Solution Polymerization of Acrylic Acid Initiated by Redox Couple Na-PS/Na-MBS: Kinetic Model and Transition to Continuous Process


Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Tests
2.1.1. Chemicals and Analytical Technique
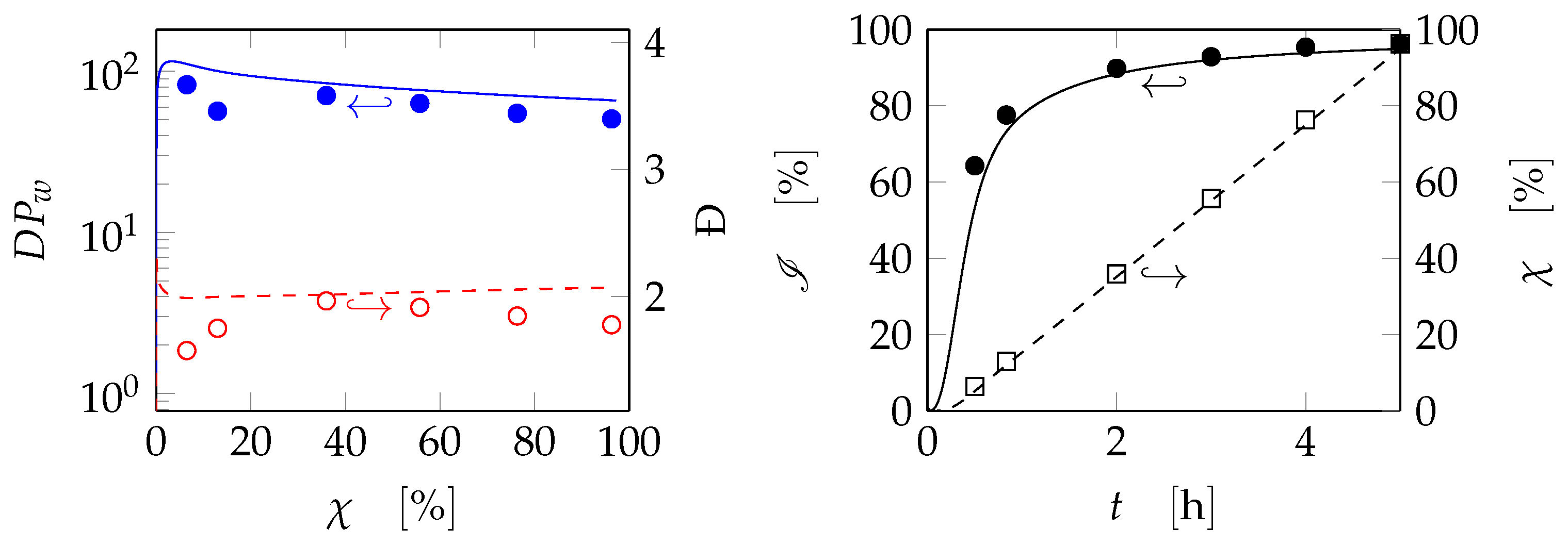
2.1.2. Discontinuous Reactor
3. Model Development
3.1. Kinetic Scheme
3.2. Governing Equations
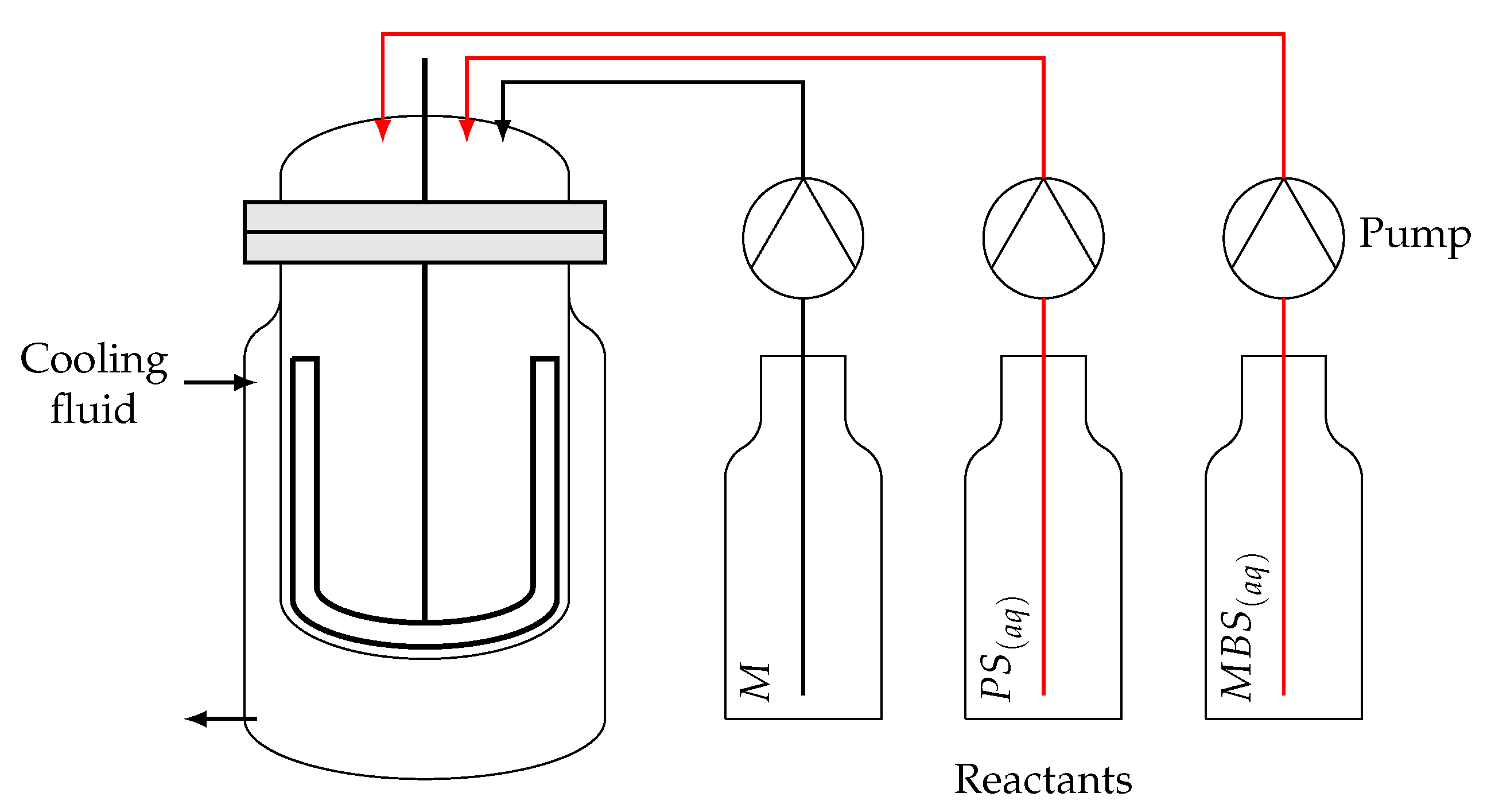
3.2.1. SBR Model
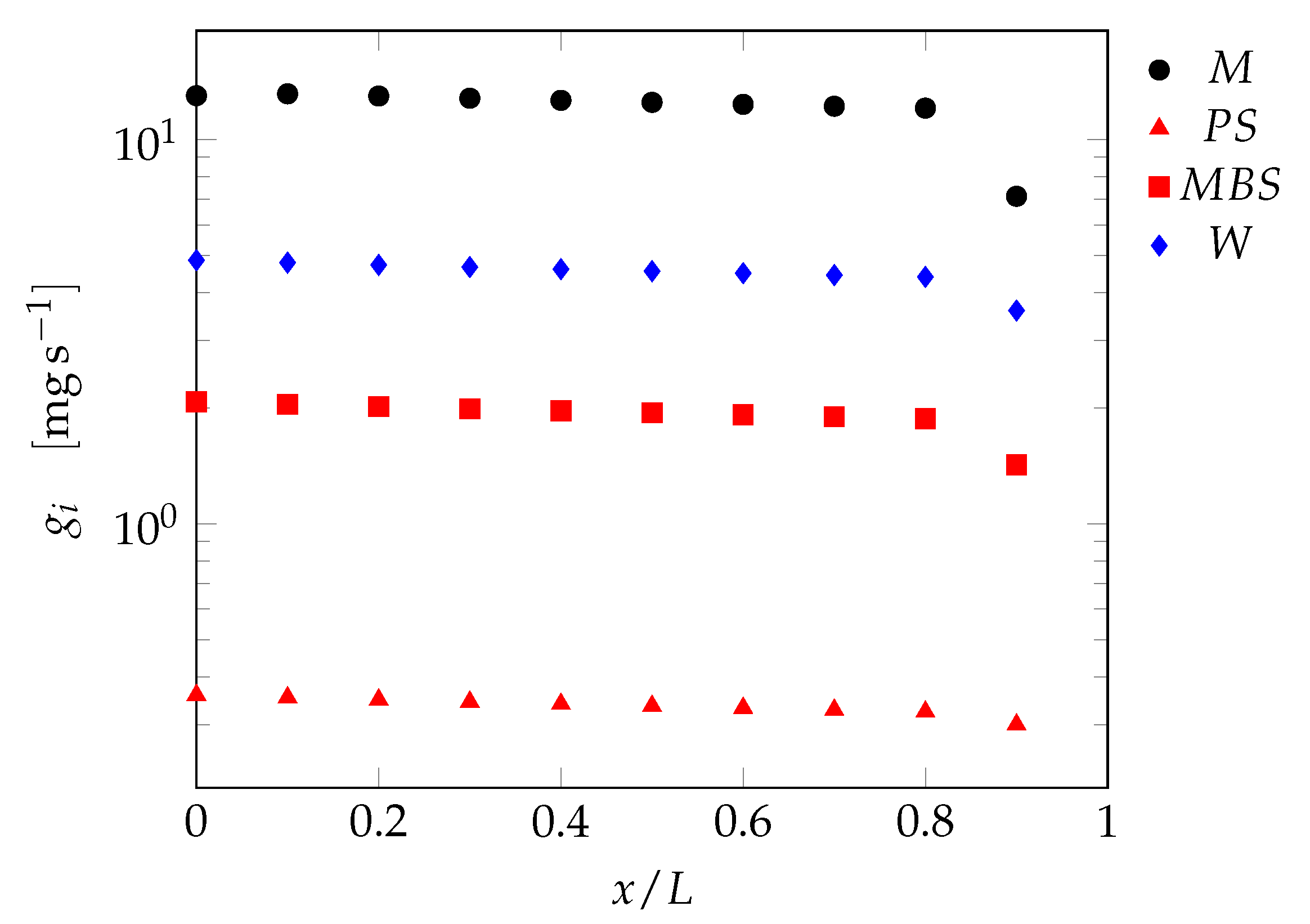
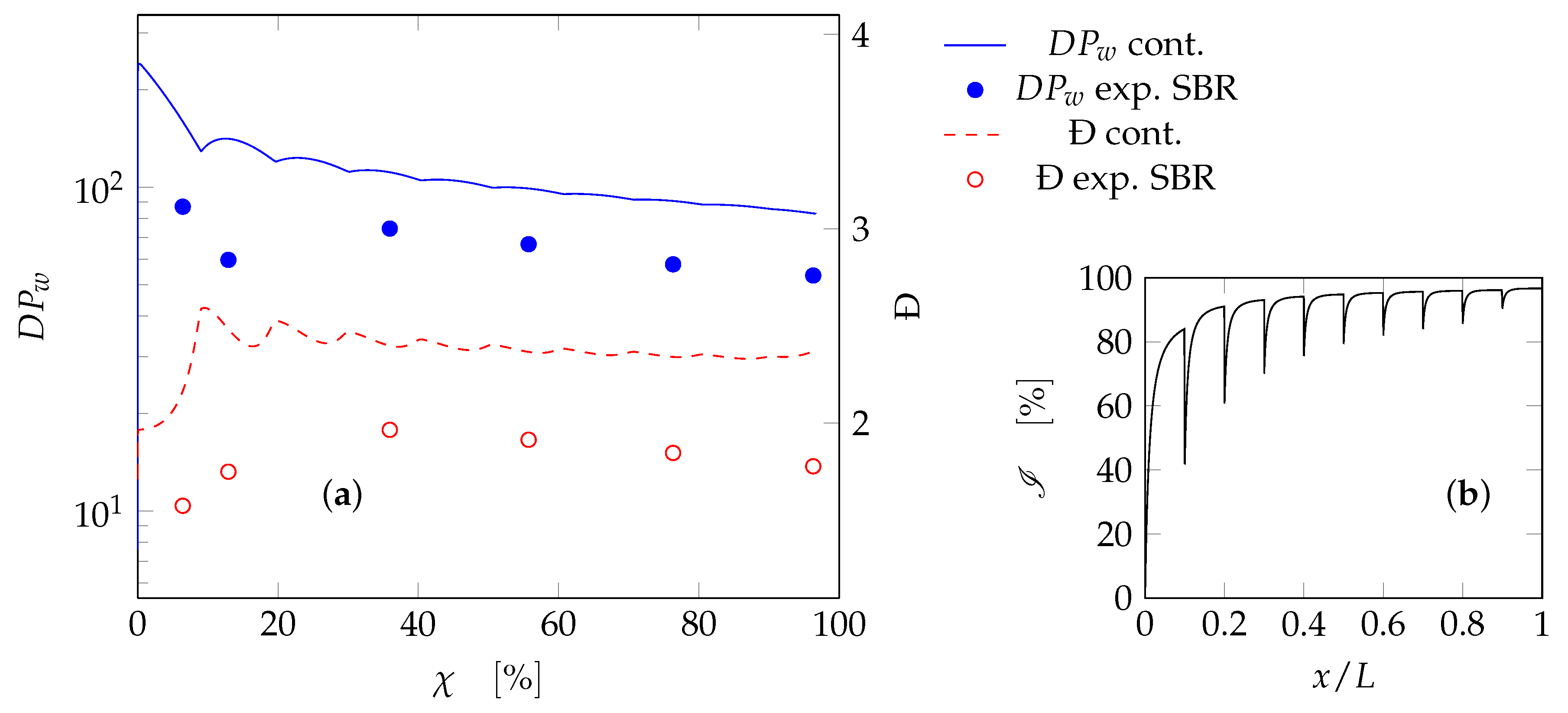
3.2.2. Tubular Reactor Model
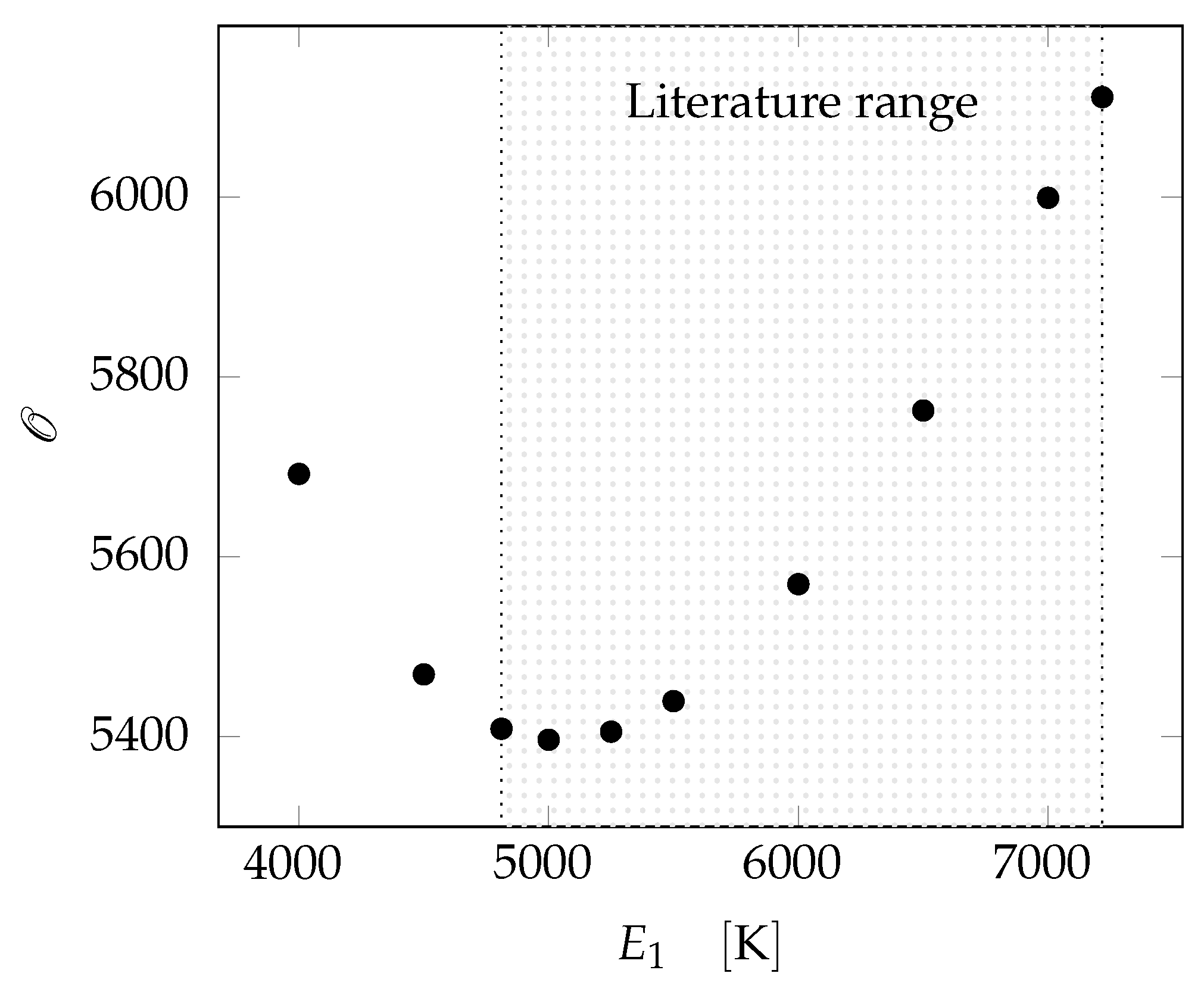
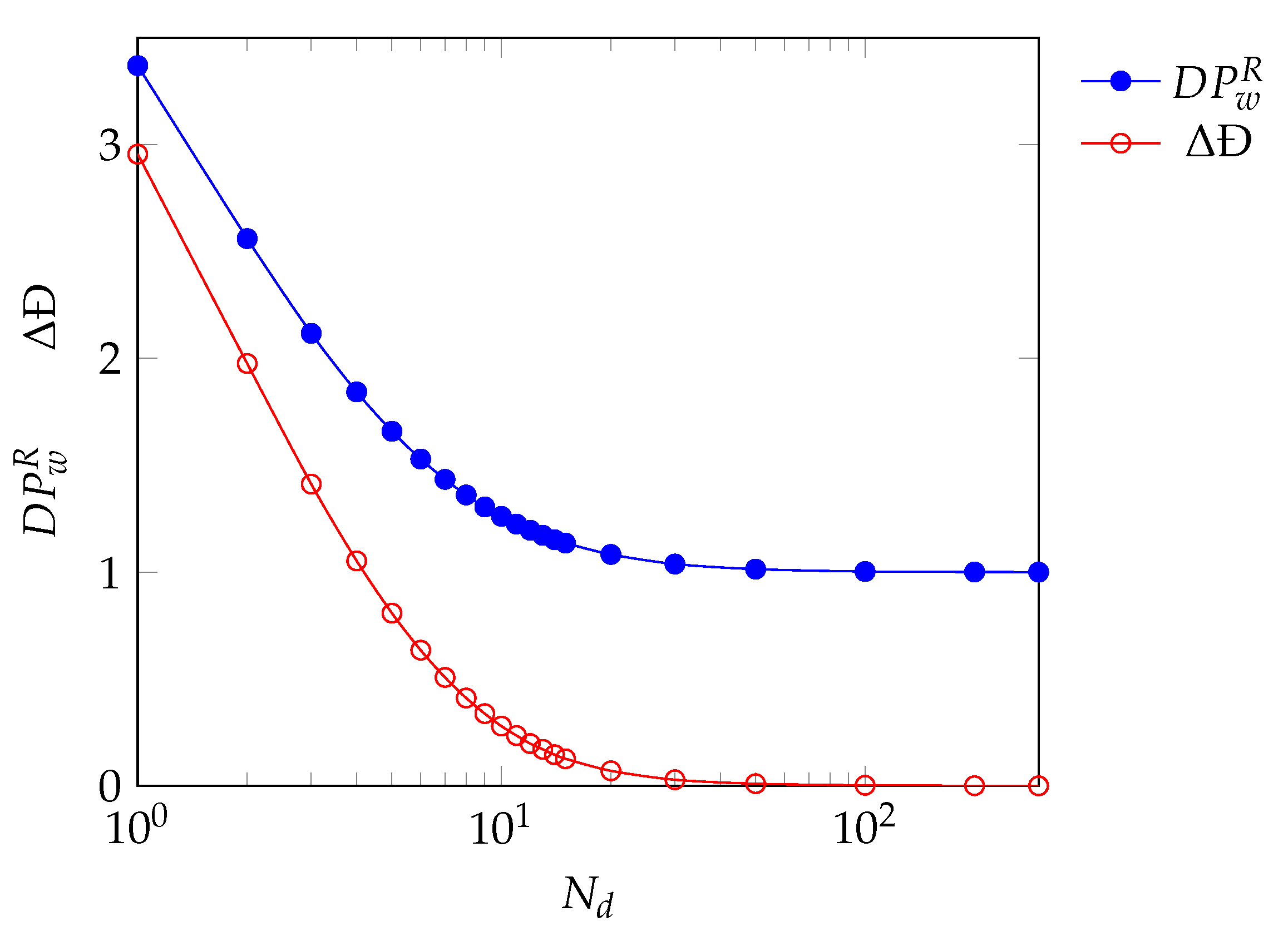
3.2.3. Parameter Evaluation
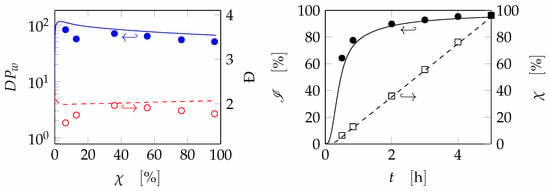
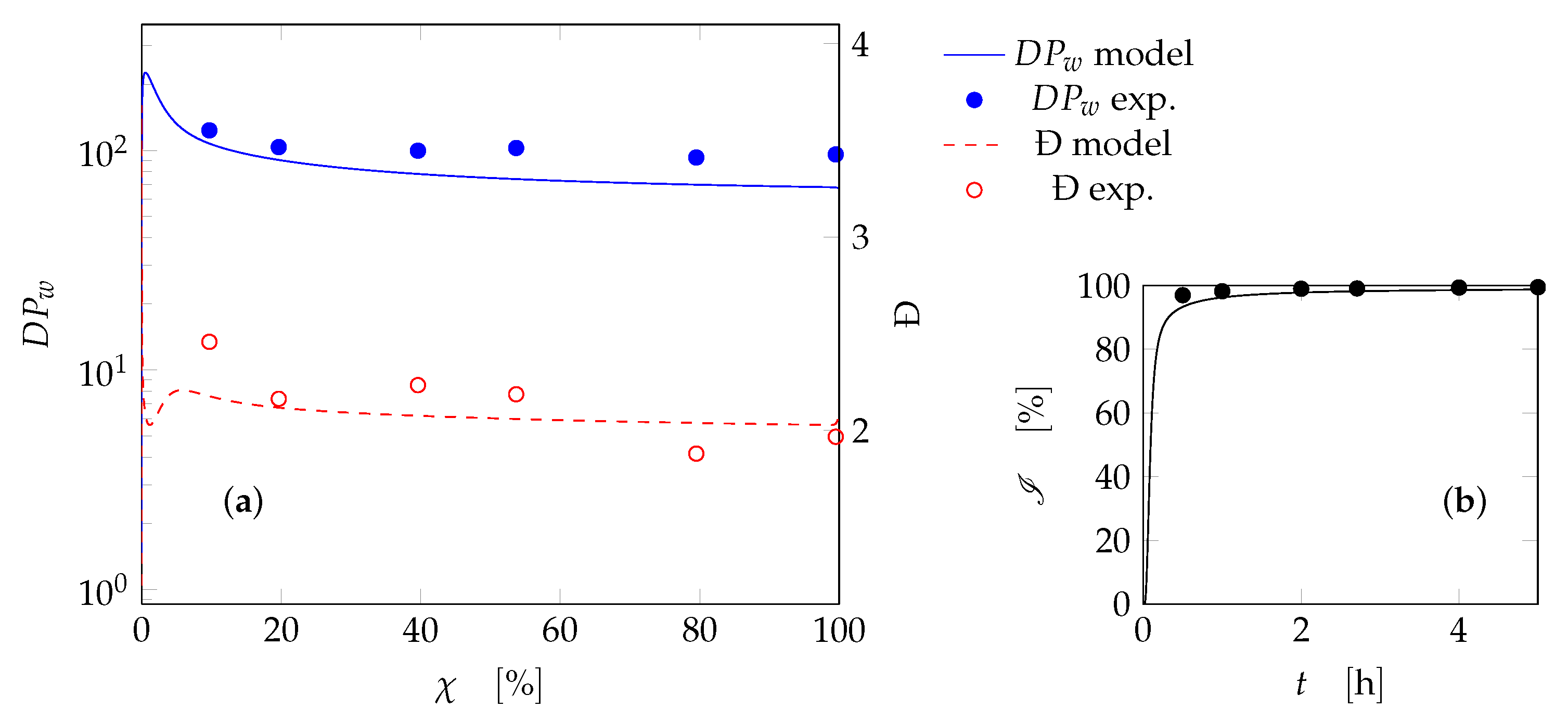
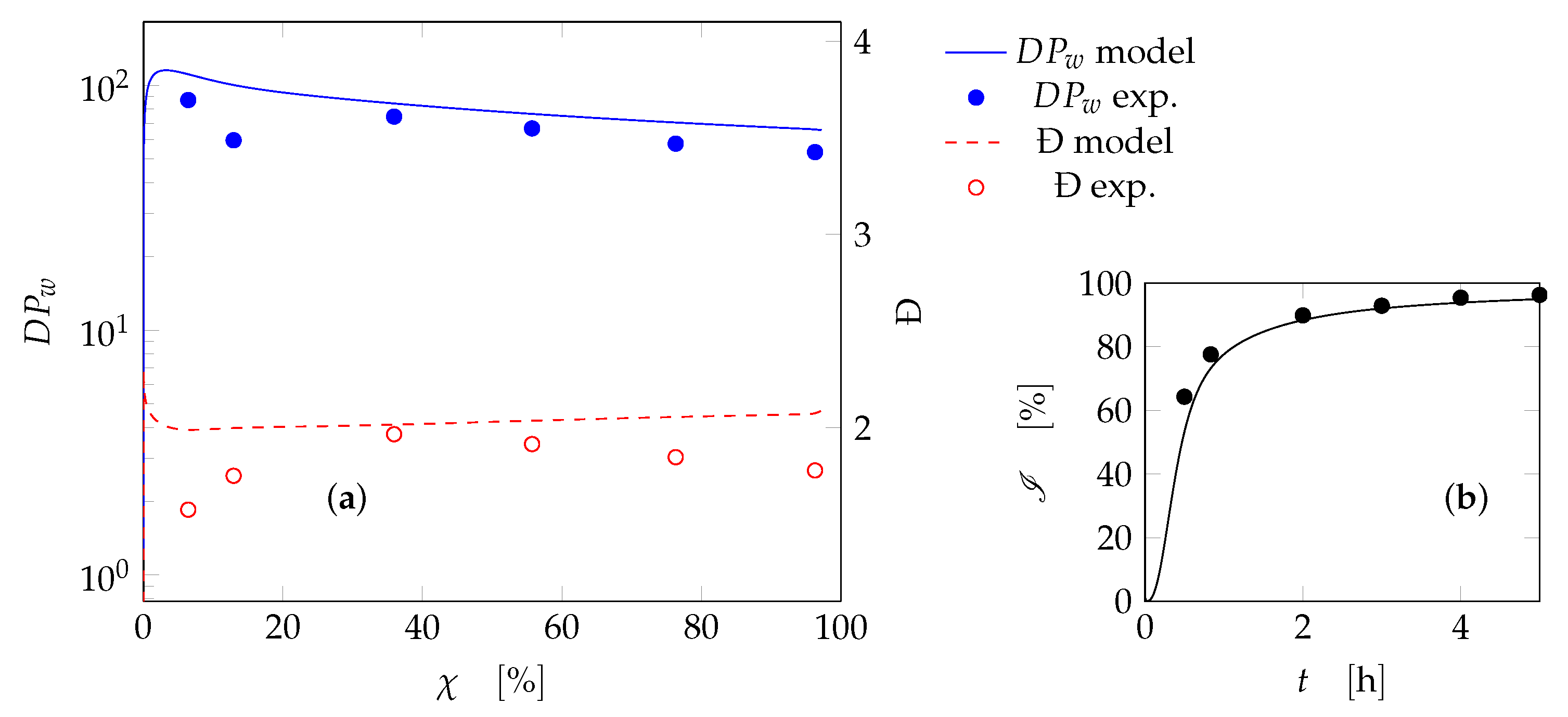
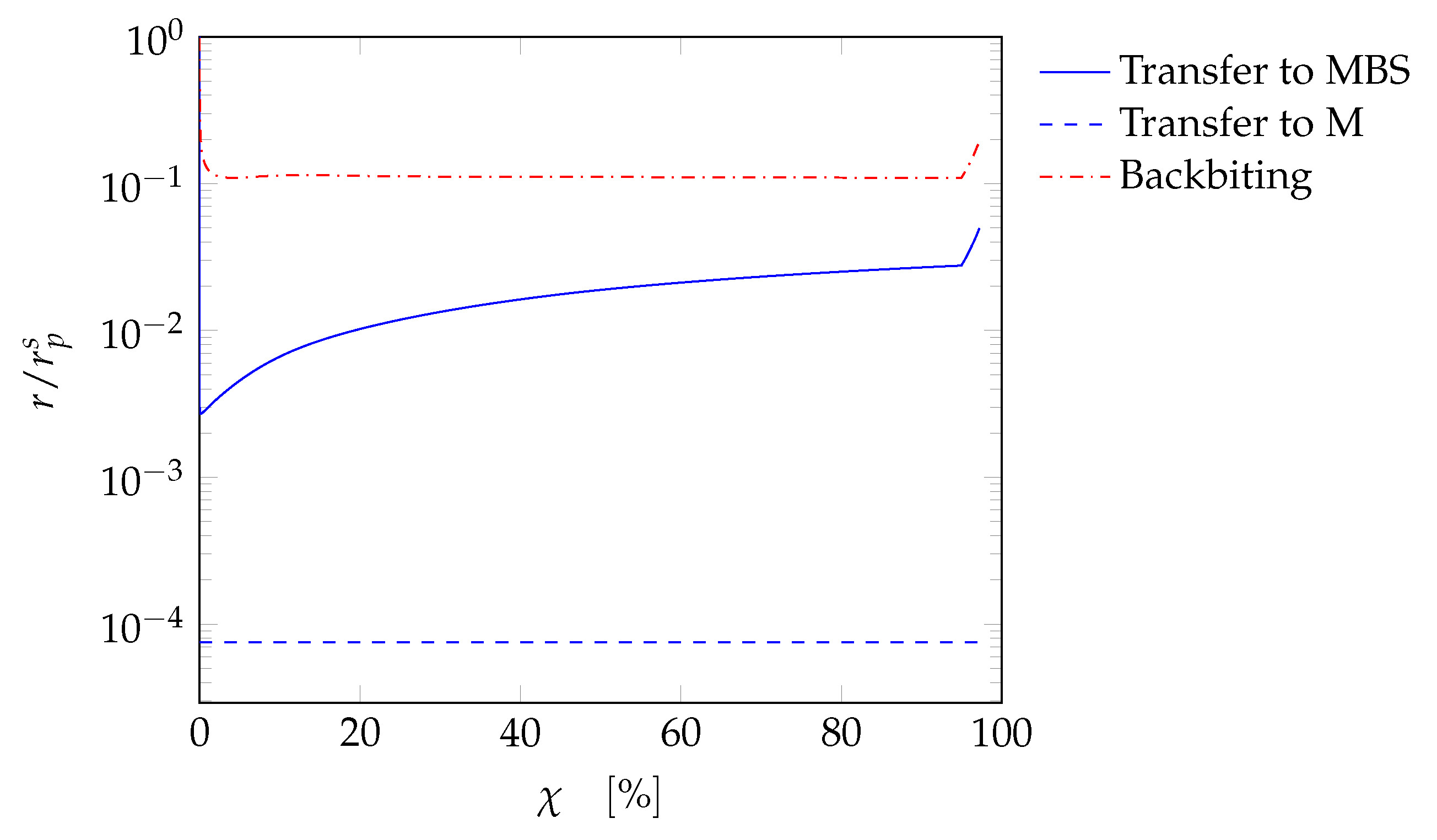
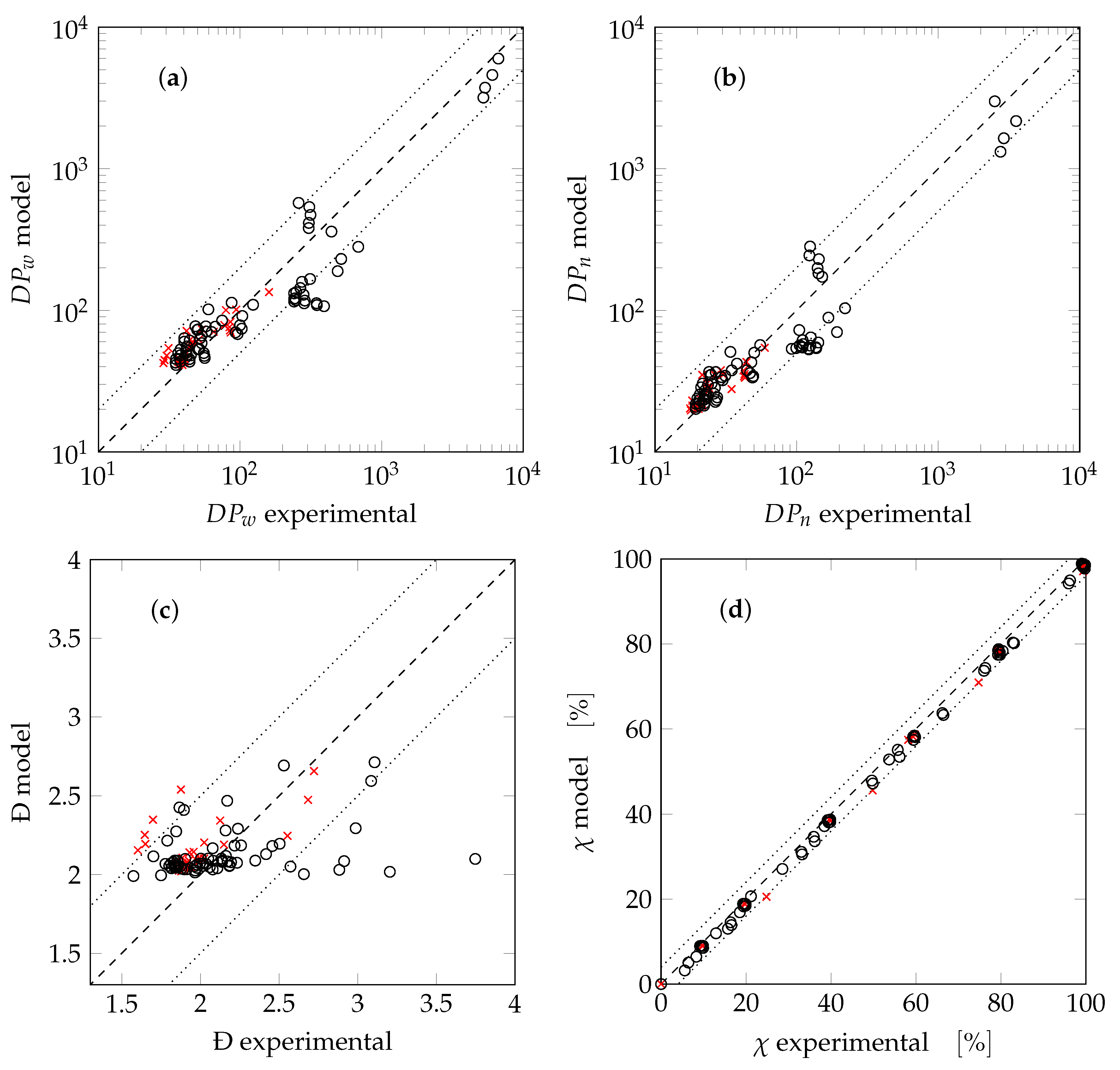
4. Results and Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | Acrylic acid |
| BR | Batch reactor |
| GPC | Gel permeation chromatography |
| HPLC | High-performance liquid chromatography |
| MBS | Metabisulfate |
| MCR | Mid-chain radicals |
| MoM | Method of moments |
