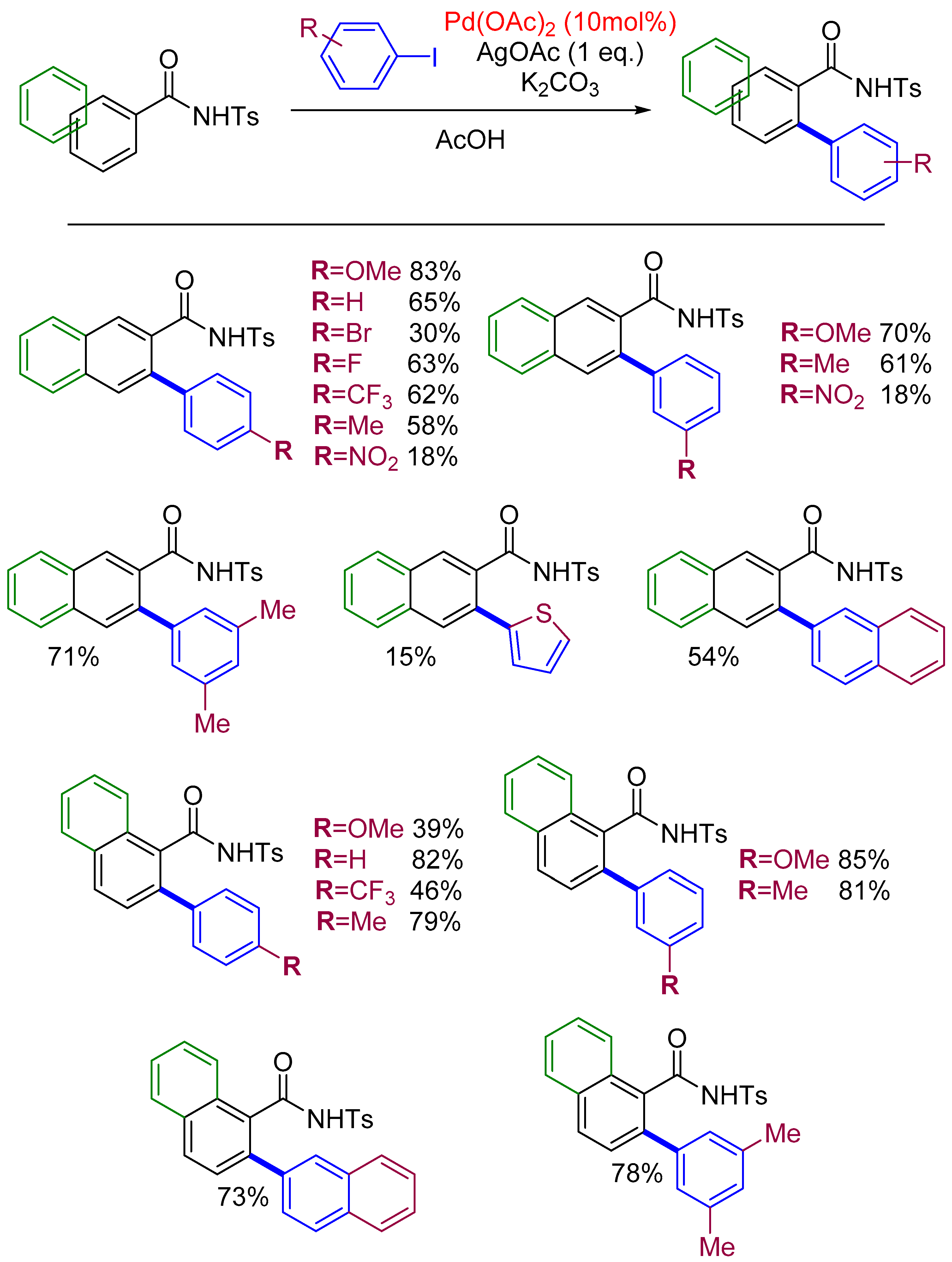
2.4. Arylation
3. Access to Six-Membered Rings through C–H Functionalization and Subsequent Transformation of Tosylbenzamides
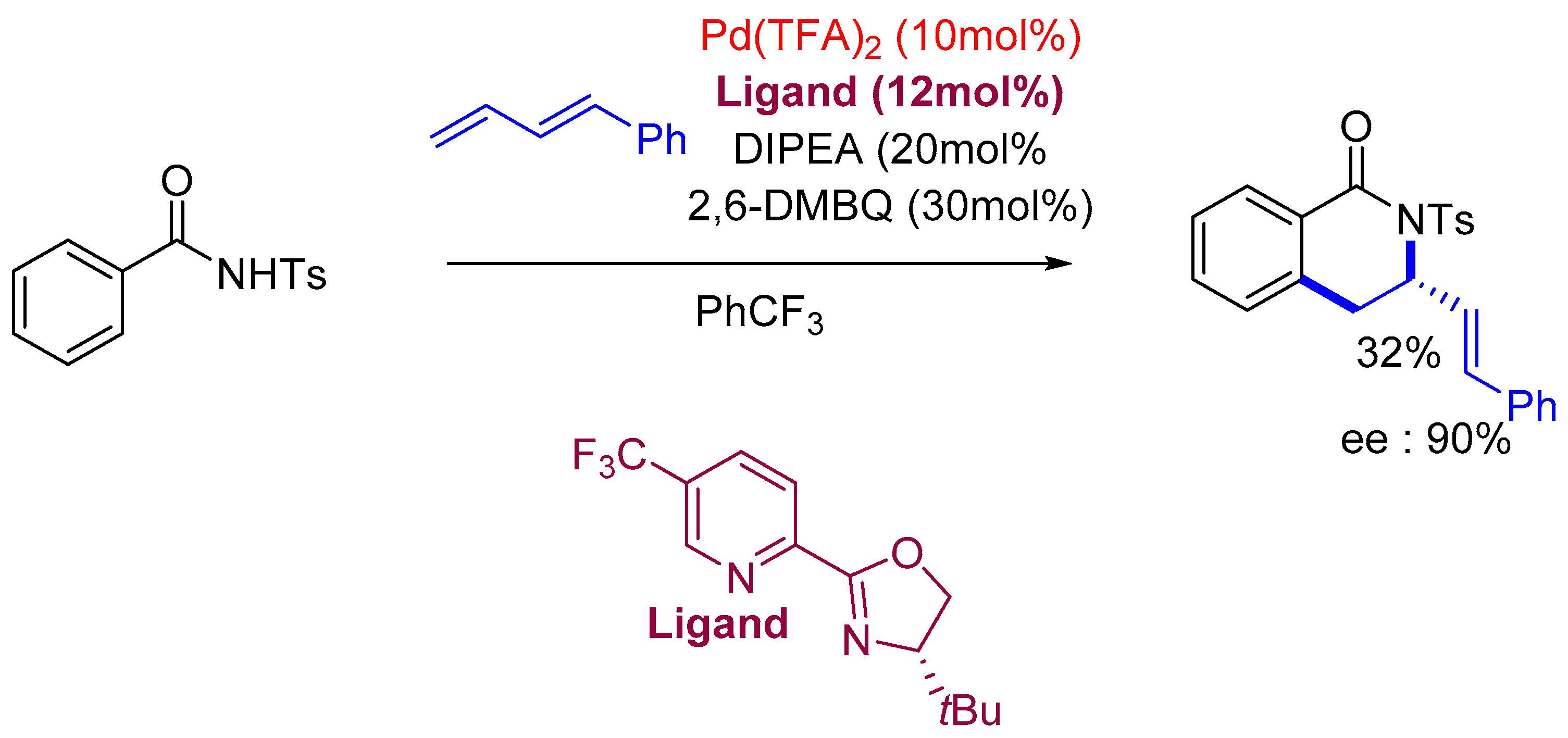
3.1. Sequential Approach
3.2. One-Pot Sequence
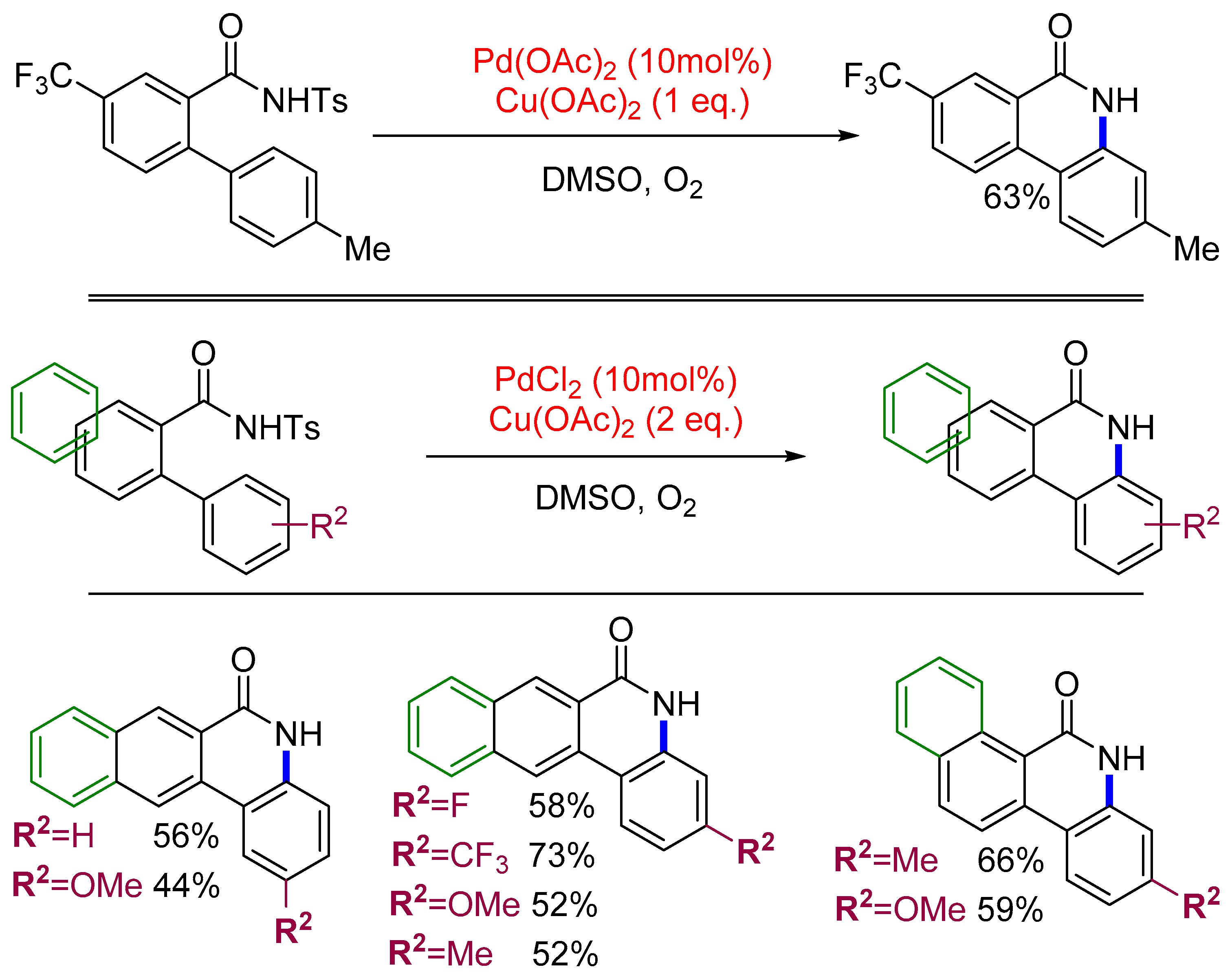
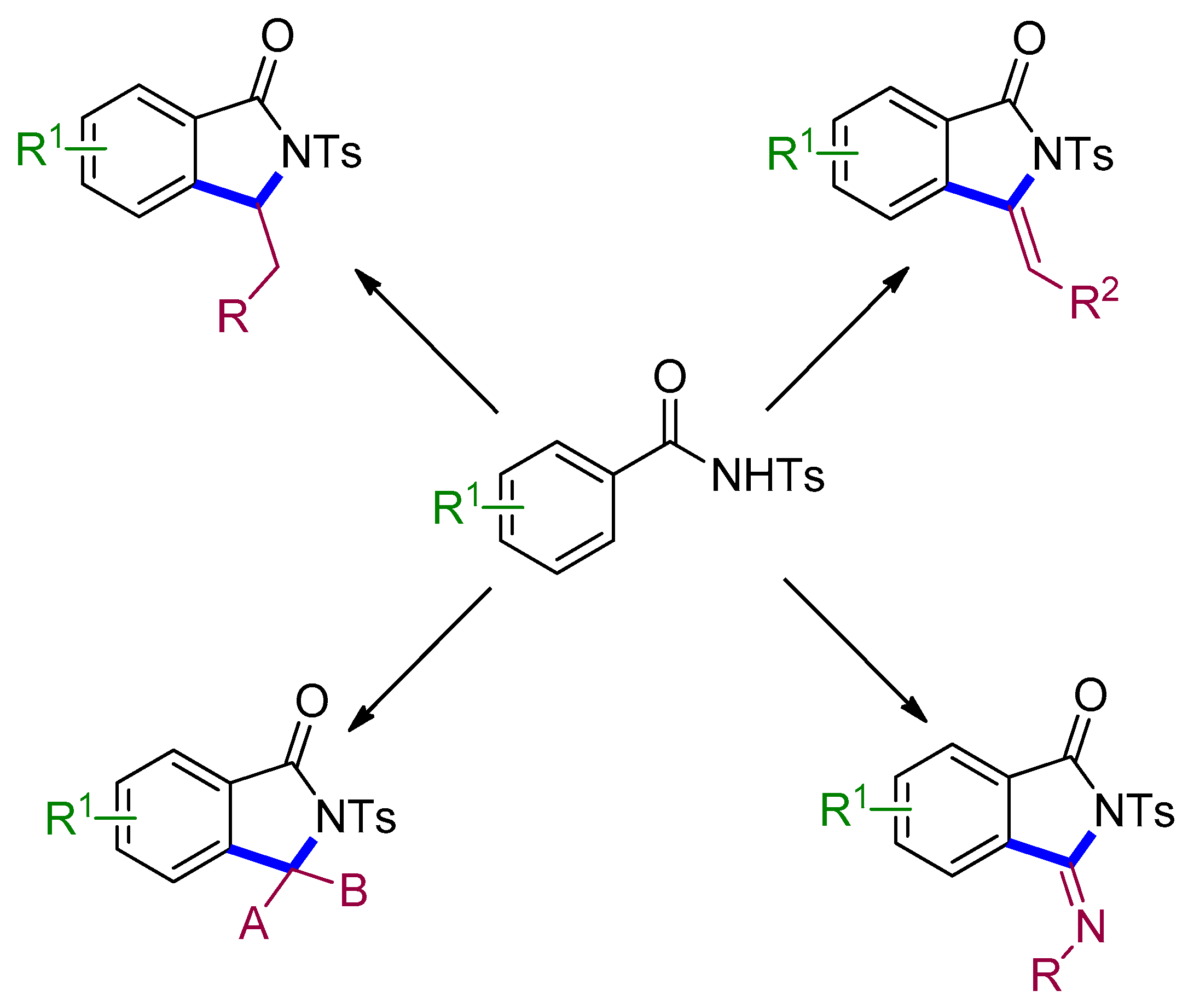
4. Access to Five-Membered Rings through C–H Functionalization and Subsequent Transformation of Tosylbenzamides
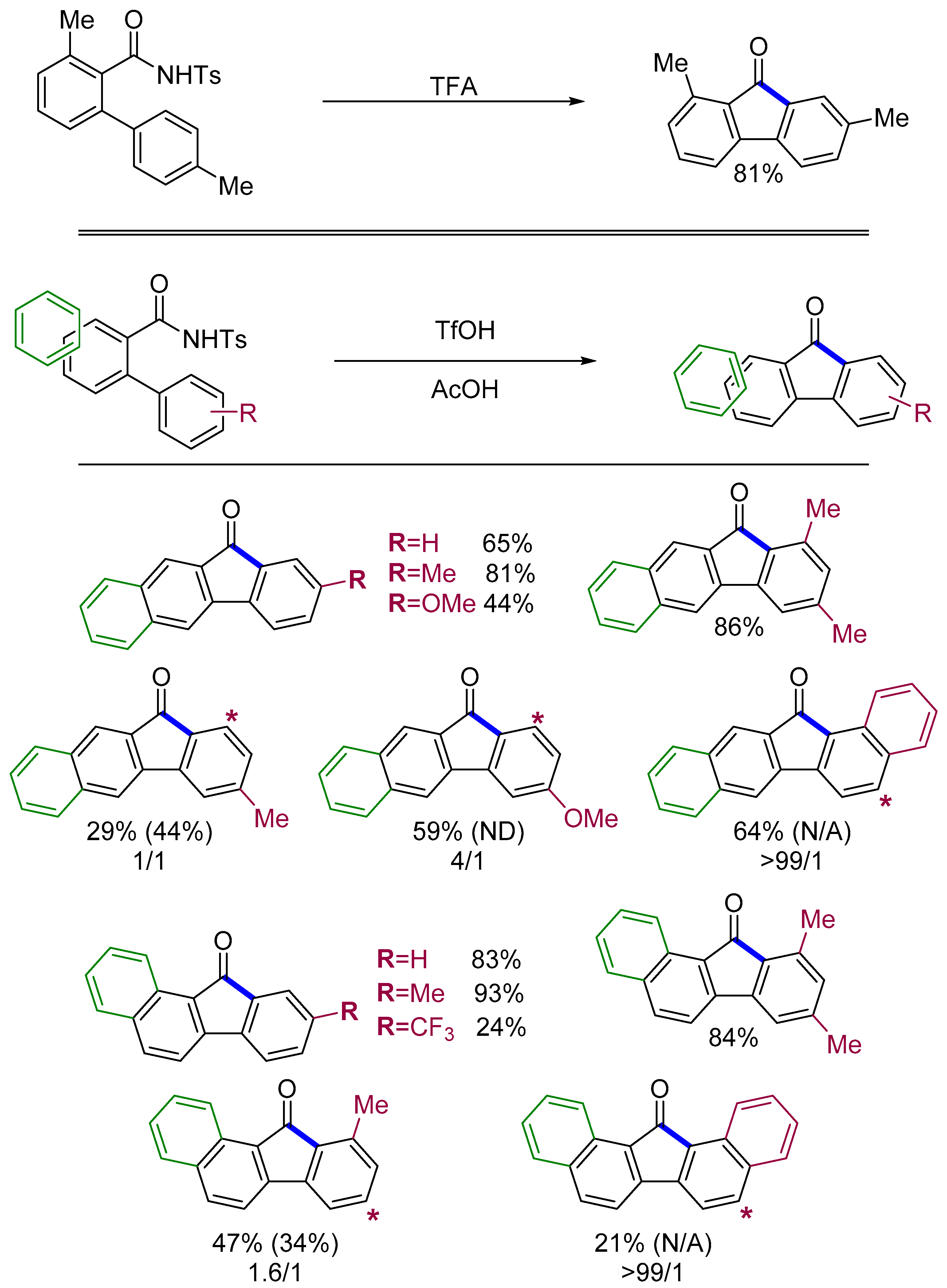
4.1. Fluorenones
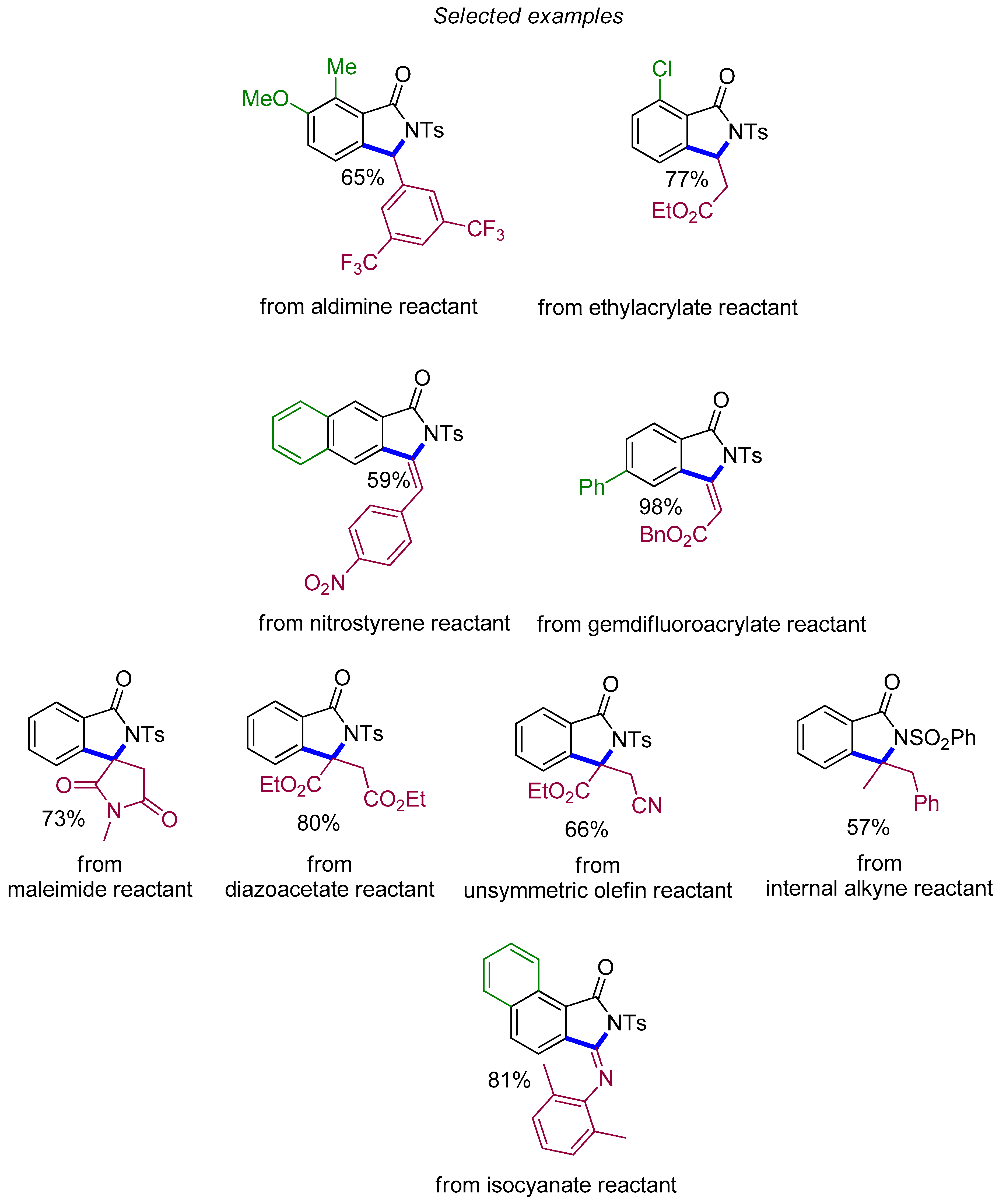
4.2. Isoindolinones
4.3. Naphthalene-Fused Isoindolinones
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rosen, B.R.; Simke, L.R.; Thuy-Boun, P.S.; Dixon, D.D.; Yu, J.-Q.; Baran, P.S. C–H Functionalization Logic Enables Synthesis of (+)-Hongoquercin A and Related Compounds. Angew. Chem. Int. Ed. 2013, 52, 7317–7320. [Google Scholar] [CrossRef] [PubMed]
- Gandeepan, P.; Müller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. 3d Transition Metals for C–H Activation. Chem. Rev. 2019, 119, 2192–2452. [Google Scholar] [CrossRef]
