Abstract
Oxidative stress is considered one of the factors that cause dysfunction and damage of neurons, causing diseases such as amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD), and Parkinson’s disease (PD).Recently, natural antioxidant sources have emerged as one of the main research areas for the discovery of potential neuroprotectants that can be used to treat neurological diseases. In this research, we assessed the neuroprotective effect of a 70% ethanol Salvia miltiorrhiza Radix (SMR) extract and five of its constituent compounds (tanshinone IIA, caffeic acid, salvianolic acid B, rosmarinic acid, and salvianic acid A) in HT-22 hippocampal cells. The experimental data showed that most samples were effective in attenuating the cytotoxicity caused by glutamate in HT-22 cells, except for rosmarinic acid and salvianolic acid B. Of the compounds tested, tanshinone IIA (TS-IIA) exerted the strongest effect in protecting HT-22 cells against glutamate neurotoxin. Treatment with 400 nM TS-IIA restored HT-22 cell viability almost completely. TS-IIA prevented glutamate-induced oxytosis by abating the accumulation of calcium influx, reactive oxygen species, and phosphorylation of mitogen-activated protein kinases. Moreover, TS-IIA inhibited glutamate-induced cytotoxicity by reducing the activation and phosphorylation of p53, as well as by stimulating Akt expression. This research suggested that TS-IIA is a potential neuroprotective component of SMR, with the ability to protect against neuronal cell death induced by excessive amounts of glutamate.
1. Introduction
At present, the world population is aging more rapidly than ever before. According to World Population Prospects 2019, one person in 11 is over 65 years old in 2019, and this rate was predicted to increase one in six people by 2050 worldwide []. The increase of population aging contributes to making the neurologic diseases associated with age, which consist of amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD), and Parkinson’s disease (PD), become more common, and this increases the psychological, physical, economic, and social burdens to not only patients with the disease but also their families and society as a whole [,]. Even though the mechanisms of these nervous system disorders have not been completely revealed yet, oxidative stress and excitotoxicity are considered factors promoting the dysfunction and damage of the nerve cells [,].
Glutamate is one of the primary neurotransmitters in the cerebral cortex and various hippocampal tracts; consequently, it has a key role in learning and memory []. However, excess glutamate is considered a neurotoxin associated with various neurological diseases, such as AD and PD [,,]. This neurotoxicity consists of two distinct types called excitotoxicity and oxidative stress. The excitotoxicity occurs when excessive glutamate encounters ionotropic glutamate receptors (iGluRs) such as N-methyl-D-aspartate receptors (NMDARs), leading it to allow a high level of Ca2+ to enter into the cell, which triggers a series of downstream changes associated with cell death process including endoplasmic reticulum (ER) stress, production of reactive oxygen species (ROS) and nitric oxide, activation of calpain, caspase-mediated programmed cell death, and apoptosis inducing factor (AIF). Besides excitotoxicity, a high extracellular concentration glutamate promotes oxidative stress in neurons via the disfunction of cystine glutamate antiporters that induce a decrease of intracellular glutathione (GSH, an important antioxidant that takes the role in neutralizing intracellular ROS). The consequence of excessive ROS accumulation is the production of protein-mediated apoptosis, such as BID (BH3 interacting-domain death agonist) and AIF [,,].
The hippocampal neuronal HT-22 cells do not express iGluRs; for this reason, the glutamate-induced excitotoxicity in this cell line is negligible [,]. Because of this characteristic of the HT-22 cell line, this line was chosen to study nerve cell death mechanism related to oxidative stress [,,,,]. In HT-22 cells, the glutamate promotes oxytosis by increasing intracellular Ca2+ influx, ROS production, ER stress, and mitochondrial dysfunction. This process is mediated through inhibiting PI3K/Akt signaling and triggering mitogen-activated protein kinases (MAPKs) [].
Natural compounds are considered as a potential antioxidant source to develop new neurodegenerative drugs with strong clinical effectiveness and fewer side effects for long term treatment []. Salvia miltiorrhiza Radix (SMR), also known as Danshen, comprises the roots and rhizomes of Salvia miltiorrhiza Bunge and is one of the most famous herbs applied to treat cerebrovascular and cardiovascular diseases in Chinese traditional medicine []. Furthermore, the protection effects of the components of SMR against neurodegenerative diseases have been detected through various pathways, including the activation of Nrf2 and the inhibition of apoptosis and inflammation [,]. However, there has been little discussion on the protection effect of SMR and its constituents from glutamate-induced HT-22 cell death. In our previous study, we analyzed and examined the anticancer effects of tanshinone IIA (TS-IIA), caffeic acid (CA), salvianolic acid B (SA-B), rosmarinic acid (RA), and salvianic acid A (SA-A) in a 70% ethanol SMR extract in a human colorectal carcinoma cell HCT-116 []. In this study, we determine the ability of these compounds to protect against HT-22 hippocampal cell death by inhibiting glutamate neurotoxicity.
2. Materials and Methods
2.1. Cell Culture and Treatment
The HT-22 Mouse Hippocampal Neuronal Cell Line was obtained from Dr. David Schubert (Salk Institute, La Jolla, CA.). HT-22 cells were propagated in Dulbecco’s Modified Eagle Medium (DMEM; Corning, Manassas, VA, USA) containing 1% streptomycin-penicillin (Gibco, Grand Island, NY, USA) and 10% fetal bovine serum (FBS; Atlas, Fort Collins, CO, USA) at 37 °C in a humid atmosphere containing 5% CO2. Stock solutions (100 mM) of SA-A, SA-B, CA, RA (Sigma-Aldrich, St. Louis, MO, USA), TS-IIA (10 mM; R&D systems, Minneapolis, MN, USA), and a 70% ethanol SMR extract (100 mg/mL) were prepared in dimethyl sulfoxide (DMSO; Biosesang, Gyeonggi, Korea). The final percentage of DMSO was maintained less than 1‰, in which the cytotoxicity of vehicle (DMSO) could not be detected compared to the nontreated cells. For experiments, the density of 3 × 104 cells/cm2 was chosen to seed into multiwell plates. Subsequently, the HT-22 cells were allowed to adhere for 24 h and then treated with specific concentrations of compounds in either the presence or absence of 5 mM glutamate (Sigma-Aldrich, St. Louis, MO, USA).