| PAA | Poly(acrylic acid) |
| PFR | Plug-flow reactor |
| PS | Persulfate |
| SBR | Semi-batch reactor |
References
- Herth, G.; Schornick, G.; Buchholz, F.L. Polyacrylamides and Poly(Acrylic Acids). In Ullmann’s Encyclopedia of Industrial Chemistry; American Cancer Society: Atlanta, GA, USA, 2015; pp. 1–16. [Google Scholar]
- Roberts, D. Heats of Polymerization. A Summary of Published Values and Their Relation to Structure. J. Res. Natl. Bur. Stand. 1950, 44, 221–232. [Google Scholar] [CrossRef]
- Stankiewicz, A.I.; Yan, P. 110th Anniversary: The Missing Link Unearthed: Materials and Process Intensification. Ind. Eng. Chem. Res. 2019, 58, 9212–9222. [Google Scholar] [CrossRef]
- Anxionnaz, Z.; Cabassud, M.; Gourdon, C.; Tochon, P. Transposition of an Exothermic Reaction From a Batch Reactor to an Intensified Continuous One. Heat Transf. Eng. 2010, 31, 788–797. [Google Scholar] [CrossRef]
- Goerke, T.; Kohlmann, D.; Engell, S. Transfer of Semibatch Processes to Continuous Processes with Side Injections—Opportunities and Limitations. Macromol. React. Eng. 2016, 10, 364–388. [Google Scholar] [CrossRef]
