A Review of Methane Activation Reactions by Halogenation: Catalysis, Mechanism, Kinetics, Modeling, and Reactors

 , ,
, ,  ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Homogeneous Gas Phase Reactions
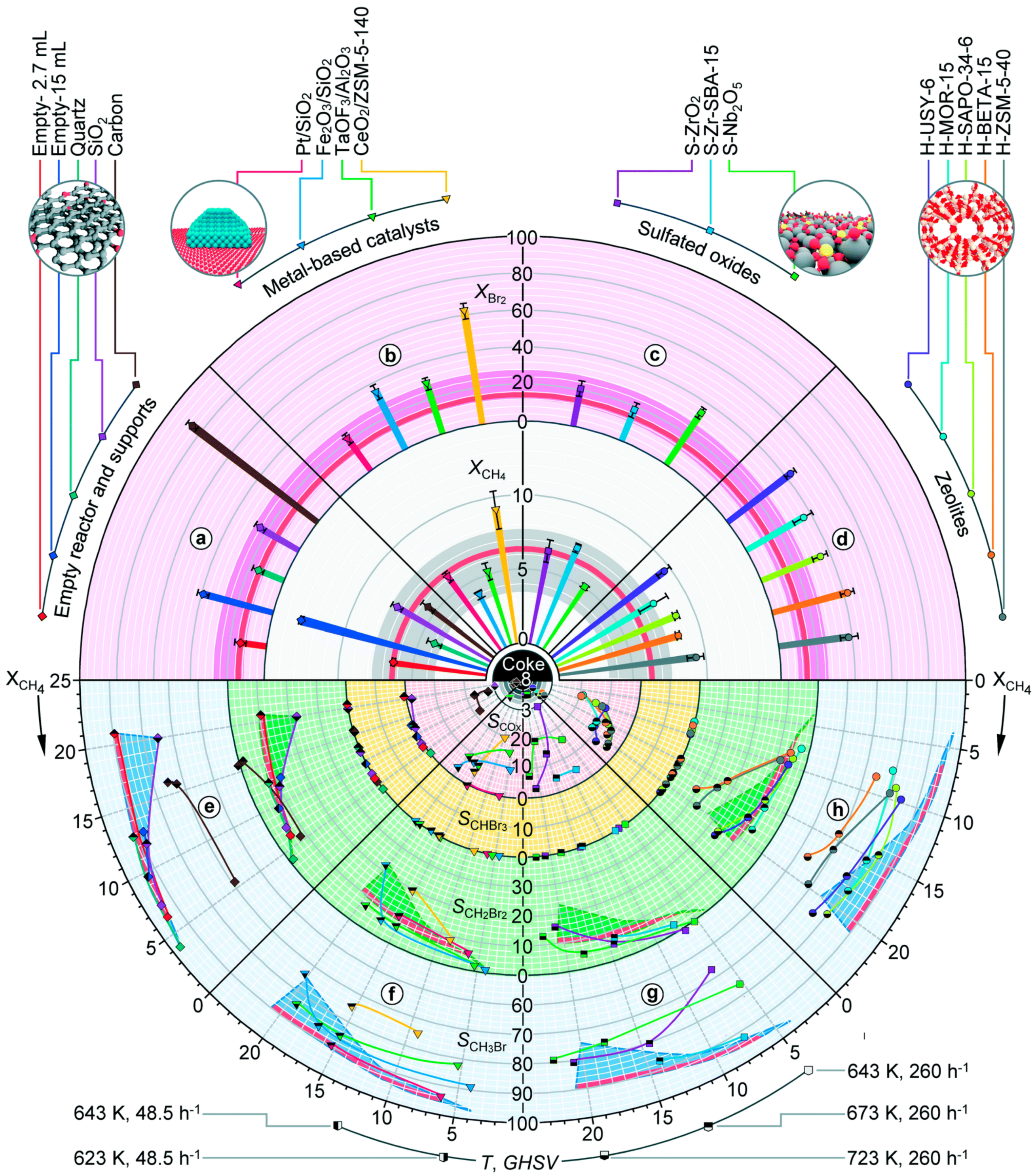
3. Catalytic Halogenation
3.1. Non-Oxidative Catalytic Halogenation
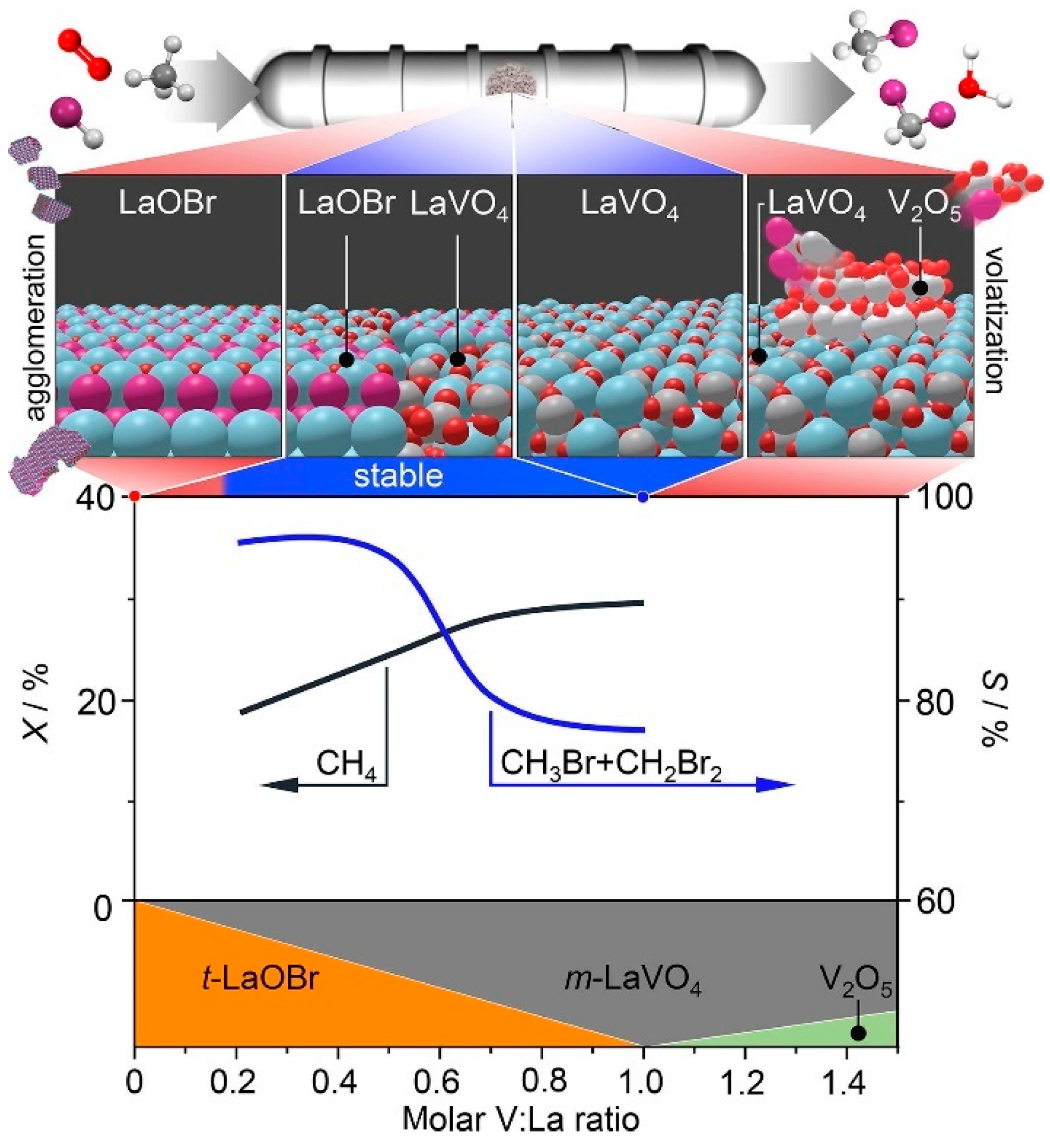
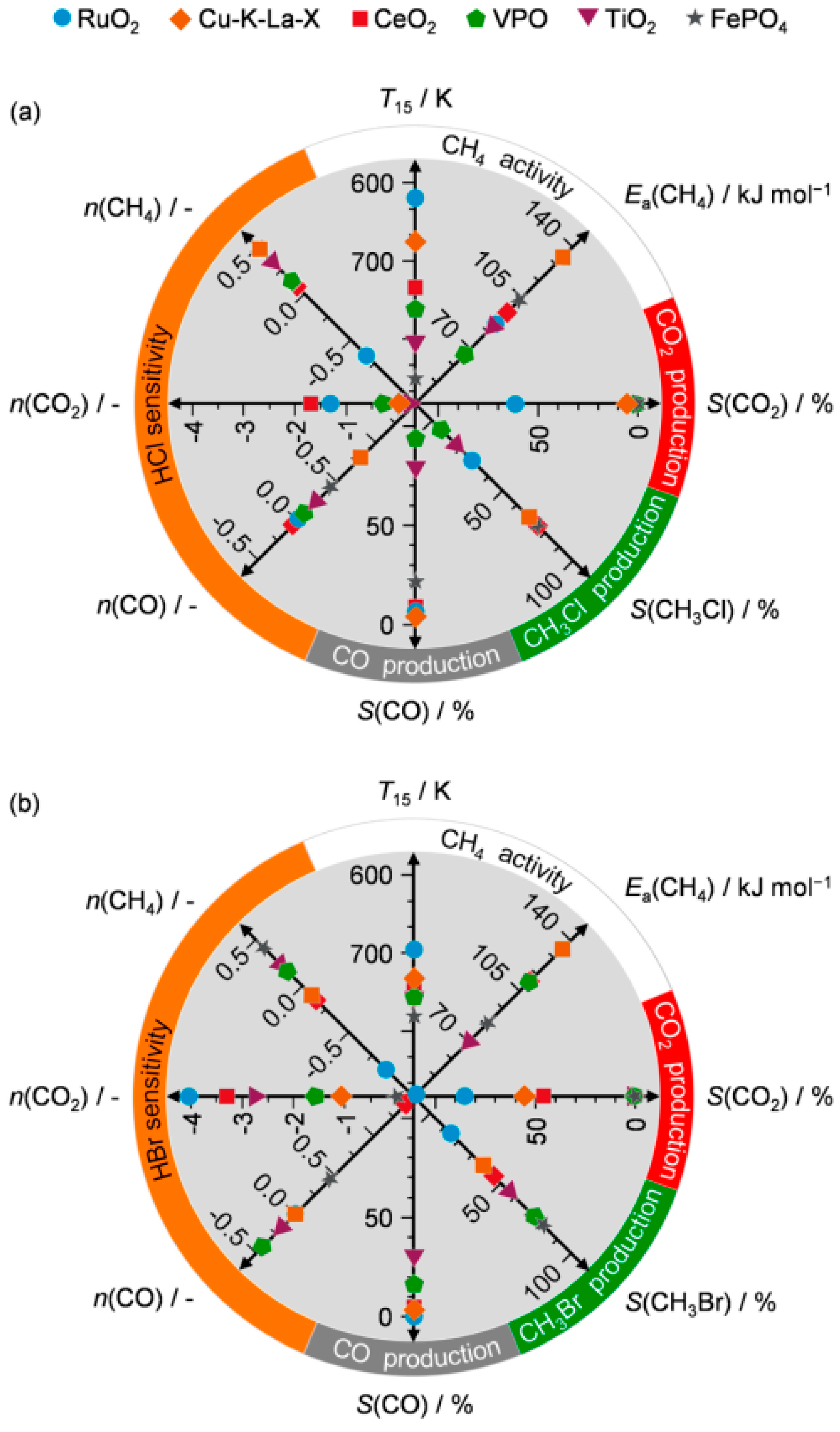
3.2. Oxidative Catalytic Halogenation
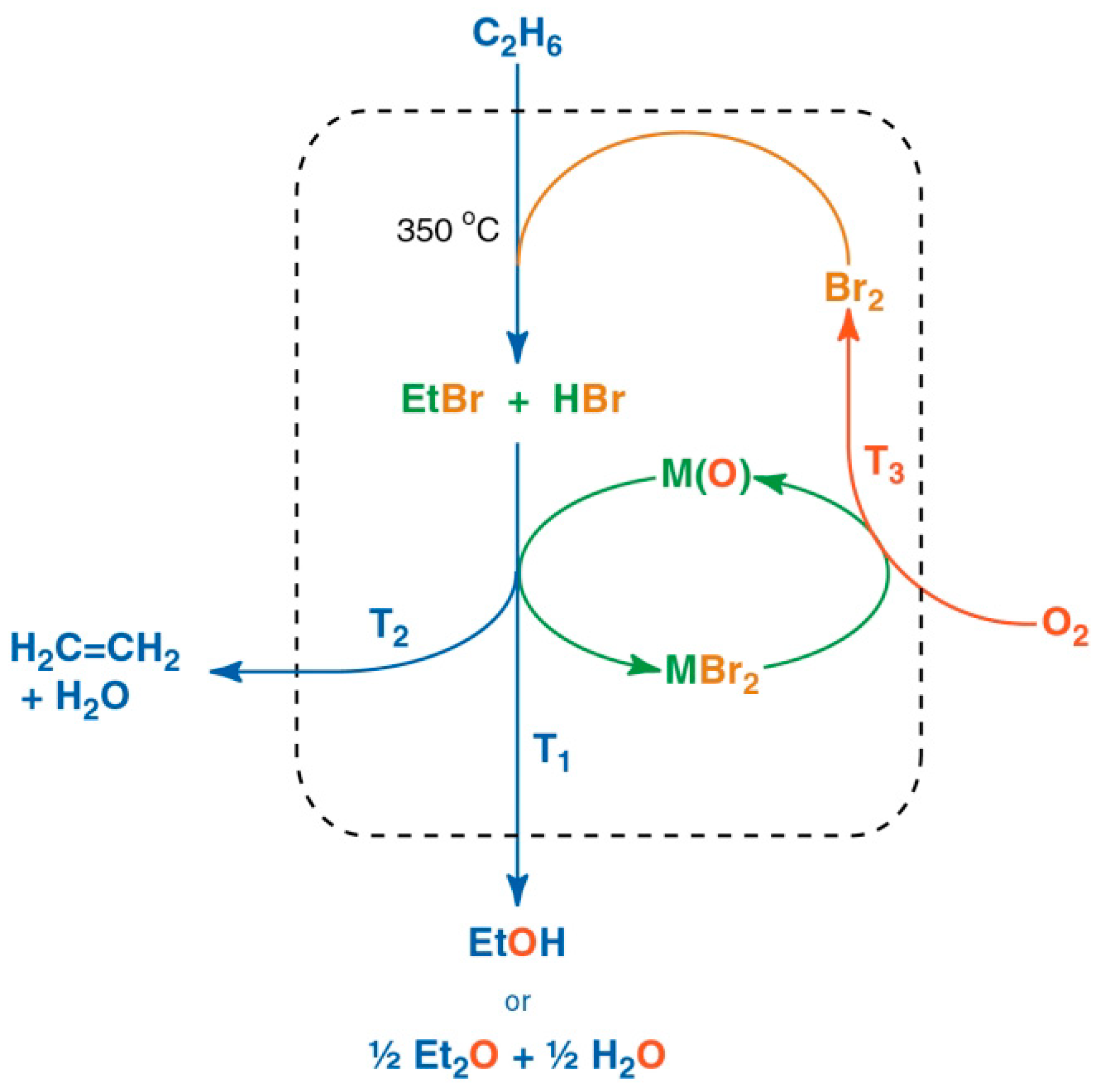
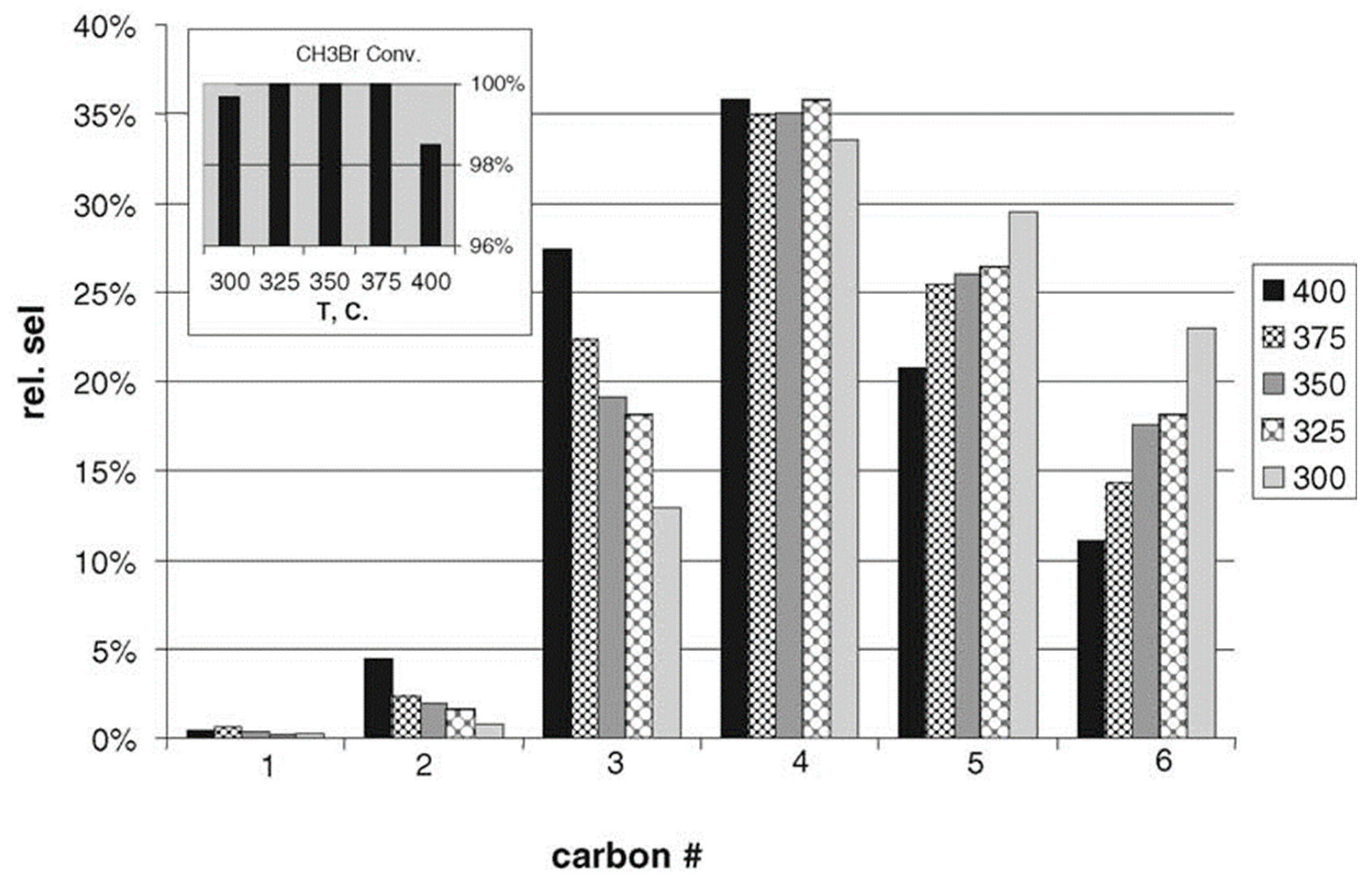
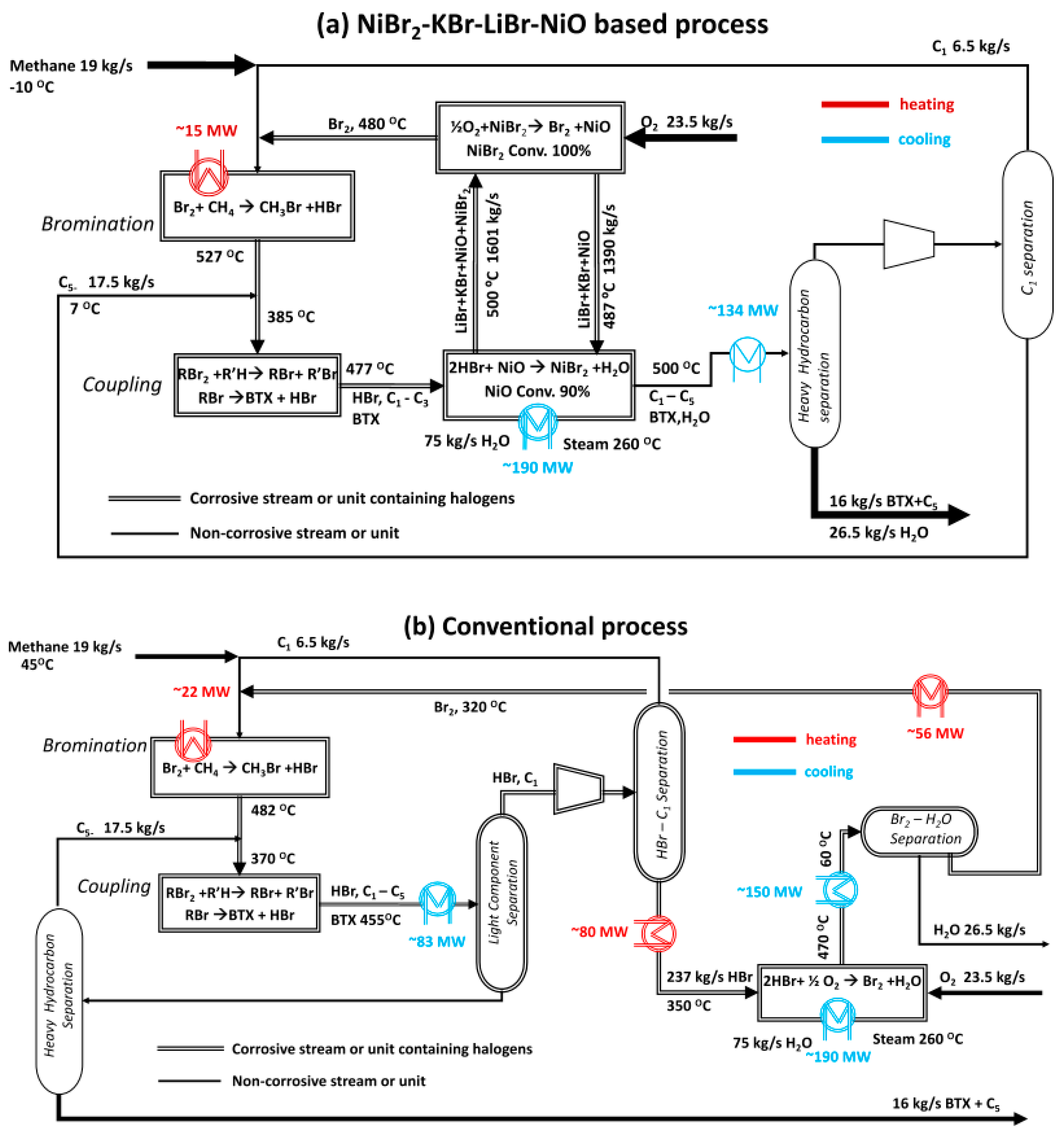
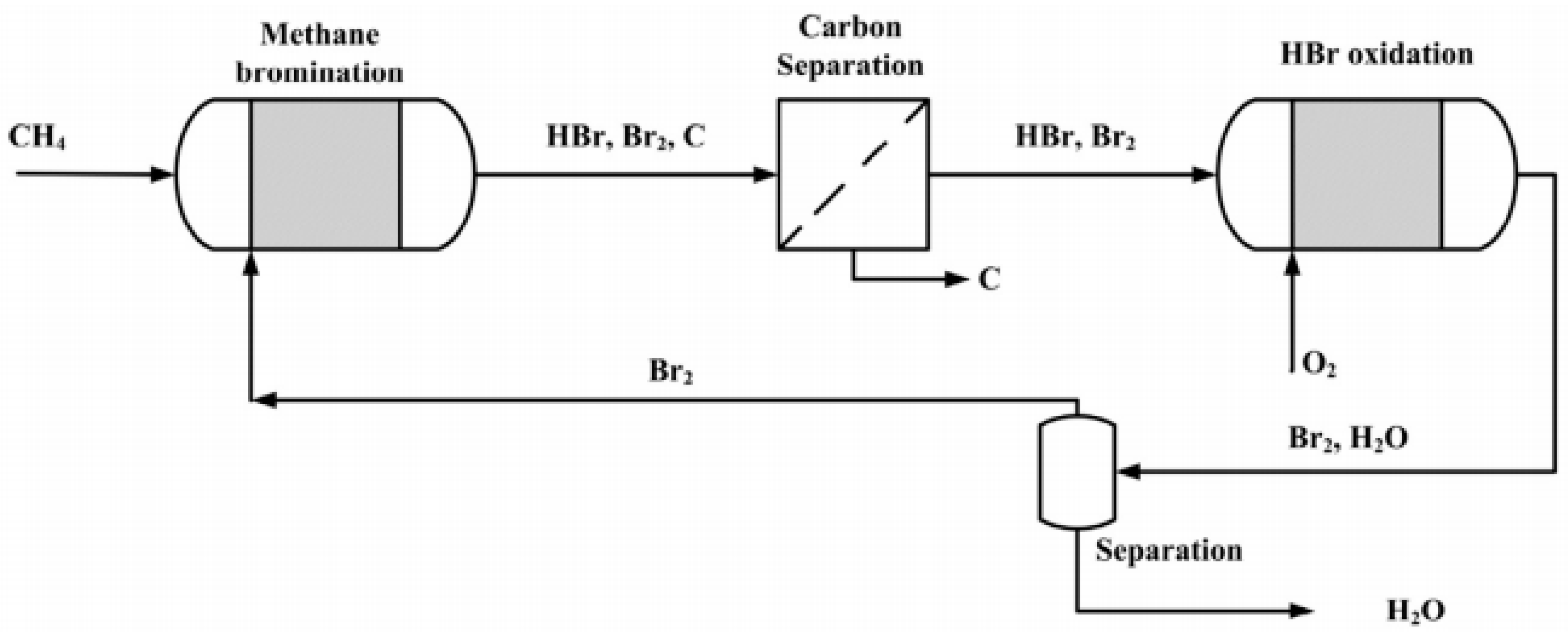
4. Multi-Step Processes
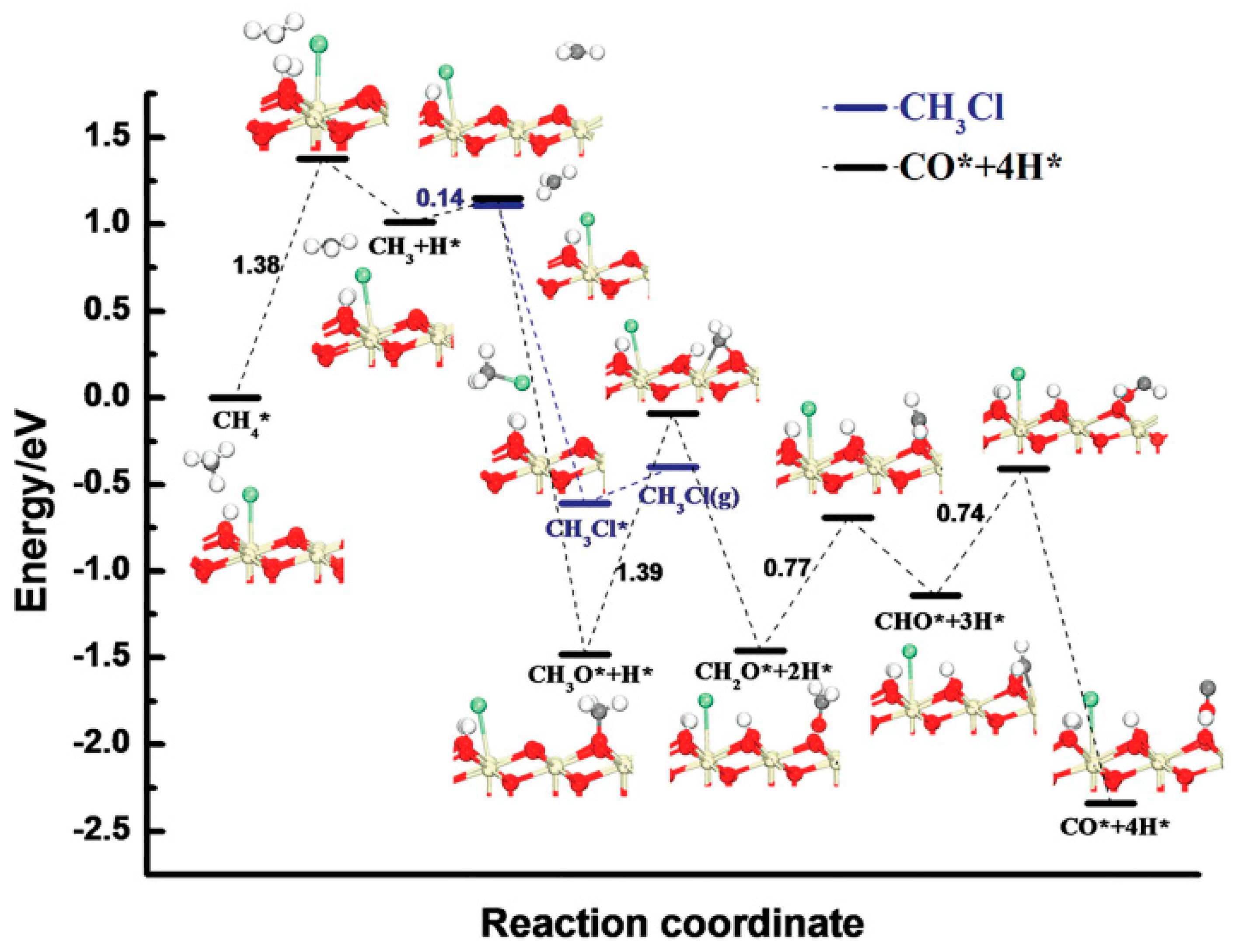
5. Density Functional Theory (DFT)
6. Green Processes and Fuel
7. Unconventional Technologies
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Anderson, R.B.; Kölbel, H.; Rálek, M. The Fischer-Tropsch Synthesis; Academic Press: Waltham, MA, USA, 1984; Volume 16. [Google Scholar]
- Ma, Y.; Ge, Q.; Li, W.; Xu, H. Methanol synthesis from sulfur-containing syngas over Pd/CeO2 catalyst. Appl. Catal. B Environ. 2009, 90, 99–104. [Google Scholar] [CrossRef]
- Song, C. Global challenges and strategies for control, conversion and utilization of CO2 for sustainable development involving energy, catalysis, adsorption and chemical processing. Catal. Today 2006, 115, 2–32. [Google Scholar] [CrossRef]
- Tian, P.; Wei, Y.; Ye, M.; Liu, Z. Methanol to olefins (MTO): From fundamentals to commercialization. ACS Catal. 2015, 5, 1922–1938. [Google Scholar] [CrossRef]
- Bjørgen, M.; Joensen, F.; Holm, M.S.; Olsbye, U.; Lillerud, K.-P.; Svelle, S. Methanol to gasoline over zeolite H-ZSM-5: Improved catalyst performance by treatment with NaOH. Appl. Catal. A Gen. 2008, 345, 43–50. [Google Scholar] [CrossRef]
- Zaidi, H.A.; Pant, K.K. Catalytic conversion of methanol to gasoline range hydrocarbons. Catal. Today 2004, 96, 155–160. [Google Scholar] [CrossRef]
- Conte, M.; Lopez-Sanchez, J.A.; Zhou, W.; Morgan, D.J.; Ryabenkova, Y.; Bartley, J.K.; Carley, A.F.; Taylor, S.H.; Kiely, C.J.; Khalid, K.; et al. Modified zeolite ZSM-5 for the methanol to aromatics reaction. Catal. Sci. Technol. 2012, 2, 105–112. [Google Scholar] [CrossRef]
- Schwach, P.; Pan, X.; Bao, X. Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects. Chem. Rev. 2017, 117, 8497–8520. [Google Scholar] [CrossRef]
- Galadima, A.; Muraza, O. Revisiting the oxidative coupling of methane to ethylene in the golden period of shale gas: A review. J. Ind. Eng. Chem. 2016, 37, 1–13. [Google Scholar] [CrossRef]
- Sun, K.; Ginosar, D.M.; He, T.; Zhang, Y.; Fan, M.; Chen, R. Progress in Nonoxidative Dehydroaromatization of Methane in the Last 6 Years. Ind. Eng. Chem. Res. 2018, 57, 1768–1789. [Google Scholar] [CrossRef]
- Xie, P.; Pu, T.; Nie, A.; Hwang, S.; Purdy, S.C.; Yu, W.; Su, D.; Miller, J.T.; Wang, C. Nanoceria-Supported Single-Atom Platinum Catalysts for Direct Methane Conversion. ACS Catal. 2018, 8, 4044–4048. [Google Scholar] [CrossRef]
