Abstract
Poor aqueous solubility limits the therapeutic efficacy of many marketed and investigational drugs. Synthesis of new drugs with improved solubility is challenging due to time constraint and expenses involved. Therefore, finding the solubility enhancers for existing drugs is an attractive and profitable strategy. In this study, PEGylated oleic acid (OA-mPEG5000), a conjugate of oleic acid and mPEG5000 was synthesized and evaluated as a solubilizer for furosemide. OA-mPEG5000 was evaluated as a nanocarrier for furosemide by formulating polymersomes. Solubility of furosemide in milli-Q water and aqueous OA-mPEG5000 solution was determined using shake flask method. At 37 °C, the solubility of furosemide in OA-mPEG5000 (1% w/w) and milli-Q water was 3404.7 ± 254.6 µg/mL and 1020.2 ± 40.9 µg/mL, respectively. Results showed there was a 3.34-fold increase in solubility of furosemide in OA-mPEG5000 compared to water at 37 °C. At 25 °C, there was a 3.31-fold increase in solubilization of furosemide in OA-mPEG5000 (1% w/w) (90.0 ± 1.45 µg/mL) compared to milli-Q water (27.2 ± 1.43 µg/mL). Size, polydispersity index and zeta potential of polymersomes ranged from 85–145.5 nm, 0.187–0.511 and −4.0–12.77 mV, respectively. In-vitro release study revealed a burst release (71%) within 1 h. Significant enhancement in solubility and formation of polymersomes suggested that OA-mPEG5000 could be a good solubilizer and nanocarrier for furosemide.
Keywords:
oleic acid; mPEG; furosemide; OA-mPEG5000; solubility; polymersomes; BCS class IV; PEGylated oleic acid 1. Introduction
All active pharmaceutical ingredients (APIs) irrespective of the mode of drug administration should have at least limited aqueous solubility for therapeutic efficacy as water is the major component in all body fluids [1]. Poor aqueous solubility of APIs leads to suboptimal patient outcomes due to poor oral bioavailability and variable pharmacokinetics [2,3,4]. The maximum amount of a substance that will dissolve in a given amount of solvent at a specified temperature is known as solubility and is a characteristic property of a specific solute–solvent combination [2,5,6]. Each API differs in terms of solubility profile [1,5]. Nearly 40% of drugs in the market and about 90% of molecules in the discovery pipeline have poor water solubility [7,8]. According to the Biopharmaceutics Classification (BCS) system, an increasing percentage of drug molecules under development by pharmaceutical industries can be classified as Class II compounds, compounds with high permeability but poor solubility. Also, a significant percentage of molecules under investigation have poor solubility and permeability (BCS Class IV) [4]. Therefore, improving the solubility of BCS Class II and Class IV APIs can enhance the therapeutic efficacy, and it is a promising area of research in pharmaceutical industry. Formulation strategies such as encapsulating powder in capsules and conventional tablets cannot address solubility and permeability issues [4]. Various solubilization techniques are used to improve the solubility and bioavailability of poorly soluble drugs. Some of the solubilization techniques include: (i) modifying pH and salt formation; (ii) conversion into amorphous forms; (iii) making solid dispersions; (iv) Co-crystallization; (v) polymeric micelles formulation; (vi) solid lipid nanoparticles formulation; (vii) inclusion complexation; (iii) size reduction and nanonization; (ix) liposomes and pro-liposomes formulation; and (x) microemulsions and self-emulsifying drug delivery systems formulation [7].
The development of novel formulations for existing drugs is a good strategy for pharmaceutical industry as the cost associated with drug discovery and development for new molecules is substantially high [6]. As the majority of the API’s available in the market have solubility issues, filing new drug applications (NDA) under 505(b)(2) for formulations with rapid dissolution and enhanced bioavailability is advantageous for the pharmaceutical industry [7,9].