- Kohlmann, D.; Chevrel, M.C.; Hoppe, S.; Meimaroglou, D.; Chapron, D.; Bourson, P.; Schwede, C.; Loth, W.; Stammer, A.; Wilson, J.; et al. Modular, Flexible, and Continuous Plant for Radical Polymerization in Aqueous Solution. Macromol. React. Eng. 2016, 10, 339–353. [Google Scholar] [CrossRef]
- Florit, F.; Busini, V.; Storti, G.; Rota, R. From semi-batch to continuous tubular reactors: A kinetics-free approach. Chem. Eng. J. 2018, 354, 1007–1017. [Google Scholar] [CrossRef]
- Florit, F.; Busini, V.; Storti, G.; Rota, R. Kinetics-free transformation from non-isothermal discontinuous to continuous tubular reactors. Chem. Eng. J. 2019, 373, 792–802. [Google Scholar] [CrossRef]
- Omidian, H.; Zohuriaan-Mehr, M.; Bouhendi, H. Aqueous solution polymerization of neutralized acrylic acid using Na2S2O5/(NH4)2S2O8 redox pair system under atmospheric conditions. Int. J. Polym. Mater. Polym. Biomater. 2003, 52, 307–321. [Google Scholar] [CrossRef]
- Ebdon, J.; Huckerby, T.; Hunter, T. Free-radical aqueous slurry polymerizations of acrylonitrile: 1. End-groups and other minor structures in polyacrylonitriles initiated by ammonium persulfate/sodium metabisulfite. Polymer 1994, 35, 250–256. [Google Scholar] [CrossRef]