2.2. Evaluation of Cell Viability
The HT-22 cells were exposed to the indicated compounds in the presence or absence of 5 mM glutamate in a 96-well plate. After 24 h, the medium in the plate was replaced by a 10% EZ-Cytox (Dogen, Seoul, Korea) solution, and the plate was incubated at 37 °C in a humid atmosphere with 5% CO2 for 30 min. The OD450 of each well was measured using a microplate reader (SPARK 10M; Tecan, Männedorf, Switzerland), and viability of cell was determined as a percentage relative to the viability of the nontreated group.
2.3. Analysis of ROS Accumulation
To assess the increase in ROS, we applied the method of Soo Yeon Choi et al. (2018) with a few adjustments []. After treatment with the specified concentrations of TS-IIA in the presence or absence of 5 mM glutamate for 10 h, the cells were exposed to 10 µM 2′, 7′-dichlorofluorescein diacetate (DCFDA; Sigma-Aldrich, St. Louis, MO, USA) for 30 min and then washed three times with phosphate buffered saline (PBS) to remove the unbound dye. The fluorescent intensity (excitation = 485 nm; emission = 530 nm) was analyzed using a microplate reader (SPARK 10 M; Tecan, Männedorf, Switzerland). In addition, we also observed the fluorescence intensity of each group using the fluorescence microscope IX51 (Olympus, Tokyo, Japan) connected to a charge-coupled device (CCD) camera.
2.4. Intracellular Ca2+ Influx Assay
The determination of intracellular Ca2+ concentration was examined using the fluorescent dye Fluo-4 AM (Abcam, Cambridge, UK). HT-22 cells were treated with the specified concentrations of TS-IIA in the presence or absence of 5 mM glutamate for 10 h. After this step, the cells were stained with 2.5 µM Fluo-4 AM for 30 min. Subsequently, the cells were washed with PBS in triplicate, and the fluorescence intensity (excitation = 485 nm; emission = 530 nm) was analyzed using the microplate reader (SPARK 10 M; Tecan, Männedorf, Switzerland). Furthermore, the fluorescence intensity of each group was captured using an IX51 fluorescence microscopy (Olympus, Tokyo, Japan) connected to a CCD camera.
2.5. Hoechst 33,258 Staining
HT-22 cells were imaged to determine the presence of condensed pyknotic nuclei, which is indicative of apoptosis, after 12 h of treatment with the specified concentrations of TS-IIA in the presence or absence of 5 mM glutamate. The cells were exposed to 10 µM Hoechst 33,258 (Abcam, Cambridge, UK) for 10 min in the dark and washed with PBS. Images of the cell nuclei were assessed by an IX51 fluorescence microscope (Olympus, Tokyo, Japan) connected to a CCD camera. The ImageJ software (Version 1.51 J, National Institutes of Health, Bethesda, MD, USA) was used to calculate the percentage of nuclear abnormalities from the fluorescence images.
2.6. Propidium Iodide and Annexin V Staining
The assessment of apoptosis in HT-22 cells was performed using the Tali® Apoptosis Kit (Thermo Fisher Scientific, Eugene, OR, USA). After exposure to the indicated doses of TS-IIA in the presence or absence of 5 mM glutamate for 12 h, the cells were harvested, washed with PBS, dyed with annexin V, and protected from light. After 20 min, the cells were continuously washed with PBS, stained with propidium iodide, and left to stand in the dark for 5 min. The images were captured using a Tali® Image-Based Cytometer (Invitrogen, Carlsbad, CA, USA). The number of apoptotic cells was computed using TaliPCApp software, version 1.0.
2.7. Immunoblotting Analysis
The immunoblotting analysis was performed in accordance with the method of Jang et al. (2018), with some modifications []. After treatment for 12 h, HT-22 cells were washed with PBS and lysed in a 1× radioimmunoprecipitation assay buffer (1× RIPA buffer; Tech & Innovation, Gangwon, Korea), supplemented with a phosphatase inhibitor cocktail 2 (Sigma-Aldrich, St. Louis MO, USA) and a protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN, USA). The whole cell lysate was centrifuged at 16,000× g for 20 min at 4 °C to collect the supernatant. The protein samples were quantified using a BCA Protein Assay Kit (Abcam, Cambridge, UK) and mixed with a 4× NuPAGE LDS Sample Buffer (Thermo Fisher Scientific, Rockford, IL, USA) containing 10% 2-mercaptoethanol (Bio-Rad Laboratories; Inc., Hercules, CA, USA) and 1× RIPA. The mixtures were heated to 95 °C for 10 min and left to cool at room temperature prior to gel electrophoresis, and then separated on a polyacrylamide gel with equal amounts of proteins (10 µg/lane). The resolved proteins were transferred to a polyvinylidene difluoride membrane (Merck Millipore, Darmstadt, Germany) activated by methanol. Nonspecific binding to the membranes was blocked by incubation of the membrane with 5% nonfat milk in tris-buffered saline supplemented with 1‰ Alkest TW 20 (TBS-T) for 1 h. After three washes with TBS-T, the membranes were probed with the first antibodies of interest (Table S1—Supplementary Materials) in an immunoreaction enhancer solution 1 (Can get signal, Toyobo, Osaka, Japan) overnight at 4 ℃. The unbound primary antibodies were removed by washing the membranes with TBS-T, and the membranes were then incubated with suitable secondary antibodies (Table S1—Supplementary Materials) in an immunoreaction enhancer solution 2 (Can get signal, Toyobo, Osaka, Japan) for 1 h. Subsequently, the membranes were exposed to a mixture of solution A and solution B in a 1:1 ratio (EZ-Western Lumi Femto Kit, Dogen, Seoul, Korea), and the immunoreactive bands were developed using the Azure 300 Chemiluminescent Western Blot Imaging System (Dublin, CA, USA). The signal intensity was assessed and normalized to that of the control using AzureSpot software (Dublin, CA, USA).