There are several molecules currently being used in clinics which fall under BCS Class IV. Examples of BCS class IV drugs include Acetazolamide, amphotericin B, paclitaxel, ritonavir, and furosemide [10]. Chemically, furosemide is 5-(aminosulfonyl)-4-chloro-2-((2-furanylmethyl)amino) benzoic acid (Figure 1) [11] and is a loop diuretic used for treating edema associated with cardiovascular diseases such as heart failure, infarction and hypertension, liver disease, and nephrotic syndrome [11,12,13,14]. Reported pKa1 and pKa2 of furosemide are 3.8 (carboxylic acid) and 7.5 (amino) [15], respectively with log P (n-octanol/water) values of 2.29 [16] and 1.81 [17].
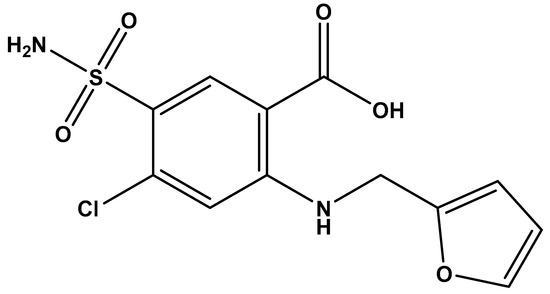
Figure 1.
Chemical structure of furosemide. Reproduced with permission from Kalhapure, International Journal of Pharmaceutics; published by Elsevier. 2012 [11].
Literature reports that aqueous solubility of furosemide varies from 0.01825 mg/mL [18] to 0.04 mg/mL [19] and it has extremely variable bioavailability due to its low solubility [20,21]. Despite suboptimal biopharmaceutical properties, furosemide is the most commonly prescribed diuretic [22]. Several formulations such as solid dispersion [21,23,24], self micro-emulsifying drug delivery systems, nanosuspension [14,25], micelles [19], solid lipid nanoparticles [26], polymeric microcontainers [27], and nano-cocrystal [28] have been proposed for enhancing the dissolution, solubility and bioavailability of furosemide. Among all the formulation strategies, amphiphilic polymers for the enhancement of solubility is a promising strategy as these polymers can form nano-carriers. Polymeric micelles were proposed for the first time as possible drug carriers in 1984 [29]. Since then, they have emerged as promising carriers for poorly water-soluble drugs [30]. Polymeric micelles from poly (ethylene glycol)-b-poly(e-caprolactone-co-trimethylene carbonate) [19,31] and diblock copolymers of ethylene oxide and styrene oxide [32] have been evaluated as solubilizers for furosemide. There has been continuous effort to explore new polymers and excipients for enhancing the solubility of furosemide. OA-mPEG5000 is a pegylated oleic acid prepared by conjugation of oleic acid with monomethoxy polyethylene glycol (mPEG) (MW 5000). This polymer can form polymersomes and has proven to be a promising nanocarrier for enhancing antibacterial activity of vancomycin [33]. Due to the 18-fold higher molecular weight of hydrophilic segment (mPEG) than hydrophobic segment (OA), this polymer can self-assemble in water to form polymersomes [33]. Amphiphilic polymers forming polymersomes could enhance aqueous solubility of poorly soluble drugs. OA-mPEG5000 polymer was synthesized using a one step process and has been made up of biocompatible materials such as oleic acid and polyethylene glycol. Despite its interesting properties such as easy synthesis and non-toxicity to mammalian cells, it has not been studied extensively for pharmaceutical applications such as drug carrier and solubilizer. Therefore, we evaluated the potential of recently reported polymer, OA-mPEG5000, as a solubilizer and nanocarrier for furosemide. To the best of our knowledge, this is the first report on the applicability of OA-mPEG5000 as a solubility enhancer and polymersome-forming material for furosemide.
2. Materials and Methods
2.1. Materials
Furosemide was purchased from TCI America (Portland, OR, USA) and oleic acid from Alfa Aesar (Ward Hill, MA, USA). Monomethoxy polyethylene glycol (mPEG) (MW 5000), diethyl ether and tetrahydrofuran (THF; 99.9% pure) were purchased from Sigma Aldrich (St. Louis, MO, USA), Fisher Chemicals (Fairlawn, NJ, USA) and Acros Organics (Fairlawn, NJ, USA), respectively. An in-house Elix water purification system (Millipore Corp., Burlington, MA, USA) was used to obtain milli-Q water (resistivity: 18.2 MΩ).