- Barth, J.; Meiser, W.; Buback, M. SP-PLP-EPR Study into Termination and Transfer Kinetics of Non-Ionized Acrylic Acid Polymerized in Aqueous Solution. Macromolecules 2012, 45, 1339–1345. [Google Scholar] [CrossRef]
- Anseth, K.S.; Scott, R.A.; Peppas, N.A. Effects of Ionization on the Reaction Behavior and Kinetics of Acrylic Acid Polymerizations. Macromolecules 1996, 29, 8308–8312. [Google Scholar] [CrossRef]
- Çatalgil Giz, H.; Giz, A.; Alb, A.M.; Reed, W.F. Absolute online monitoring of acrylic acid polymerization and the effect of salt and pH on reaction kinetics. J. Appl. Polym. Sci. 2004, 91, 1352–1359. [Google Scholar] [CrossRef]
- Wittenberg, N.F.G.; Preusser, C.; Kattner, H.; Stach, M.; Lacík, I.; Hutchinson, R.A.; Buback, M. Modeling Acrylic Acid Radical Polymerization in Aqueous Solution. Macromol. React. Eng. 2016, 10, 95–107. [Google Scholar] [CrossRef]
- Ebdon, J.; Huckerby, T.; Hunter, T. Free-radical aqueous slurry polymerizations of acrylonitrile: 2. End-groups and other minor structures in polyacrylonitriles initiated by potassium persulfate/sodium bisulfite. Polymer 1994, 35, 4659–4664. [Google Scholar] [CrossRef]