- Yi, H.; Zhang, G.; Wang, H.; Huang, Z.; Wang, J.; Singh, A.K.; Lei, A. Recent Advances in Radical C–H Activation/Radical Cross-Coupling. Chem. Rev. 2017, 117, 9016–9085. [Google Scholar] [CrossRef] [PubMed]
- Large, B.; Prim, D. C–H Functionalization Strategies in the Naphthalene Series: Site Selections and Functional Diversity. Synthesis 2020. [Google Scholar] [CrossRef]
- Cheng, G.; Wang, P.; Yu, J.Q. Meta-C−H Arylation and Alkylation of Benzylsulfonamide Enabled by a Palladium(II)/Isoquinoline Catalyst. Angew. Chem. Int. Ed. 2017, 56, 8183–8186. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Ajitha, M.J.; Lee, Y.; Ryu, J.; Kim, J.; Lee, Y.; Jung, Y.; Chang, S. Hydrogen-Bond-Assisted Controlled C–H Functionalization via Adaptive Recognition of a Purine Directing Group. J. Am. Chem. Soc. 2014, 136, 1132–1140. [Google Scholar] [CrossRef]
- Sarkar, D.; Gulevich, A.V.; Melkonyan, F.S.; Gevorgyan, V. Synthesis of Multisubstituted Arenes via PyrDipSi-Directed Unsymmetrical Iterative C–H Functionalizations. ACS Catal. 2015, 5, 6792–6801. [Google Scholar] [CrossRef]