2.8. Statistical Methods
Experimental results were presented as the mean ± standard error of mean (S.E.M). Statistical differences were assessed using one-way analysis of variance (ANOVA) with Tukey’s honest significance test. The statistical significance was set at p-values of less than 0.05, 0.01, and 0.001.
3. Results and Discussion
3.1. Effect of SMR and Its Constituent Compounds on Glutamate Neurotoxicity in HT-22 Cells
In our previous study, the HPLC analysis of the SMR (70% ethanol extract) identified five markers, as shown in Figure 1, at the following concentrations: 3.72 mg/g RA, 0.12 mg/g CA, 1.27 mg/g SA-A, 64.36 mg/g SA-B, and 4.96 mg/g TS-IIA. These compounds are typical antioxidant marker components found in SMR [,], and several previous studies have mentioned their neuroprotective benefits. For example, SA, RA, and TS-IIA exert preventive action against AD by inhibiting the generation and aggregation of Aβ in PC-12 cells [,]. In addition, Spencer et al. showed the potential of CA for the inhibition of Aβ25–35-induced memory deficits and cognition impairment in a mouse model of AD [].
Figure 1.
Five bioactive marker compounds found in Salviae miltiorrhizae Radix.
In present study, we assessed the effect of SMR and its constituents on the neutralization of glutamate toxicity in HT-22 hippocampal cells. N-Acetylcysteine (NAC) was used as the reference drug []. As shown in Figure 2, treatment with 5 mM glutamate reduced the cell viability to 30.60% ± 0.86% (N = 3, p = 0.001) compared with nontreated cells. Of the compounds tested, TS-IIA showed by far the strongest neuroprotection effect against glutamate-induced cytotoxicity. Specifically, exposure to 400 nM TS-IIA restored cell viability to 96.25% ± 4.03% (N = 3, p = 0.001) of the control, whereas NAC concentration of 50 µM restored the cell viability to 102.65% ± 0.42% (N = 3, p = 0.001). In addition to TS-IIA, SMR also strongly inhibited glutamate neurotoxicity, increasing HT-22 cell viability to 96.35% ± 3.87% (N = 3, p = 0.001) at 3 µM. At 100 µM, SA-A and CA increased cell viability to 70.94% ± 1.63% (N = 3, p = 0.001) and 57.49% ± 4.81% (N = 3, p = 0.001), respectively. The neuroprotective effects of SA-B and RA were not significant. These results showed the outstanding potential of TS-IIA in protecting HT-22 cells against glutamate toxicity; consequently, we performed mechanistic assessments to determine how this compound exerted its neuroprotective effects.
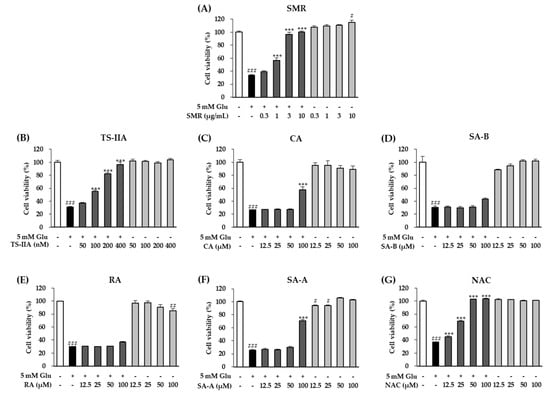
Figure 2.
Comparison of protective effects of (A) 70% ethanol extract of SMR, (B) TS-IIA, (C) CA, (D) SA-B, (E) RA, (F) SA-A, and (G) NAC (reference drug). The bar graphs depict the viability of HT-22 cells after treatment with the specified concentrations of compounds in the presence or absence of 5 mM glutamate for 24 h (N = 3). Notes: # p < 0.05, ## p < 0.01, and ### p < 0.001 compared with the untreated group, *** p < 0.001 compared with the glutamate-treated group. (Glu: glutamate, SMR: Salviae miltiorrhizae Radix, TS-IIA: tanshinone IIA, CA: caffeic acid, SA-B: salvianolic acid B, RA: rosmarinic acid, SA-A: salvianic acid A, NAC: N-Acetylcysteine).
3.2. Effect of TS-IIA on Inhibition of ROS Production and Calcium Influx
Excessive ROS levels is one of the main factors that promote aging and neurodegenerative disorders. Increased accumulation of ROS stimulates various forms of intracellular damage, including DNA damage, protein denaturation, lipid peroxidation, ER stress, calcium homeostasis, and membrane destabilization, inducing the loss of synaptic activity and neurotransmission in nerve cells or apoptosis, which contributes to cognitive dysfunction []. Accompanying ROS is an abnormal increase in intracellular calcium influx that also contributes to nerve cell death in several neurodegenerative diseases []. Massive intracellular calcium influx can trigger cell death process through the activation of calpain and the translocation of AIF [,].