- Riddick, J.A.; Bunger, W.B.; Sakano, T.K. Organic Solvents: Physical Properties and Methods of Purification, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 1986; p. 376. [Google Scholar]
- Misra, G.; Bajpai, U. Redox polymerization. Prog. Polym. Sci. 1982, 8, 61–131. [Google Scholar] [CrossRef]
- Odian, G. Radical Chain Polymerization. In Principles of Polymerization; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2004; Chapter 3; pp. 198–349. [Google Scholar]
- Costa, C.; Santos, V.; Araujo, P.; Sayer, C.; Santos, A.; Fortuny, M. Microwave-assisted rapid decomposition of persulfate. Eur. Polym. J. 2009, 45, 2011–2016. [Google Scholar] [CrossRef]
- Kuchta, F.D.; van Herk, A.M.; German, A.L. Propagation Kinetics of Acrylic and Methacrylic Acid in Water and Organic Solvents Studied by Pulsed-Laser Polymerization. Macromolecules 2000, 33, 3641–3649. [Google Scholar] [CrossRef]
- Thakur, R.; Vial, C.; Nigam, K.; Nauman, E.; Djelveh, G. Static mixers in the process industries—A review. Chem. Eng. Res. Des. 2003, 81, 787–826. [Google Scholar] [CrossRef]