- Guo, X.; Fang, G.; Li, G.; Ma, H.; Fan, H.; Yu, L.; Ma, C.; Wu, X.; Deng, D.; Wei, M.; et al. Direct, nonoxidative conversion of methane to ethylene, aromatics, and hydrogen. Science 2014, 344, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Taifan, W.; Baltrusaitis, J. CH4 conversion to value added products: Potential, limitations and extensions of a single step heterogeneous catalysis. Appl. Catal. B Environ. 2016, 525–547. [Google Scholar] [CrossRef]
- Wei, y.; Zhang, D.; Liu, Z.; Su, B.-L. Methyl halide to olefins and gasoline over zeolites and SAPO catalysts: A new route of MTO and MTG. Chin. J. Catal. 2012, 33, 11–21. [Google Scholar] [CrossRef]
- Zhang, A.; Sun, S.; Komon, Z.J.A.; Osterwalder, N.; Gadewar, S.; Stoimenov, P.; Auerbach, D.J.; Stucky, G.D.; McFarland, E.W. Improved light olefin yield from methyl bromide coupling over modified SAPO-34 molecular sieves. Phys. Chem. Chem. Phys. 2011, 13, 2550–2555. [Google Scholar] [CrossRef]
- Zhang, D.; Wei, Y.; Xu, L.; Chang, F.; Liu, Z.; Menga, S.; Su, B.-L.; Liu, Z. MgAPSO-34 molecular sieves with various Mg stoichiometries: Synthesis, characterization and catalytic behavior in the direct transformation of chloromethane into light olefins. Microporous Mesoporous Mater. 2008, 116, 684–692. [Google Scholar] [CrossRef]
- Ding, K.; Derk, A.R.; Zhang, A.; Hu, Z.; Stoimenov, P.; Stucky, G.D.; Metiu, H.; McFarland, E.W. Hydrodebromination and Oligomerization of Dibromomethane. ACS Catal. 2012, 2, 479–486. [Google Scholar] [CrossRef]
- Upham, D.C.; Snodgrass, Z.R.; Tabatabaei, M.; McConnaughy, T.B.; Gordon, M.J.; Metiu, H.; McFarland, E.W. Molten salt chemical looping for reactive separation of HBr in a halogen- based natural gas conversion process. Chem. Eng. Sci. 2017, 160, 245–253. [Google Scholar] [CrossRef]
- Pérez-Ramírez, J.; Mondelli, C.; Schmidt, T.; Schlüter, O.F.-K.; Wolf, A.; Mleczko, L.; Dreier, T. Sustainable chlorine recycling via catalysed HCl oxidation: From fundamentals to implementation. Energy Environ. Sci. 2011, 4, 4786–4799. [Google Scholar] [CrossRef]
- Horn, R.; Schlögl, R. Methane Activation by Heterogeneous Catalysis. Catal. Lett. 2015, 145, 23–39. [Google Scholar] [CrossRef]
- Kistiakowsky, G.B.; Van Artsdalen, E.R. Bromination of Hydrocarbons. I. Photochemical and Thermal Bromination of Methane and Methyl Bromine. Carbon-Hydrogen Bond Strength in Methane. J. Chem. Phys. 1944, 12, 469–478. [Google Scholar] [CrossRef]
- Lorkovic, I.M.; Sun, S.; Gadewar, S.; Breed, A.; Macala, G.S.; Sardar, A.; Cross, S.E.; Sherman, J.H.; Stucky, G.D.; Ford, P.C. Alkane bromination revisited: “reproportionation” in gas-phase methane bromination leads to higher selectivity for CH3Br at moderate temperatures. J. Phys. Chem. A 2006, 110, 8695–8700. [Google Scholar] [CrossRef] [PubMed]
- McBee, E.T.; Hass, H.B.; Neher, C.M.; Strickland, H. Chlorination of methane. Ind. Eng. Chem. 1942, 34, 296–300. [Google Scholar] [CrossRef]
- Golden, D.M.; Walsh, R.; Benson, S.W. The Thermochemistry of the Gas Phase Equilibrium I2 + CH4 ↔ CH3I + HI and the Heat of Formation of the Methyl Radical1. J. Am. Chem. Soc. 1965, 87, 4053–4057. [Google Scholar] [CrossRef]