2.2. Methods
2.2.1. Synthesis of Pegylated Oleic Acid (OA-mPEG5000)
A literature reported procedure with some minor modifications was followed for the synthesis of OA-mPEG5000 polymer [33]. OA (16 g, 56.3 mmoL) and mPEG (10 g, 2.0 mmoL) were mixed together in a 100 mL round bottom flask and heated in an oil bath at 170 °C for 5 h under inert atmosphere. The reaction mixture was allowed to cool to 25 °C and was then added to 200 mL cold diethyl ether. This diethyl layer was centrifuged at 10,000 rpm at 20 °C for 20 min and supernatant was decanted to remove the solvent and excess of oleic acid. Isolated solid was again dispersed in fresh ice-cold diethyl ether and re-centrifuged. This procedure was repeated three times to ensure complete removal of unreacted excess oleic acid. Obtained solid white mass was then dried in a vacuum desiccator for 24 h to obtain OA-mPEG5000 as off-white solid (9.7 g, 91.8%). The conjugate was extensively characterized using Fourier-transform infrared spectroscopy (FT-IR) and nuclear magnetic resonance (NMR) imaging (1H and 13C) [33].
FT-IR: 2881.45, 1733.27, 1341.51, 1103.23, 958.58, 842.23 cm−1. 0.82 (t; 3H; —CH3), 1.19–1.23 (m; 22H; —CH2—), 1.55 (q; 2H; —CH2CH2COO—), 1.94 (m; 4H; —CH2—CH=CH—CH2—), 2.25 (t; 2H; —CH2CO—), 3.31 (s; 3H; —OCH3), 3.38–3.49 (m; 224H; —OCH2—CH2O—), 3.75 (t; 2H; —CH2COOCH2CH2—) 4.15 (t; 2H; —CH2COOCCH2—), 5.26 (m; 2H; —CH=CH—). 13C NMR (CDCl3) δ (ppm): 14.10, 22.65, 24.79, 24.87, 27.15, 29.09, 29.29, 31.84, 59.00, 57.12, 66.89, 70.53, 71.90, 129.73, 173.81 [33].
2.2.2. Solubility Study
Saturation solubility of furosemide in OA-mPEG5000 was determined at 25° and 37 °C using a shake flask method. Excess furosemide was mixed with aqueous solution of OA-mPEG5000 (1% w/w) and kept at constant temperature for 6 days in a shaking water bath rotating at 100 rpm. The insolubilized drug was filtered through 0.45 μm membrane filter (Millipore, Burlington, MA, USA), and the clear liquid obtained was analyzed for furosemide content at a λmax of 277 nm [14] using a UV-spectrophotometer (Shimadzu UV 1800, Kyoto, Japan). The regression equation for the calibration curve was y = 0.0573x + 0.0678 with a high R2 value of 0.994. Blank experiments were conducted using milli-Q water without OA-mPEG5000 to obtain aqueous solubility of furosemide. All measurements were performed in triplicate.
2.2.3. Formulation of Polymersomes
Polymersomes were formulated to evaluate the ability of OA-mPEG5000 as a nanocarrier for furosemide, a BCS class IV molecule. Polymersomes were prepared using an o/w emulsion solvent evaporation method [33]. A solution of furosemide (10 mg) in THF (5 mL) was added dropwise to the aqueous solution of OA-mPEG5000 (100 mg) in milli-Q water (20 mL) under stirring. The formed emulsion was stirred for 24 h at room temperature to ensure the complete evaporation of the THF.
2.2.4. Dynamic Light Scattering
Dynamic light scattering technique was used to measure the size, PI and ZP of furosemide loaded polymersomes. ZP was determined by measuring the electrophoretic mobility. Polymersomes formulation (200 µL) was diluted in 10 mL of milli-Q water and size; PDI and ZP were determined using a Zetasizer Nano ZS90 (Malvern Instruments Ltd., Malvern, UK) equipped with a laser beam at 633 nm and 173° scattering angle.