- Gutierrez, C.G.; Cáceres Montenegro, G.; Minari, R.J.; Vega, J.R.; Gugliotta, L.M. Scale Inhibitor and Dispersant Based on Poly(Acrylic Acid) Obtained by Redox-Initiated Polymerization. Macromol. React. Eng. 2019, 13, 1900007. [Google Scholar] [CrossRef]
- Minari, R.J.; Caceres, G.; Mandelli, P.; Yossen, M.M.; Gonzalez-Sierra, M.; Vega, J.R.; Gugliotta, L.M. Semibatch Aqueous-Solution Polymerization of Acrylic Acid: Simultaneous Control of Molar Masses and Reaction Temperature. Macromol. React. Eng. 2011, 5, 223–231. [Google Scholar] [CrossRef]
- Florit, F.; Busini, V.; Rota, R. Kinetics-free process intensification: From semi-batch to series of continuous chemical reactors. Chem. Eng. Process. Process. Intensif. 2020, 154, 108014. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test | T | N Data | |||||||
|---|---|---|---|---|---|---|---|---|---|
| [g] | [g] | [g] | [g] | [g] | [g] | [C] | [h] | ||
| SBR1 | 280 | 7.8 | 44.44 | 37.8 | 66.66 | 137.8 | 90 | 5 | 6 |
| SBR2 | 280 | 7.8 | 44.44 | 37.8 | 66.66 | 137.8 | 90 | 0.5 | 4 |
| SBR3 | 252 | 7.0 | 40.00 | 34.0 | 60.00 | 124.0 | 75 | 5 | 6 |
| SBR4 | 280 | 7.8 | 22.24 | 37.8 | 33.36 | 137.8 | 70 | 5 | 6 |