- Pease, R.N.; Walz, G.F. Kinetics of the thermal chlorination of methane. J. Am. Chem. Soc. 1931, 53, 3728–3737. [Google Scholar] [CrossRef]
- Rashid, H.U.R.; Yu, K.; Zhou, J. Advances in the Development of Methane Bromination. J. Chem. Soc. Pak. 2011, 33, 922–928. [Google Scholar]
- Aglulin, A.G.; Bakshi, Y.M. Kinetic studies of the mechanism of direct chlorination of methane in the presence of porous fillers. Kinet. Catal. 1983, 24, 80–86. [Google Scholar]
- Pritchard, H.O.; Pyke, J.B.; Trotman-Dickenson, A.F. The study of Chlorine Atom Reactions in the Gas Phase. J. Am. Chem. Soc. 1955, 77, 2629–2633. [Google Scholar] [CrossRef]
- Gordon, A.R.; Barnes, C. The Free Energy, Entropy and Heat Capacity of Bromine and of Hydrogen Bromide from Spectroscopic Data. J. Chem. Phys. 1933, 1, 692–696. [Google Scholar] [CrossRef]
- Degirmenci, V.; Uner, D.; Yilmaz, A. Methane to higher hydrocarbons via halogenation. Catal. Today 2005, 106, 252–255. [Google Scholar] [CrossRef]
- Arai, K.K.Z.; Yoshida, T.M.; Shinoda, K. Rate of consumption of chlorine in thermal chlorination of methane. Kogyo Kagaku Zassi 1958, 61, 1231–1233. [Google Scholar] [CrossRef]
- Rozanov, V.N.; Treger, Y.A. Kinetics of the Gas Phase Thermal Chlorination of Methane. Kinet. Catal. 2010, 51, 661–669. [Google Scholar] [CrossRef]
- Tirtowidjo, M. Fundamental Kinetic Modeling of Industrial Reactors. In Proceedings of the AIChE Annual Meeting, Los Angeles, CA, USA, 16–21 November 1997. Paper 78b. [Google Scholar]
- Shah, J.J.; Fox, R.O. Computational Fluid Dynamics Simulation of Chemical Reactors: Application of in Situ Adaptive Tabulation to Methane Thermochlorination Chemistry. Ind. Eng. Chem. Res. 1999, 38, 4200–4212. [Google Scholar] [CrossRef]
- Raman, V.; Fox, R.O.; Harvey, A.D.; West, D.H. CFD Analysis of Premixed Methane Chlorination Reactors with Detailed Chemistry. Ind. Eng. Chem. Res. 2001, 40, 5170–5176. [Google Scholar] [CrossRef]
- Upham, D.C.; Kristoffersen, H.H.; Snodgrass, Z.R.; Gordon, M.J.; Metiu, H.; Mcfarland, E.W. Bromine and iodine for selective partial oxidation of propane and methane. Appl. Catal. A Gen. 2019, 580, 102–110. [Google Scholar] [CrossRef]
- Larsen, P.; Ulin, J.; Dahlstrøm, K.; Jensen, M. Synthesis of [11C] iodomethane by iodination of [11C] methane. Appl. Radiat. Isot. 1997, 48, 153–157. [Google Scholar] [CrossRef]
- Olah, G.A.; Gupta, B.; Felberg, J.D.; Ip, W.M.; Husain, A.; Karpeles, R.; Lammertsma, K.; Melhotra, A.K.; Trivedi, N.J. Electrophilic reactions at single bonds. 20. Selective monohalogenation of methane over supported acidic or platinum metal catalysts and hydrolysis of methyl halides over gamma-alumina-supported metal oxide/hydroxide catalysts. A feasible path for the oxidative conversion of methane into methyl alcohol/dimethyl ether. J. Am. Chem. Soc. 1985, 107, 7097–7105. [Google Scholar] [CrossRef]
- Gorin, E.; Fontana, C.M.; Kidder, G.A. Chlorination of Methane with Copper Chloride Melts Rate of Chlorination. Ind. Eng. Chem. 1948, 40, 2128–2134. [Google Scholar] [CrossRef]