As presented above, due to the lack of iGluRs, glutamate mainly induces oxidative toxicity rather than excitotoxicity, causing a fast increase of intracellular Ca2+ and neuron death via the effect of excessive extracellular glutamate concentration on iGluRs in HT-22 cells. Murphy et al. (1989) reported that the high extracellular glutamate concentration inhibited cystine glutamate antiporters xc−, inducing a decrease of the important antioxidant GSH []. When the concentration of GSH decreased to below about 20% (6–8 h after glutamate exposure), the intracellular ROS concentration increased exponentially []. Furthermore, the depletion of GSH also leads to glutathione peroxidase 4 (GPx4) inhibition, which needs a stable GSH source for normal activity []. Under reduced GSH and inhibited GPx4, 12/15-lipoxygenase (12/15-LOX) is triggered [,], known as a mediated factor for increase of intracellular Ca2+ concentration [,]. In detail, the activation of 12/15-ROX induces 12 and 15-HETE production, which promotes translocation of Ca2+ from outside to inside of the cell via the Ca2+ channel related to store-operated Ca2+ entry (SOCE). Moreover, 12/15-LOX also combines with the membrane of ER and mitochondria, inducing additional accumulation of ROS and possibly Ca2+ release []. Therefore, rebalancing ROS and calcium levels may be a significant factor in the prevention of damage to HT-22 cells.
Thus, the cells were exposed to DCFDA and Fluo-4 AM to observe the effect of TS-IIA on the inhibition of ROS generation and calcium influx, respectively. We found that the cells treated with TS-IIA tended to show a marked decrease in the fluorescence intensity of DCFDA and Fluo-4 AM, compared with the glutamate-only treated group (Figure 3A). The quantitative data obtained from the microplate reader illustrated that exposure to 5 mM glutamate increased the fluorescent intensity of DCFDA and Fluo-4 AM by 5.21 ± 0.19 fold (N = 3, p = 0.001) and 2.80 ± 0.11 (N = 3, p = 0.001) fold, respectively, whereas treatment with TS-IIA reduced it in a dose-dependent manner (Figure 3B,C). These data demonstrated the ability of TS-IIA to reverse the process of glutamate-induced HT-22 hippocampal cell death by attenuating ROS production and calcium influx.
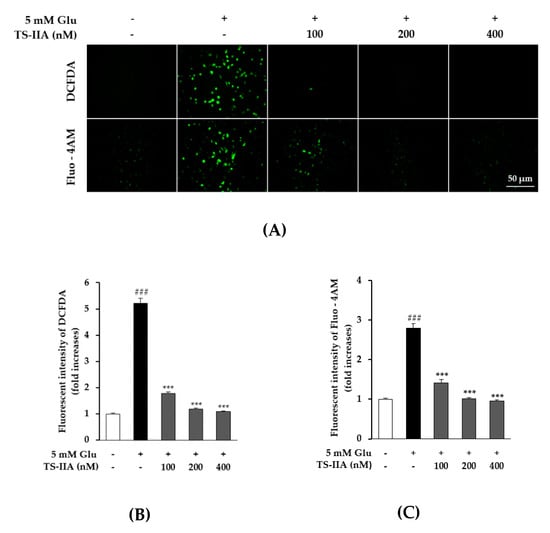
Figure 3.
Inhibitory effect of tanshinone IIA (TS-IIA) on intracellular reactive oxygen species (ROS) production and calcium accumulation following glutamate treatment. (A) A total of 10,000 cells were seeded in each well of a 96-well plate and incubated for 24 h. Subsequently, the cells were exposed to 100, 200, and 400 nM TS-IIA in the presence or absence of 5 mM glutamate for 10 h and stained with DCFDA or Fluo-4 AM. Fluorescent images were captured by fluorescence microscopy (20×). (B) TS-IIA attenuated production of ROS induced by glutamate (N = 3). (C) TS-IIA decreased calcium concentration caused by glutamate (N = 3). Notes: ### p < 0.001 compared with the untreated group, *** p < 0.001 compared with the glutamate-treated group. (TS- IIA: tanshinone IIA, Glu: glutamate).
3.3. Effect of TS-IIA on Glutamate-Induced Phosphorylation of MAPKs
MAPKs consist of mitogen-activated protein kinases (p38), extracellular signal-regulated kinases (ERK), and c-Jun N-terminal kinases (JNK) that contribute to various cellular functions (apoptosis, differentiation, mitosis, proliferation, cell survival, and gene expression) and responses (proinflammatory cytokines, heat shock, mitogens, and osmotic stress) []. The phosphorylation of MAPKs associated with oxidative stress promotes several neurological disorders such as ALS, PD, and AD []. Furthermore, the previous study showed that MAPKs also were phosphorylated, following a glutamate-induced oxytosis in HT-22 cells []. In addition, Fukui et al. (2009, 2010) indicated that treatment with SB202190 (p38 inhibitor), SP600125 (JNK inhibitor), and U0126 (ERK inhibitor) inhibited cytotoxicity in a dose-dependent manner caused by glutamate in HT-22 cells [,]. Therefore, the attenuation of MAPKs phosphorylation could be considered a typical modulator of neuroprotective effect related in glutamate-induced oxidative stress. An immunoblotting analysis was used to study the effect of TS-IIA on the phosphorylation of MAPKs consisting of p38, ERK, and JNK. After exposure to the indicated concentrations of TS-IIA and 5 mM glutamate for 12 h, HT-22 cells were harvested, and western blotting was performed to determine the relative levels of proteins. The obtained data indicated that the phosphorylation of p38, JNK, and ERK was increased significantly in cells treated with glutamate but reduced substantially in cells treated with TS-IIA (Figure 4). These figures confirmed that the inhibition of MAPKs phosphorylation was the basis of the protection effect of TS-IIA against oxytosis caused by excess glutamate.