- Chen, X.-Y.; Ozturk, S.; Sorensen, E.J. Pd-Catalyzed Ortho C–H Hydroxylation of Benzaldehydes Using a Transient Directing Group. Org. Lett. 2017, 19, 6280–6283. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, S.Y.; He, G.; Ai, Z.; Nack, W.A.; Chen, G. Copper-Catalyzed Carboxamide-Directed Ortho Amination of Anilines with Alkylamines at Room Temperature. Org. Lett. 2014, 16, 1764–1767. [Google Scholar] [CrossRef]
- Chen, X.Y.; Ozturk, S.; Sorensen, E.J. Synthesis of Fluorenones from Benzaldehydes and Aryl Iodides: Dual C–H Functionalizations Using a Transient Directing Group. Org. Lett. 2017, 19, 1140–1143. [Google Scholar] [CrossRef]
- Guan, Z.; Chen, S.; Huang, Y.; Yao, H. Rhodium(III)-Catalyzed Intramolecular Olefin Hydroarylation of Aromatic Aldehydes Using a Transient Directing Group. Org. Lett. 2019, 21, 3959–3962. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-Y.; Sorensen, E.J. Pd-Catalyzed, Ortho C–H Methylation and Fluorination of Benzaldehydes Using Orthanilic Acids as Transient Directing Groups. J. Am. Chem. Soc. 2018, 140, 2789–2792. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.H.; Park, H.; Hu, J.H.; Hu, Y.; Zhang, Q.L.; Wang, B.L.; Sun, B.; Yeung, K.S.; Zhang, F.L.; Yu, J.Q. Diverse Ortho-C(Sp2)-H