- Paunović, V.; Pérez-Ramírez, J. Catalytic halogenation of methane: A dream reaction with practical scope? Catal. Sci. Technol. 2019, 9, 4515–4530. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, J.; Kim, H.W.; Kim, T.-W.; Kim, H.J.; Chang, H.; Park, M.B.; Chae, H.-J. Sulfated Tin Oxide as Highly Selective Catalyst for the Chlorination of Methane to Methyl Chloride. ACS Catal. 2019, 9, 9398–9410. [Google Scholar] [CrossRef]
- Degirmenci, V.; Yilmaz, A.; Uner, D. Selective methane bromination over sulfated zirconia in SBA-15 catalysts. Catal. Today. 2009, 142, 30–33. [Google Scholar] [CrossRef]
- Bucsi, I.; Olah, G.A. Selective monochlorination of methane over solid acid and zeolite catalysts. Catal. Lett. 1992, 16, 27–38. [Google Scholar] [CrossRef]
- Joo, H.; Kim, D.; Lim, K.S.; Choi, Y.N.; Na, K. Selective methane chlorination to methyl chloride by zeolite Y-based catalysts. Solid State Sci. 2018, 77, 74–80. [Google Scholar] [CrossRef]
- Kwon, S.; Chae, H.-J.; Na, K. Control of methane chlorination with molecular chlorine gas using zeolite catalysts: Effects of Si/Al ratio and framework type. Catal. Today 2020. [Google Scholar] [CrossRef]
- Paunovic, V.; Mitchell, S.; Verel, R.; Lee, S.S.; Pérez-Ramírez, J. Aluminum Redistribution in ZSM-5 Zeolite upon Interaction with Gaseous Halogens and Hydrogen Halides and Implications in Catalysis. J. Phys. Chem. C 2020, 124, 722–733. [Google Scholar] [CrossRef]
- Batamack, P.T.D.; Mathew, T.; Prakash, G.K.S. One-Pot Conversion of Methane to Light Olefins or Higher Hydrocarbons through H-SAPO-34-Catalyzed in Situ Halogenation. J. Am. Chem. Soc. 2017, 139, 18078–18083. [Google Scholar] [CrossRef]
- Centi, G.; Barbera, K.; Perathoner, S.; Gupta, N.K. Onion-Like Graphene Carbon Nanospheres as Stable Catalysts for Carbon Monoxide and Methane Chlorination. ChemCatChem 2015, 7, 3036–3046. [Google Scholar] [CrossRef]
- Paunović, V.; Zichittella, G.; Moser, M.; Amrute, A.P.; Pérez-Ramírez, J. Catalyst design for natural-gas upgrading through oxybromination chemistry. Nat. Chem. 2016, 8, 803. [Google Scholar] [CrossRef]
- Paunović, V.; Artusi, M.; Verel, R.; Krumeich, F.; Hauert, R.; Pérez-Ramírez, J. Lanthanum vanadate catalysts for selective and stable methane oxybromination. J. Catal. 2018, 363, 69–80. [Google Scholar] [CrossRef]
- Paunović, V.; Zichittella, G.; Hemberger, P.; Bodi, A.; Pérez-Ramírez, J. Selective Methane Functionalization via Oxyhalogenation over Supported Noble Metal Nanoparticles. ACS Catal. 2019, 9, 1710–1725. [Google Scholar] [CrossRef]
- Lin, R.; Ding, Y.; Gong, L.; Dong, W.; Wang, J.; Zhang, T. Efficient and stable silica-supported iron phosphate catalysts for oxidative bromination of methane. J. Catal. 2010, 272, 65–73. [Google Scholar] [CrossRef]
- Wang, R.; Lin, R.; Ding, Y. Model Iron Phosphate Catalysts for the Oxy-bromination of Methane. Catal. Lett. 2014, 1384–1392. [Google Scholar] [CrossRef]
- Wang, R.; Lin, R.; Ding, Y.; Liu, J.; Wang, J.; Zhang, T. Structure and phase analysis of one-pot hydrothermally synthesized FePO4-SBA-15 as an extremely stable catalyst for harsh oxy-bromination of methane. Appl. Catal. A Gen. 2013, 453, 235–243. [Google Scholar] [CrossRef]