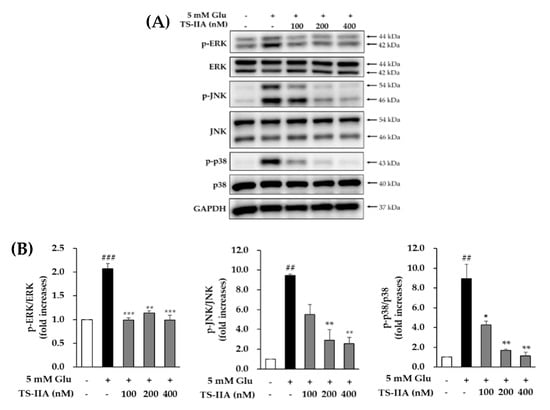
Figure 4.
Effect of tanshinone IIA (TS-IIA) on glutamate-induced phosphorylation of MAPKs. (A) HT-22 cells were exposed to 100, 200, or 400 nM TS-IIA in the presence or absence of 5 mM glutamate for 12 h, and analyzed by immunoblotting to determine the immunoreactive bands for p-JNK, JNK, p-ERK, ERK, p-p38, p38, and GAPDH (Glyceraldehyde-3-Phosphate Dehydrogenase). (B) The bar chart describes the fold-increase of the phosphorylation of MAPKs compared with the control (N = 2). Notes: ## p < 0.01 and ### p < 0.001 compared with the untreated group, * p < 0.05, ** p < 0.01, and *** p < 0.001 compared with the glutamate-treated group. (TS- IIA: tanshinone IIA, Glu: glutamate).
3.4. Antiapoptotic Effect of TS-IIA in HT-22 Cells
Previous reports indicated that synaptic loss and neuronal loss in the hippocampus and cerebral cortex are the main causes of memory impairment in AD [,,]. Apoptosis is considered as one cause of nerve cell death []. As shown above, oxidative stress causes several forms of intracellular damage, including DNA breakage.
The p53 tumor protein, also known as p53, takes an important part in the response of cells to DNA damage, leading to the mediation of apoptosis through modulation of the transcription of genes involved in cell death. In 2000, Morison and colleagues described the role of p53-induced nerve cell death through modulation of the transcription of proapoptotic genes Bax [].
In contrast to p53, Akt (protein kinase B) stimulates cell survival by inhibiting apoptosis through the phosphorylation and inactivation of several targets, including forkhead transcription factors [], c-Raf [], and caspase-9 []. The previous studies indicated that activation of PI3K/Akt signaling promoted survival of nerve cells through regulating nuclear proteins, impinging on the cytoplasmic cell death machinery, and mediating the expression of genes involved in cell death and survival []. In 2010, Fukui and colleagues showed that Resveratrol protected HT-22 cells against oxidative glutamate toxicity by activating PI3K/Akt/GSK-3β signaling the pathways-induced translocation of β-catenin, which leads to the transcription of superoxide effutase 2 (SOD2) gene. SOD2 protein contributes to neutralizing ROS caused by oxidative glutamate toxicity []. In addition, Yamaguchi et al. (2001) reported that activating Akt protected hippocampal nerve cells from programmed cell death through the inhibition of p53 transcriptional activity []. Thus, Akt activation may take an important role in the protective effects of TS-IIA in HT-22 cells.
Moreover, previous studies showed that glutamate induced apoptosis through activating p53 [] and inhibiting the PI3K/Akt signaling pathway []. Therefore, we studied the antiapoptotic effect of TS-IIA by examining the relative levels of phospho-Akt, p53, and phospho-p53. As shown in Figure 5, the signal for phospho-Akt was decreased in the glutamate-treated group and increased in the presence of TS-IIA. In contrast to the trend in the phosphorylation of Akt, glutamate increased the expression of phospho-p53 and p53, which was decreased in the TS-IIA-treated groups. These immunoblotting data proved that TS-IIA protected HT-22 hippocampal cells against glutamate-induced programed cell death through the activation of Akt, which led to the inhibition of p53 activation and phosphorylation.
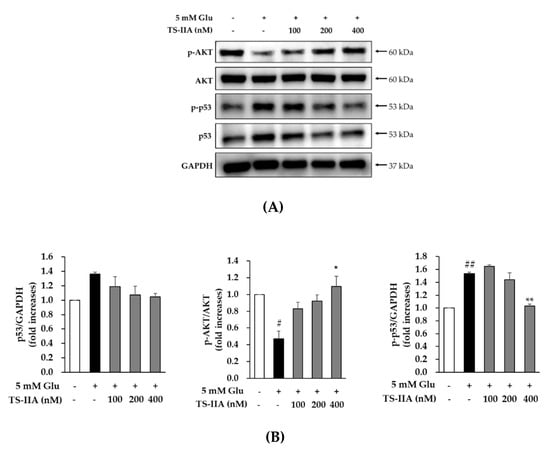
Figure 5.
The protective effect of tanshinone IIA (TS-IIA) against glutamate-induced apoptosis in HT-22 cells occurred through the activation of Akt, which led to the inhibition of p53 activation and phosphorylation. (A) HT-22 cells were exposed to 100, 200, or 400 nM TS-IIA in the presence or absence of 5 mM glutamate for 12 h, and analyzed immunoblotting to determine immunoreactive bands for p-Akt, Akt, p-p53, p53, and GAPDH. (B) The bar chart described the fold-increase in the phosphorylation of Akt and p53, together with p53 in the control (N = 2). Notes: # p < 0.05 and ## p < 0.01 compared with the untreated group, * p < 0.05 and ** p < 0.01 compared with the glutamate-treated group (TS-IIA: tanshinone IIA, Glu: glutamate).