Functionalization of Benzaldehydes Using Transient Directing Groups. J. Am. Chem. Soc. 2017, 139, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, Y.; Wang, Y.; Li, Y.; Xu, X.; Jin, Z. Pd-Catalyzed Direct Ortho -C–H Arylation of Aromatic Ketones Enabled by a Transient Directing Group. Org. Lett. 2017, 19, 1562–1565. [Google Scholar] [CrossRef] [PubMed]
- Prim, D.; Marque, S.; Gaucher, A.; Campagne, J.-M. Transition-Metal-Catalyzed α-Arylation of Enolates. In Organic Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011. [Google Scholar] [CrossRef]
- Nagamoto, M.; Fukuda, J.; Hatano, M.; Yorimitsu, H.; Nishimura, T. Hydroxoiridium-Catalyzed Hydroarylation of Alkynes and Bicycloalkenes with N -Sulfonylbenzamides. Org. Lett. 2017, 19, 5952–5955. [Google Scholar] [CrossRef] [PubMed]
- Péron, F.; Fossey, C.; Sopkova-de Oliveira Santos, J.; Cailly, T.; Fabis, F. Room-Temperature Ortho -Alkoxylation and -Halogenation of N-Tosylbenzamides by Using Palladium(II)-Catalyzed C―H Activation. Chem. Eur. J. 2014, 20, 7507–7513. [Google Scholar] [CrossRef]