- Yang, F.; Liu, Z.; Li, W.S.; Zhou, X.P. The Oxidative Bromination of Methane Over Rh/SiO2 Catalyst. Catal. Lett. 2008, 226–232. [Google Scholar] [CrossRef]
- Liu, Z.; Li, W.; Zhou, X. Product oriented oxidative bromination of methane over Rh/SiO2 catalysts. J. Nat. Gas Chem. 2010, 19, 522–529. [Google Scholar] [CrossRef]
- Lin, R.; Ding, Y.; Gong, L.; Dong, W.; Chen, W.; Lu, Y. Studies on oxy-bromination of methane and coke deposition over FePO4/SiO2 catalysts. Catal. Today 2011, 164, 34–39. [Google Scholar] [CrossRef]
- Wang, K.X.; Xu, H.F.; Li, W.S.; Zhou, X.P. Acetic acid synthesis from methane by non-synthesis gas process. J. Mol. Cat. A Chem. 2005, 1, 65–69. [Google Scholar] [CrossRef]
- Lin, R.; Ding, Y.; Gong, L.; Li, J.; Chen, W.; Yan, L.; Lu, Y. Oxidative bromination of methane on silica-supported non-noble metal oxide catalysts. Appl. Catal. A Gen. 2009, 353, 87–92. [Google Scholar] [CrossRef]
- Wang, P.; Chen, L.; Shen, S.; Au, C.-T.; Yin, S.-F. Methane oxybromination over Rh-based catalysts: Effect of supports. Chin. J. Chem. Eng. 2019. [Google Scholar] [CrossRef]
- Zichittella, G.; Paunovic, V.; Amrute, A.P.; Pe, J. Catalytic Oxychlorination versus Oxybromination for Methane Functionalization. ACS Catal. 2017. [Google Scholar] [CrossRef]
- Pieters, W.J.M.; Conner Jr, W.C.; Carlson, E.J. The oxyhydrochlorination of methane on fumed silica-based Cu + 1, K., La catalysts: I. Catalyst synthesis. Appl. Catal. 1984, 11, 35–48. [Google Scholar] [CrossRef]
- Conner Jr, W.C.; Pieters, W.J.M.; Signorelli, A.J. The oxyhydrochlorination of methane on fumed silica-based Cu, K., La catalysts: III. Bulk & surface analysis. Appl. Catal. 1984, 11, 59–71. [Google Scholar] [CrossRef]
- Conner, W.C., Jr.; Pieters, W.J.M.; Gates, W.; Wilkalis, J.E. The oxyhydrochlorination of methane on fumed silica—Based Cu+1, K., La catalysts: II. Gas phase stoichiometry. Appl. Catal. 1984, 11, 49–58. [Google Scholar] [CrossRef]
- Garcia, C.L.; Resasco, D.E. Effects of the support and the addition of a second promoter on potassium chloride-copper (II) chloride catalysts used in the oxychlorination of methane. Appl. Catal. 1989, 46, 251–267. [Google Scholar] [CrossRef]
- Garcia, C.L.; Resasco, D.E. High-temperature oxychlorination catalysts: Role of LaCl3 as an inhibitor of the segregation of active species during heating/cooling cycles. J. Catal. 1990, 122, 151–165. [Google Scholar] [CrossRef]
- Peringer, E.; Tejuja, C.; Salzinger, M.; Lemonidou, A.A.; Lercher, J.A. On the synthesis of LaCl3 catalysts for oxidative chlorination of methane. Appl. Catal. A Gen. 2008, 350, 178–185. [Google Scholar] [CrossRef]
- Peringer, E.; Podkolzin, S.G.; Jones, M.E.; Olindo, R.; Lercher, J.A. LaCl3 -based catalysts for oxidative chlorination of CH4. Top. Catal. 2006, 38, 211–220. [Google Scholar] [CrossRef]
- Peringer, E.; Salzinger, M.; Hutt, M.; Lemonidou, A.A.; Lercher, J.A. Modified Lanthanum Catalysts for Oxidative Chlorination of Methane. Top. Catal. 2009, 1220–1231. [Google Scholar] [CrossRef]