The involvement of apoptosis was further confirmed using Hoechest staining to identify chromatin condensation, which is one of the indicators of apoptotic cells [,], and the Tali assay for a quantification of the number of cells undergoing apoptosis by counting the number of annexin V-dyed cells. As described in Figure 6A, there was a gradual reduction in chromatin condensation in cells treated with TS-IIA compared with those treated with glutamate only. The quantitative results obtained from the fluorescence images showed that treatment with 5 mM glutamate enhanced the percentage of nuclear abnormalities to 38.96% ± 1.36% (N = 2, p = 0.001) while exposure to 400 nM TS-IIA decreased the percentage of nuclear abnormalities to 1.13% ± 0.11% (N = 2, p = 0.001) (Figure 6C). Similarly, TS-IIA substantially decreased the percentage of annexin V-stained cells compared with those treated with glutamate in the absence of TS-IIA (Figure 6B). The quantitative data depicted that exposure to 5 mM glutamate increased the percentage of apoptotic cells to 48.06% ± 0.41% (N = 2, p = 0.001), whereas treatment with 400 nM TS-IIA reduced the percentage of apoptotic cells to 12.33% ± 2.73% (N = 2, p = 0.001) (Figure 6D). These figures confirmed the potential of TS-IIA to prevent HT-22 cells from glutamate-induced apoptosis.
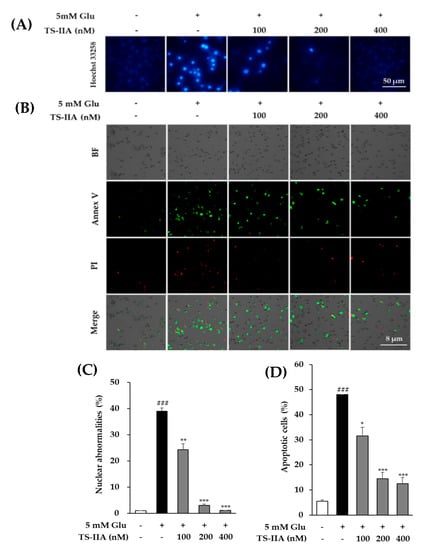
Figure 6.
Protective effect of tanshinone IIA (TS-IIA) against glutamate-induced apoptosis in HT-22 cells. (A) After treatment for 12 h with 100, 200, or 400 nM TS-IIA in the presence or absence of 5 mM glutamate, the chromatin of nucleus was stained with Hoechst 33,258 and observed by a fluorescent microscope (40×). (B) HT-22 cells were exposed to 100, 200, or 400 nM TS-IIA in the presence or absence of 5 mM glutamate for 12 h and dyed with PI and annexin V to visualize the dead and apoptotic cells using the Tali® Image-Based Cytometer. (C) The percentage of nuclear abnormalities was calculated from the fluorescence images using ImageJ software (N = 2). (D) The column graph depicts the proportion of cells undergoing apoptosis (annexin V-positive cells) quantified from the obtained images using TaliPCApp software (N = 2). Notes: ### p < 0.001 compared with the untreated group, * p < 0.05, ** p < 0.01, and *** p < 0.001 compared with the glutamate-treated group. (TS-IIA: tanshinone IIA, Glu: glutamate).
4. Conclusions
The present study examined the benefit of SMR (70% ethanol extract) and five marker compounds (SA-A, SA-B, CA, TS-IIA, and RA) from SMR in protecting hippocampal HT-22 cells against glutamate neurotoxicity. The experimental results showed that TS-IIA possessed the strongest effect in neutralizing oxidative glutamate toxicity among the compounds tested. TS-IIA prevented glutamate-induced cytotoxicity in HT-22 cells by attenuating the massive influx of calcium, the accumulation of ROS, and the phosphorylation of the MAPK signaling pathway. Moreover, TS-IIA protected HT-22 cells against glutamate-induced apoptosis by activating Akt, which led to the inhibition of p53 activation and phosphorylation. Based on the data obtained in this research, we suggest that TS-IIA is the active compound in SMR that protects neronal cells from excess glutamate.
Supplementary Materials
The following are available online at https://www.mdpi.com/2227-9717/8/8/914/s1, Figure S1: Information of antibodies used for western blotting., Table S1: Information of antibodies used for western blotting.
Author Contributions
S.L. and K.S.K. performed conceptualization; H.M.P. performed formal analysis; H.M.P. and S.L. performed investigation and data curation; H.M.P. performed writing-original draft preparation; S.L. performed writing—review and editing; K.S.K. performed project administration and funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This work was conducted with the support of the National Research Foundation of Korea (NRF-2019R1F1A1059173).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nations, U. World Population Prospects 2019: Highlights. U.N. Dep. Econ. Soc. Aff. Popul. Div. 2019. [Google Scholar] [CrossRef]
- Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 19 September 2019).