- Yang, L.; Li, S.; Cai, L.; Ding, Y.; Fu, L.; Cai, Z.; Ji, H.; Li, G. Palladium-Catalyzed C–H Trifluoroethoxylation of N -Sulfonylbenzamides. Org. Lett. 2017, 19, 2746–2749. [Google Scholar] [CrossRef]
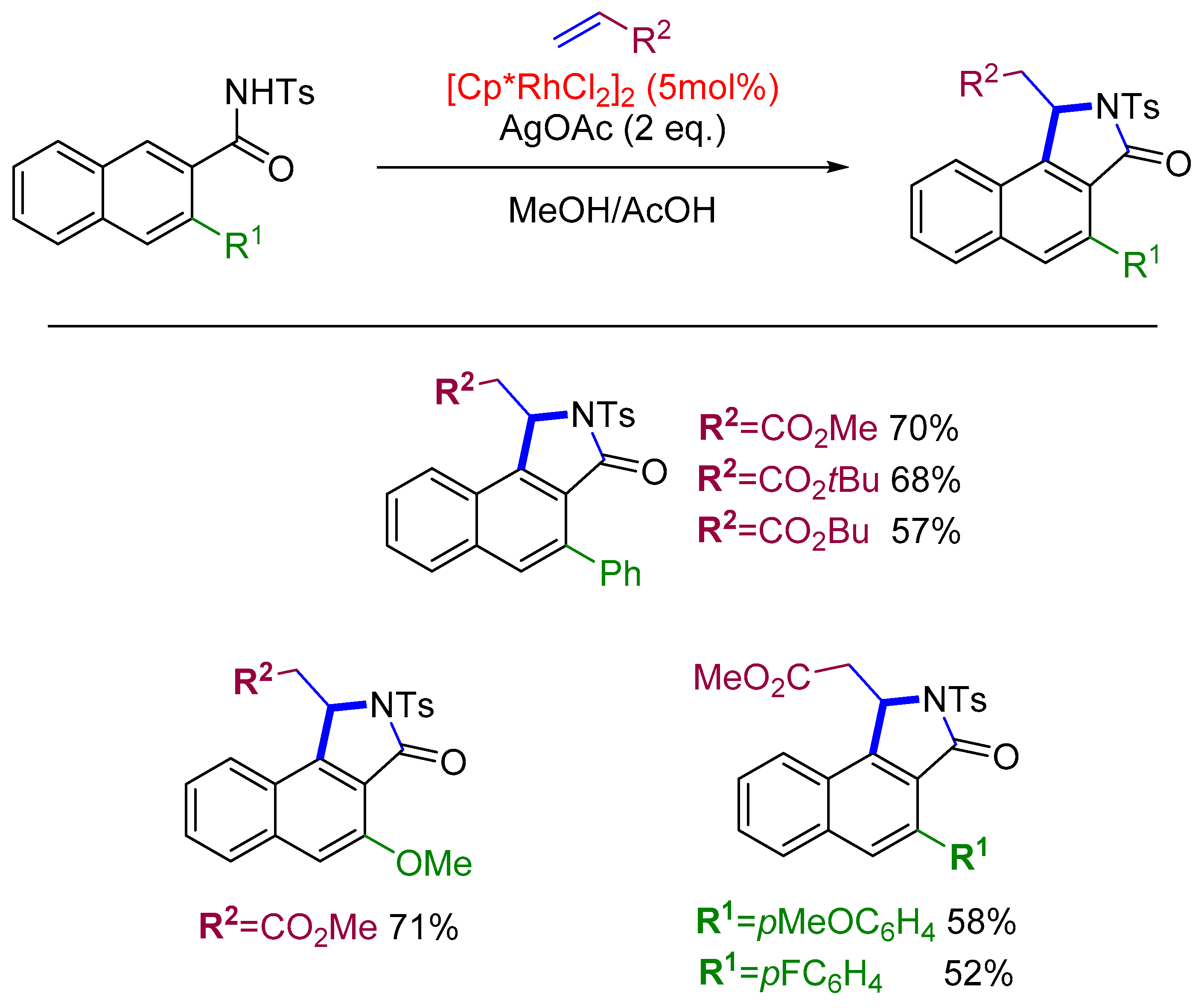
- Large, B.; Gigant, N.; Joseph, D.; Prim, D. Rhodium-Catalyzed C–H Activation of Naphthamides for the Synthesis of Substituted 3H-Benzo[e]Isoindolin-3-Ones. Eur. J. Org. Chem. 2019, 2019, 6407–6412. [Google Scholar] [CrossRef]
- Dubost, E.; Babin, V.; Benoist, F.; Hébert, A.; Barbey, P.; Chollet, C.; Bouillon, J.-P.; Manrique, A.; Pieters, G.; Fabis, F.; et al. Palladium-Mediated Site-Selective C–H Radio-Iodination. Org. Lett. 2018, 20, 6302–6305. [Google Scholar] [CrossRef]
- Péron, F.; Fossey, C.; Cailly, T.; Fabis, F. N -Tosylcarboxamide as a Transformable Directing Group for Pd-Catalyzed C–H Ortho -Arylation. Org. Lett. 2012, 14, 1827–1829. [Google Scholar] [CrossRef]
- Large, B.; Gigant, N.; Joseph, D.; Clavier, G.; Prim, D. Site-Selective Arylation of Naphthalenes: A Key Entry towards Extended Fluorenones and Phenanthridinones. Eur. J. Org. Chem. 2019, 2019, 1835–1841. [Google Scholar] [CrossRef]