- Podkolzin, S.G.; Stangland, E.E.; Jones, M.E.; Peringer, E.; Lercher, J.A. Methyl Chloride Production from Methane over Lanthanum-Based Catalysts. J. Am. Chem. Soc. 2007, 2569–2576. [Google Scholar] [CrossRef]
- Huang, J.; Wang, W.; Li, D.; Xu, S.; Liu, Q.; Chen, X.; Fei, Z.; Zhang, Z. Facile construction of non-crystalline ZrO2 as an active yet durable catalyst for methane oxychlorination. J. Sol-Gel Sci. Technol. 2019, 163–172. [Google Scholar] [CrossRef]
- Zhou, X.; Yilmaz, A.; Yilmaz, G.A.; Lorkovic, I.M.; Laverman, L.E.; Weiss, M.; Sherman, J.H.; McFarland, E.W.; Stucky, G.D.; Ford, P.C. An integrated process for partial oxidation of alkanes. Chem. Commun. 2003, 3, 2294–2295. [Google Scholar] [CrossRef] [PubMed]
- Lorkovic, I.M.; Noy, M.L.; Schenck, W.A.; Weiss, M.; Sherman, J.H.; McFarland, E.W.; Stucky, G.D.; Ford, P.C. C1 coupling via bromine activation and tandem catalytic condensation and neutralization over CaO/zeolite composites. Chem. Commun. 2004, 49, 566–567. [Google Scholar] [CrossRef] [PubMed]
- Lorkovic, I.M.; Noy, M.L.; Schenck, W.A.; Belon, C.; Weiss, M.; Sun, S.; Sherman, J.H.; McFarland, E.W.; Stucky, G.D.; Ford, P.C. C1 oxidative coupling via bromine activation and tandem catalytic condensation and neutralization over CaO/zeolite composites: II. Product distribution variation and full bromine confinement. Catal. Today 2004, 98, 589–594. [Google Scholar] [CrossRef]
- Lorkovic, I.M.; Yilmaz, A.; Yilmaz, G.A.; Zhou, X.; Laverman, L.E.; Sun, S.; Schaefer, D.J.; Weiss, M.; Noy, M.L.; Cutler, C.I.; et al. A novel integrated process for the functionalization of methane and ethane: Bromine as mediator. Catal. Today 2004, 98, 317–322. [Google Scholar] [CrossRef]
- You, Q.; Liu, Z.; Li, W.; Zhou, X. Synthesis of dimethyl ether from methane mediated by HBr. J. Nat. Gas Chem. 2009, 18, 306–311. [Google Scholar] [CrossRef]
- Xu, H.F.; Wang, K.X.; Li, W.S.; Zhou, X.P. Dimethyl ether synthesis from methane by non syngas process. Catal. Lett. 2005, 100, 53–57. [Google Scholar] [CrossRef]
- Wang, K.X.; Xu, H.F.; Li, W.S.; Au, C.T.; Zhou, X.P. The synthesis of acetic acid from methane via oxidative bromination, carbonylation, and hydrolysis. Appl. Catal. A Gen. 2006, 304, 168–177. [Google Scholar] [CrossRef]
- Marquaire, P.-M.; Al Kazzaz, M.; Muller, Y.; Saint Just, J. Methane to vinyl chloride by “Chloro-pyrolysis” of methyl chloride. Stud. Surf. Sci. Catal. 1997, 107, 269–274. [Google Scholar] [CrossRef]
- Shalygin, A.; Paukshtis, E.; Kovalyov, E.; Bal’zhinimaev, B. Light olefins synthesis from C1-C2 paraffins via oxychlorination processes. Front. Chem. Sci. Eng. 2013, 7, 279–288. [Google Scholar] [CrossRef]
- Osterwalder, N.; Stark, W.J. Direct Coupling of Bromine-Mediated Methane Activation and Carbon-Deposit Gasification. ChemPhysChem 2007, 8, 297–303. [Google Scholar] [CrossRef]
- Taylor, C.E.; Noceti, R.P.; Schehl, R.R. Direct conversion of methane to liquid hydrocarbons through chlorocarbon intermediates. Stud. Surf. Sci. Catal. 1988, 483–489. [Google Scholar] [CrossRef]