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Doble, A. The role of excitotoxicity in neurodegenerative disease: Implications for therapy. Pharmacol. Ther. 1999, 81, 163–221. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed]
- William, J.; McEnteet, T.H.C. Glutamate: Its role in learning, memory, and the aging brain. Psychopharmacology 1993, 111, 391–401. [Google Scholar]
- Blandini, F.; Porter, R.H.; Greenamyre, J.T. Glutamate and Parkinson’s disease. Mol. Neurobiol. 1996, 12, 73–94. [Google Scholar] [CrossRef]
- Choi, J.H.; Jang, M.; Nah, S.-Y.; Oh, S.; Cho, I.-H. Multitarget effects of Korean red ginseng in animal model of Parkinson’s disease: Antiapoptosis, antioxidant, antiinflammation, and maintenance of blood–brain barrier integrity. J. Ginseng Res. 2018, 42, 379–388. [Google Scholar] [CrossRef]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef]
- He, M.; Liu, J.; Cheng, S.; Xing, Y.; Suo, W.Z. Differentiation renders susceptibility to excitotoxicity in HT22 neurons. Neural Regen. Res. 2013, 8, 1297. [Google Scholar]
- Kritis, A.A.; Stamoula, E.G.; Paniskaki, K.A.; Vavilis, T.D. Researching glutamate–induced cytotoxicity in different cell lines: A comparative/collective analysis/study. Front. Cell. Neurosci. 2015, 9, 91. [Google Scholar] [CrossRef]
- Sato, K.; Yamanaka, Y.; Asakura, Y.; Nedachi, T. Glutamate levels control HT22 murine hippocampal cell death by regulating biphasic patterns of Erk1/2 activation: Role of metabolic glutamate receptor 5. Biosci. Biotechnol. Biochem. 2016, 80, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Fukui, M.; Song, J.-H.; Choi, J.; Choi, H.J.; Zhu, B.T. Mechanism of glutamate-induced neurotoxicity in HT22 mouse hippocampal cells. Eur. J. Pharmacol. 2009, 617, 1–11. [Google Scholar] [CrossRef]
- Jeong, G.S.; An, R.B.; Oh, S.H.; Kang, D.G.; Lee, H.S.; Kim, Y.C. Cytoprotective activity of Belamcanda chinensis rhizome against glutamate-induced oxidative injury in HT22 cells. Nat. Prod. Sci. 2007, 13, 101–104. [Google Scholar]
- Kim, D.H.; Kim, D.W.; Jung, B.H.; Lee, J.H.; Lee, H.; Hwang, G.S.; Kang, K.S.; Lee, J.W. Ginsenoside Rb2 suppresses the glutamate-mediated oxidative stress and neuronal cell death in HT22 cells. J. Ginseng Res. 2019, 43, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Ko, W.; Chowdhury, A.; Li, B.; Kim, S.C.; Oh, H.; Kim, Y.C.; Woo, E.R.; Baek, N.I.; Lee, D.S. Brassicaphenanthrene A from Brassica rapa protects HT22 neuronal cells through the regulation of Nrf2-mediated heme oxygenase-1 expression. Mol. Med. Rep. 2020, 21, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Takashima, M.; Ichihara, K.; Hirata, Y. Neuroprotective effects of Brazilian green propolis on oxytosis/ferroptosis in mouse hippocampal HT22 cells. Food Chem. Toxicol. 2019, 132, 110669. [Google Scholar] [CrossRef] [PubMed]
- Ratheesh, G.; Tian, L.; Venugopal, J.R.; Ezhilarasu, H.; Sadiq, A.; Fan, T.-P.; Ramakrishna, S. Role of medicinal plants in neurodegenerative diseases. Biomanuf. Rev. 2017, 2, 2. [Google Scholar] [CrossRef]
- Zhou, L.; Zuo, Z.; Chow, M.S.S. Danshen: An overview of its chemistry, pharmacology, pharmacokinetics, and clinical use. J. Clin. Pharmacol. 2005, 45, 1345–1359. [Google Scholar] [CrossRef]
- Li, G.-H.; Li, Y.-R.; Jiao, P.; Zhao, Y.; Hu, H.-X.; Lou, H.-X.; Shen, T. Therapeutic potential of salviae miltiorrhizae radix et rhizoma against human diseases based on activation of Nrf2-mediated antioxidant defense system: Bioactive constituents and mechanism of action. Oxidative Med. Cell. Longev. 2018, 2018, 7309073. [Google Scholar] [CrossRef]
- Zhang, X.-Z.; Qian, S.-S.; Zhang, Y.-J.; Wang, R.-Q. Salvia miltiorrhiza: A source for anti-Alzheimer’s disease drugs. Pharm. Biol. 2016, 54, 18–24. [Google Scholar] [CrossRef]
- Kang, B.; Lee, S.; Seo, C.-S.; Kang, K.S.; Choi, Y.-K. Analysis and Identification of Active Compounds from Salviae miltiorrhizae Radix Toxic to HCT-116 Human Colon Cancer Cells. Appl. Sci. 2020, 10, 1304. [Google Scholar] [CrossRef]
- Choi, S.Y.; Kim, J.H.; Quilantang, N.G.; Lee, S.; Cho, E.J. Acer okamotoanum inhibit the hydrogen peroxide-induced oxidative stress in C6 glial cells. Nat. Prod. Sci. 2018, 24, 148–154. [Google Scholar] [CrossRef]
- Jang, M.G.; Ko, H.C.; Kim, S.J. Effect of Sasa quelpaertensis Nakai extracts and its constituent p-coumaric acid on the apoptosis of human cancer cell lines. Nat. Prod. Sci. 2018, 24, 293–297. [Google Scholar] [CrossRef]
- Ma, X.-H.; Ma, Y.; Tang, J.-F.; He, Y.-L.; Liu, Y.-C.; Ma, X.-J.; Shen, Y.; Cui, G.-H.; Lin, H.-X.; Rong, Q.-X.; et al. The biosynthetic pathways of tanshinones and phenolic acids in Salvia miltiorrhiza. Molecules 2015, 20, 16235–16254. [Google Scholar] [CrossRef] [PubMed]
- Seong, G.-U.; Chung, S.-K. Marker compound contents and antioxidant capacities of the taproot and lateral root of Danshen (Salvia miltiorrhiza Radix). J. Appl. Biol. Chem. 2020, 63, 23–28. [Google Scholar] [CrossRef]