- Xia, X.-F.; Wang, Y.-Q.; Zhang, L.-L.; Song, X.-R.; Liu, X.-Y.; Liang, Y.-M. Palladium-Catalyzed C–H Activation and Intermolecular Annulation with Allenes. Chem. Eur. J. 2014, 20, 5087–5091. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Wu, H.; Xia, X.; Chen, W.; Wang, Z.; Yang, J. Asymmetric Palladium-Catalyzed C–H Functionalization Cascade for Synthesis of Chiral 3,4-Dihydroisoquinolones. J. Org. Chem. 2019, 84, 12835–12847. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Falck, J.R. N-Acylsulfonamide Assisted Tandem C−H Olefination/Annulation: Synthesis of Isoindolinones. Org. Lett. 2011, 13, 1214–1217. [Google Scholar] [CrossRef]
- Bechtoldt, A.; Tirler, C.; Raghuvanshi, K.; Warratz, S.; Kornhaaß, C.; Ackermann, L. Ruthenium Oxidase Catalysis for Site-Selective C–H Alkenylations with Ambient O 2 as the Sole Oxidant. Angew. Chem. Int. Ed. 2016, 55, 264–267. [Google Scholar] [CrossRef]
- Miura, H.; Kimura, Y.; Terajima, S.; Shishido, T. Ruthenium-Catalyzed Synthesis of Isoindolinones via Amide-Directed Addition of Aromatic C–H Bonds to Aldimines. Eur. J. Org. Chem. 2019, 2019, 2807–2811. [Google Scholar] [CrossRef]