| SBR5 | 280 | 7.8 | 44.44 | 37.8 | 66.66 | 137.8 | 50 | 5 | 6 |
| SBR6 | 280 | 7.8 | 22.24 | 37.8 | 33.36 | 137.8 | 90 | 5 | 6 |
| SBR7 | 28 | 0.78 | 4.444 | 3.78 | 6.666 | 231.8 | 90 | 5 | 6 |
| SBR8 | 315 | 8.8 | 50.00 | 42.5 | 75.00 | 155.0 | 90 | 2.5 | 6 |
| SBR9 | 252 | 7.0 | 40.00 | 34.0 | 60.00 | 124.0 | 90 | 1.5 | 6 |
| SBR10 | 280 | 7.8 | 44.44 | 37.8 | 66.66 | 137.8 | 90 | 5 | 6 |
| SBR11 | 280 | 7.8 | 44.44 | 37.8 | 66.66 | 137.8 | 90 | 2.5 | 6 |
| SBR12 | 280 | 7.8 | 44.44 | 37.8 | 66.66 | 137.8 | 90 | 1 | 6 |
| SBR13 | 280 | 7.8 | 11.12 | 37.8 | 16.68 | 137.8 | 90 | 5 | 6 |
| SBR14 | 28 | 0.78 | 0.00 | 3.78 | 0.00 | 231.8 | 90 | 5 | 6 |
| SBR15 | 280 | 7.8 | 0.00 | 37.8 | 0.00 | 275.6 | 70 | 5 | 4 |
| SBR16 | 280 | 7.8 | 11.12 | 37.8 | 16.68 | 137.8 | 70 | 5 | 6 |
| SBR17 | 280 | 7.8 | 11.12 | 37.8 | 16.68 | 137.8 | 60 | 5 | 6 |
| Name | Reaction | Rate |
|---|---|---|
| Initiation | ||
| , efficiency f | ||
| Propagation | ||
| Backbiting | ||
| Transfer to monomer | ||
| Transfer to MBS | ||
| Termination by | ||
| combination | ||
| Termination by | ||
| disproportionation | ||
| Species | Density [g cm] |