- Yin, L.; Lu, G. A DFT + U study on the oxidative chlorination of CH4 at ceria: The role of HCl. Catal. Sci. Technol. 2017, 2498–2505. [Google Scholar] [CrossRef]
- Litvinenko, S.L.; Rudakov, E.S. DFT analysis of the mechanism for the gas-phase chlorination of methane in the HOCl–H2O system. Theor. Exp. Chem. 2012, 48, 212–216. [Google Scholar] [CrossRef]
- Zhang, Q. Methanesulfonyl Chloride (CH3SO2Cl) Decomposition as a Key Step for Low-Temperature Methane Conversions. Chem. Eng. Technol. 2017, 40, 656–662. [Google Scholar] [CrossRef]
- Paunović, V.; Hemberger, P.; Bodi, A.; López, N.; Pérez-Ramírez, J. Evidence of radical chemistry in catalytic methane oxybromination. Nat. Catal. 2018, 1, 363–370. [Google Scholar] [CrossRef]
- Chistyakov, A.L.; Stankevich, I.V.; Gambaryan, N.P.; Akhrem, I.S. Nucleophilic Assistance in Methane Activation by Superelectrophiles with Halogen-Centered Cationic Sites. Russ. J. Org. Chem. 2006, 42, 1606–1614. [Google Scholar] [CrossRef]
- Al-Douri, A.; Sengupta, D.; El-Halwagi, M.M. Shale gas monetization: A review of downstream processing to chemicals and fuels. J. Nat. Gas Sci. Eng. 2017, 45, 436–455. [Google Scholar] [CrossRef]
- Wood, D.A.; Nwaoha, C.; Towler, B.F. Gas-to-liquids (GTL): A review of an industry offering several routes for monetizing natural gas. J. Nat. Gas Sci. Eng. 2012, 9, 196–208. [Google Scholar] [CrossRef]
- Olsbye, U.; Svelle, S.; Bjørgen, M.; Beato, P.; Janssens, T.V.W.; Joensen, F.; Bordiga, S.; Lillerud, K.P. Conversion of Methanol to Hydrocarbons: How Zeolite Cavity and Pore Size Controls Product Selectivity. Angewandte 2012, 51, 5810–5831. [Google Scholar] [CrossRef]
- Wang, B.; Albarracín-Suazo, S.; Pagán-torres, Y.; Nikolla, E. Advances in methane conversion processes. Catal. Today 2017, 285, 147–158. [Google Scholar] [CrossRef]
- Lombard, C.; Marquaire, P.-M. New study of methane to vinyl chloride process. Stud. Surf. Sci. Catal. 2004, 147, 529–534. [Google Scholar] [CrossRef]
- Ding, K.; Metiu, H.; Stucky, G.D. The Selective High-Yield Conversion of Methane Using Iodine-Catalyzed Methane Bromination. ACS Catal. 2013, 3, 474–477. [Google Scholar] [CrossRef]
- Rebordinos, J.G.; Salten, A.H.J.; Agar, D.W. BrOx cycle: A novel process for CO2 -free energy production from natural gas. Int. J. Hydrogen Energy 2017, 42, 4710–4720. [Google Scholar] [CrossRef]
- Gary, J.H.; Handwerk, G.E.; Kaiser, M.J.; Geddes, D. Petroleum Refining—Technology and Economics, 5th ed.; Taylor & Francis Group: Oxfordshire, UK, 2007. [Google Scholar] [CrossRef]
- Van Iersel, M.M.; Van Schilt, M.A.; Benes, N.E.; Keurentjes, J.T.F. Ultrasonics Sonochemistry Controlled methyl chloride synthesis at mild conditions using ultrasound irradiation. Ultrason. Sonochem. 2010, 17, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Okumoto, M.; Mizuno, A. Conversion of methane for higher hydrocarbon fuel synthesis using pulsed discharge plasma method. Catal. Today 2001, 71, 211–217. [Google Scholar] [CrossRef]
- Batamack, P.; Bucsi, I.; Molnar, A.; Olah, G.A. Electrophilic chlorination of methane over superacidic sulfated zirconia. Catal. Lett. 1994, 25, 11–19. [Google Scholar] [CrossRef]