- Youn, S.W.; Ko, T.Y.; Kim, Y.H.; Kim, Y.A. Pd(II)/Cu(II)-Catalyzed Regio- and Stereoselective Synthesis of (E)-3-Arylmethyleneisoindolin-1-Ones Using Air as the Terminal Oxidant. Org. Lett. 2018, 20, 7869–7874. [Google Scholar] [CrossRef]
- Ji, W.-W.; Lin, E.; Li, Q.; Wang, H. Heteroannulation Enabled by a Bimetallic Rh(III)/Ag(I) Relay Catalysis: Application in the Total Synthesis of Aristolactam BII. Chem. Commun. 2017, 53, 5665–5668. [Google Scholar] [CrossRef]
- Liu, H.; Song, S.; Wang, C.-Q.; Feng, C.; Loh, T.-P. Redox-Neutral Rhodium-Catalyzed [4+1] Annulation through Formal Dehydrogenative Vinylidene Insertion. ChemSusChem 2017, 10, 58–61. [Google Scholar] [CrossRef]
- Zhu, C.; Falck, J.R. Rhodium Catalyzed Synthesis of Isoindolinones via C–H Activation of N-Benzoylsulfonamides. Tetrahedron 2012, 68, 9192–9199. [Google Scholar] [CrossRef]
- Zhu, C.; Falck, J.R. Rhodium Catalyzed C–H Olefination of N-Benzoylsulfonamides with Internal Alkenes. Chem. Commun. 2012, 48, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Miura, H.; Terajima, S.; Tsutsui, K.; Shishido, T. Ruthenium-catalyzed Addition of Aromatic Amides to Internal Alkynes and Subsequent Intramolecular Cyclization