- Dong, H.; Mao, S.; Wei, J.; Liu, B.; Zhang, Z.; Zhang, Q.; Yan, M. Tanshinone IIA protects PC12 cells from β-amyloid 25–35-induced apoptosis via PI3K/Akt signaling pathway. Mol. Biol. Rep. 2012, 39, 6495–6503. [Google Scholar] [CrossRef]
- Habtemariam, S. Molecular pharmacology of rosmarinic and salvianolic acids: Potential seeds for Alzheimer’s and vascular dementia drugs. Int. J. Mol. Sci. 2018, 19, 458. [Google Scholar] [CrossRef]
- Kim, J.H.; Wang, Q.; Choi, J.M.; Lee, S.; Cho, E.J. Protective role of caffeic acid in an Aβ25–35-induced Alzheimer’s disease model. Nutr. Res. Pract. 2015, 9, 480–488. [Google Scholar] [CrossRef]
- Tardiolo, G.; Bramanti, P.; Mazzon, E. Overview on the effects of N-acetylcysteine in neurodegenerative diseases. Molecules 2018, 23, 3305. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Wojda, U.; Salinska, E.; Kuznicki, J. Calcium ions in neuronal degeneration. Iubmb Life 2008, 60, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Maher, P.; van Leyen, K.; Dey, P.N.; Honrath, B.; Dolga, A.; Methner, A. The role of Ca2+ in cell death caused by oxidative glutamate toxicity and ferroptosis. Cell Calcium 2018, 70, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.H.; Miyamoto, M.; Sastre, A.; Schnaar, R.L.; Coyle, J.T. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 1989, 2, 1547–1558. [Google Scholar] [CrossRef]
- Tan, S.; Sagara, Y.; Liu, Y.; Maher, P.; Schubert, D. The regulation of reactive oxygen species production during programmed cell death. J. Cell Biol. 1998, 141, 1423–1432. [Google Scholar] [CrossRef]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent-and AIF-mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef]
- Li, Y.; Maher, P.; Schubert, D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron 1997, 19, 453–463. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M. The cystine/glutamate antiporter system x(c)(−) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.-e.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar]
- Kim, E.K.; Choi, E.-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef]
- Stanciu, M.; Wang, Y.; Kentor, R.; Burke, N.; Watkins, S.; Kress, G.; Reynolds, I.; Klann, E.; Angiolieri, M.R.; Johnson, J.W. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J. Biol. Chem. 2000, 275, 12200–12206. [Google Scholar] [CrossRef]
- Fukui, M.; Choi, H.J.; Zhu, B.T. Mechanism for the protective effect of resveratrol against oxidative stress-induced neuronal death. Free Radic. Biol. Med. 2010, 49, 800–813. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Terry, R.D.; Alford, M.; DeTeresa, R.; Hansen, L. Cortical and subcortical patterns of synaptophysinlike immunoreactivity in Alzheimer’s disease. Am. J. Pathol. 1991, 138, 235. [Google Scholar] [PubMed]
- Masliah, E.; Terry, R.D.; DeTeresa, R.M.; Hansen, L.A. Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neurosci. Lett. 1989, 103, 234–239. [Google Scholar] [CrossRef]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1991, 30, 572–580. [Google Scholar] [CrossRef]
- Barinaga, M. Is apoptosis key in Alzheimer’s disease? Science 1998, 281, 1303–1304. [Google Scholar] [CrossRef]
- Morrison, R.; Kinoshita, Y. The role of p53 in neuronal cell death. Cell Death Differ. 2000, 7, 868–879. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Zimmermann, S.; Moelling, K. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science 1999, 286, 1741–1744. [Google Scholar] [CrossRef]
- Cardone, M.H.; Roy, N.; Stennicke, H.R.; Salvesen, G.S.; Franke, T.F.; Stanbridge, E.; Frisch, S.; Reed, J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar] [CrossRef]
- Ahn, J.-Y. Neuroprotection signaling of nuclear akt in neuronal cells. Exp. Neurobiol. 2014, 23, 200–206. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Tamatani, M.; Matsuzaki, H.; Namikawa, K.; Kiyama, H.; Vitek, M.P.; Mitsuda, N.; Tohyama, M. Akt activation protects hippocampal neurons from apoptosis by inhibiting transcriptional activity of p53. J. Biol. Chem. 2001, 276, 5256–5264. [Google Scholar] [CrossRef] [PubMed]
- Uberti, D.; Grilli, M.; Memo, M. Induction of p53 in the glutamate-induced cell death program. Amino Acids 2000, 19, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Molz, S.; Dal-Cim, T.; Budni, J.; Martín-de-Saavedra, M.; Egea, J.; Romero, A.; del Barrio, L.; Rodrigues, A.L.; López, M.G.; Tasca, C.I. Neuroprotective effect of guanosine against glutamate-induced cell death in rat hippocampal slices is mediated by the phosphatidylinositol-3 kinase/Akt/glycogen synthase kinase 3β pathway activation and inducible nitric oxide synthase inhibition. J. Neurosci. Res. 2011, 89, 1400–1408. [Google Scholar] [CrossRef]
- Bratton, D.L.; Fadok, V.A.; Richter, D.A.; Kailey, J.M.; Guthrie, L.A.; Henson, P.M. Appearance of phosphatidylserine on apoptotic cells requires calcium-mediated nonspecific flip-flop and is enhanced by loss of the aminophospholipid translocase. J. Biol. Chem. 1997, 272, 26159–26165. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).