|---|---|
| W | |
| M | |
| P |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Florit, F.; Rodrigues Bassam, P.; Cesana, A.; Storti, G. Solution Polymerization of Acrylic Acid Initiated by Redox Couple Na-PS/Na-MBS: Kinetic Model and Transition to Continuous Process. Processes 2020, 8, 850. https://doi.org/10.3390/pr8070850
Florit F, Rodrigues Bassam P, Cesana A, Storti G. Solution Polymerization of Acrylic Acid Initiated by Redox Couple Na-PS/Na-MBS: Kinetic Model and Transition to Continuous Process. Processes. 2020; 8(7):850. https://doi.org/10.3390/pr8070850
Chicago/Turabian StyleFlorit, Federico, Paola Rodrigues Bassam, Alberto Cesana, and Giuseppe Storti. 2020. "Solution Polymerization of Acrylic Acid Initiated by Redox Couple Na-PS/Na-MBS: Kinetic Model and Transition to Continuous Process" Processes 8, no. 7: 850. https://doi.org/10.3390/pr8070850
APA StyleFlorit, F., Rodrigues Bassam, P., Cesana, A., & Storti, G. (2020). Solution Polymerization of Acrylic Acid Initiated by Redox Couple Na-PS/Na-MBS: Kinetic Model and Transition to Continuous Process. Processes, 8(7), 850. https://doi.org/10.3390/pr8070850