for the Atom-Economical Synthesis of Isoindolinones. J. Org. Chem. 2017, 82, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Xie, W.; Falck, J.R. Rhodium-Catalyzed Annulation of N-Benzoylsulfonamide with Isocyanide through C―H Activation. Chem. Eur. J. 2011, 17, 12591–12595. [Google Scholar] [CrossRef] [PubMed]














© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Large, B.; Terrasson, V.; Prim, D. N-Tosylcarboxamide in C–H Functionalization: More than a Simple Directing Group. Processes 2020, 8, 981. https://doi.org/10.3390/pr8080981
Large B, Terrasson V, Prim D. N-Tosylcarboxamide in C–H Functionalization: More than a Simple Directing Group. Processes. 2020; 8(8):981. https://doi.org/10.3390/pr8080981
Chicago/Turabian StyleLarge, Benjamin, Vincent Terrasson, and Damien Prim. 2020. "N-Tosylcarboxamide in C–H Functionalization: More than a Simple Directing Group" Processes 8, no. 8: 981. https://doi.org/10.3390/pr8080981
APA StyleLarge, B., Terrasson, V., & Prim, D. (2020). N-Tosylcarboxamide in C–H Functionalization: More than a Simple Directing Group. Processes, 8(8), 981. https://doi.org/10.3390/pr8080981