- Vilenchich, R.; Hodgins, J.W. Gamma Initiated Iodination of Methane in the Gas Phase. Can. J. Chem. Eng. 1970, 48, 588–590. [Google Scholar] [CrossRef]
- Cabrera, A.E.C.M.I.; Alfano, O.M. Product yield and selectivity studies in photoreactor design. Theory and experiments for the chlorination of methane. Chem. Eng. Sci. 1990, 45, 2439–2446. [Google Scholar] [CrossRef]
- Liebov, N.S.; Goldberg, J.M.; Boaz, N.C.; Coutard, N. Selective Photo-Oxygenation of Light Alkanes Using Iodine Oxides and Chloride. ChemCatChem 2019, 11, 5045–5054. [Google Scholar] [CrossRef]
- Bilke, M.; Losch, P.; Vozniuk, O.; Bodach, A.; Schuth, F. Methane to Chloromethane by Mechanochemical Activation: A Selective Radical Pathway. J. Am. Chem. Soc. 2019, 141, 11212–11218. [Google Scholar] [CrossRef]
- Rossberg, M.; Wilhelm, P.; Gerhard, P.; Tögel, A.; Theodore, R.T.; Klaus, K.B. Ullman’s Encyclopedia of Industrial Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2012; Volume 9, pp. 15–39. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
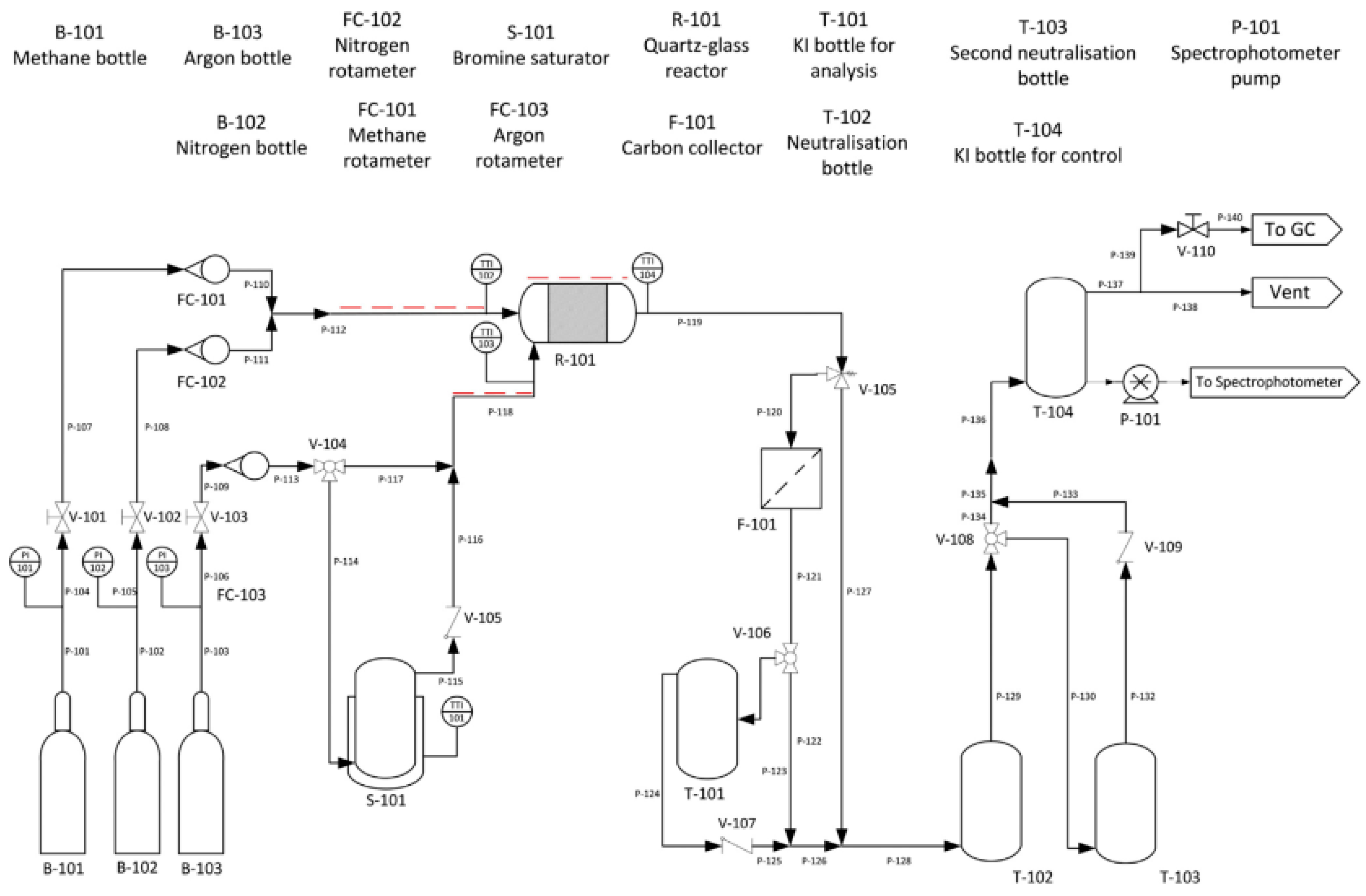{kind=link}
| Methane oxidative bromination results using the Ru/SiO2 catalyst | |||||||
| T (°C) | CH4 (mL/min) | O2 (mL/min) | CH4 conversion (%) | Selectivity (%) | |||
| CH3Br | CH2Br2 | CO | |||||
| 530 | 5 | 15 | 27.6 | 80 | 1.9 | 18.2 | |
| 530 | 5 | 20 | 24.4 | 89.4 | 1.7 | 8.8 | |
| 560 | 5 | 20 | 31.8 | 78.8 | 2.3 | 18.9 | |
| Methanol and DME synthesis over RuCl3 catalyst | |||||||
| T (°C) | H2O (g) | CH3Br (g) | CH3Br conversion (%) | Selectivity (%) | |||
| DME | CH3OH | ||||||
| 150 | 0.7 | 0.5 | 66.1 | 22.2 | 77.8 | ||
| 170 | 0.7 | 0.5 | 83.2 | 55 | 45 | ||
| 180 | 0.7 | 0.5 | 97 | 58.7 | 41.3 | ||
| 180 | 0.4 | 0.5 | 60.5 | 43.7 | 56.2 | ||
| 180 | 0.6 | 0.5 | 87 | 62.7 | 37.3 | ||
| 180 | 0.7 | 0.5 | 71.8 | 48.7 | 51.3 | ||
| 180 | 0.7 | 0.5 | 98.3 | 68.9 | 31 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bajec, D.; Grom, M.; Lašič Jurković, D.; Kostyniuk, A.; Huš, M.; Grilc, M.; Likozar, B.; Pohar, A. A Review of Methane Activation Reactions by Halogenation: Catalysis, Mechanism, Kinetics, Modeling, and Reactors. Processes 2020, 8, 443. https://doi.org/10.3390/pr8040443
Bajec D, Grom M, Lašič Jurković D, Kostyniuk A, Huš M, Grilc M, Likozar B, Pohar A. A Review of Methane Activation Reactions by Halogenation: Catalysis, Mechanism, Kinetics, Modeling, and Reactors. Processes. 2020; 8(4):443. https://doi.org/10.3390/pr8040443
Chicago/Turabian StyleBajec, David, Matic Grom, Damjan Lašič Jurković, Andrii Kostyniuk, Matej Huš, Miha Grilc, Blaž Likozar, and Andrej Pohar. 2020. "A Review of Methane Activation Reactions by Halogenation: Catalysis, Mechanism, Kinetics, Modeling, and Reactors" Processes 8, no. 4: 443. https://doi.org/10.3390/pr8040443
APA StyleBajec, D., Grom, M., Lašič Jurković, D., Kostyniuk, A., Huš, M., Grilc, M., Likozar, B., & Pohar, A. (2020). A Review of Methane Activation Reactions by Halogenation: Catalysis, Mechanism, Kinetics, Modeling, and Reactors. Processes, 8(4), 443. https://doi.org/10.3390/pr8040443