2.2.5. Determination of Encapsulation Efficiency (%EE)
%EE was determined by using ultrafiltration method using Amicon® Ultra-4 centrifugal filter tubes (Millipore Corp., USA) of 10 kDa pore size [34,35,36]. Polymersomes (2 mL) were placed in ultracentrifugal filter tube and centrifuged at 10,000 rpm for 20 min at 20 °C to separate unentrapped furosemide. The amount of free furosemide in the filtrate was determined using UV spectrophotometer at 277 nm (Shimadzu UV 1800, Kyoto, Japan). The %EE was calculated using the following equation:
%EE = (weight of furosemide in polymersomes/weight of furosemide added) × 100%
2.2.6. X-ray Diffraction
PANanlytical’s X-ray diffractometer (PANanlytical’s X’pert Pro Tokyo, Japan) equipped with X’Celerator high speed detector was used to determine crystalline properties of furosemide and furosemide polymersomes. The samples were placed on an aluminum sample holder and uniformly packed with a glass slide. CuKα was used as radiation source and operated at 45 kV and 40 mA. All scans were recorded with a continuous scanning mode over a 2θ range of 5° to 70°.
2.2.7. In Vitro Release Study
The in-vitro furosemide release studies from polymersomes were performed using dialysis bag technique. Furosemide polymersomes and their respective blanks (2 mL) were loaded in dialysis bags (pore size: 12,400 MWCO) and dialyzed against PBS of pH 7.4 (30 mL) at 37 °C in a shaking water bath at 100 rpm. Samples (3 mL) were withdrawn from the receiver solution at predetermined time intervals, and an equal amount of fresh PBS was added to maintain sink conditions. The furosemide quantity in each sample was measured spectrophotometrically at 277 nm using blank polymersomes as a reference.
2.2.8. Statistical Analysis
Data was statistically analyzed using non-parametric t-test using GraphPad Prism® (Graph Pad Software Inc., Version 5, San Diego, CA, USA). A p-value < 0.05 was considered statistically significant. All data are presented as mean ± SD.
3. Results and Discussion
3.1. Solubility Study
Log solubility of furosemide at 25 °C and 37 °C in OA-mPEG5000 and milli-Q water are presented in Figure 2. Furosemide solubility was significantly higher at 37 °C compared to 25 °C in both OA-mPEG5000 (1% w/w) and milli-Q water. At 37 °C, the solubility of furosemide in OA-mPEG5000 (1% w/w) and milli-Q water was 3404.7 ± 254.6 µg/mL and 1020.2 ± 40.9 µg/mL, respectively. These results showed there was a 3.34-fold increase in the solubility of furosemide in OA-mPEG5000 (1% w/w) solution compared to plain milli-Q water. Results at 25 °C showed that there was a 3.31-fold increase in solubilization of furosemide in OA-mPEG5000 (1% w/w) solution compared to water as solubilization values for OA-mPEG5000 (1% w/w) and milli-Q water, which were 90.0 ± 1.45 µg/mL and 27.2 ± 1.43 µg/mL, respectively. There was a significant difference (p < 0.05) between the solubility of furosemide in OA-mPEG5000 (1% w/w) aqueous solution and milli-Q water at both 25 °C and 37 °C. Solubilization of furosemide also increased significantly (p < 0.05) both in milli-Q water and OA-mPEG5000 (1% w/w) solution as the temperature increased from 25 °C to 37 °C. Literatures reported that the solubility of furosemide in milli-Q water at 25 °C and 37 °C is 0.6 mg/dL and 0.8 mg/dL, respectively after performing the study for 3–5 days [32]. In our case, the solubilization of furosemide in milli-Q water at 25 °C and 37 °C was found to be 2.72 ± 0.14 mg/dL and 102.02 ± 4.09 mg/dL at the end of 6 days. In another report, the solubility of furosemide in milli-Q water at room temperature after 24 h was reported to be 0.04 mg/mL (4 mg/dL) [17]. This solubility is 2.25-fold less than the solubility of furosemide in OA-mPEG5000 (9.00 ± 0.14 mg/dL). Overall results of the solubility study suggest the potential of OA-mPEG5000 as a solubility enhancer of furosemide, a BCS Class IV drug.
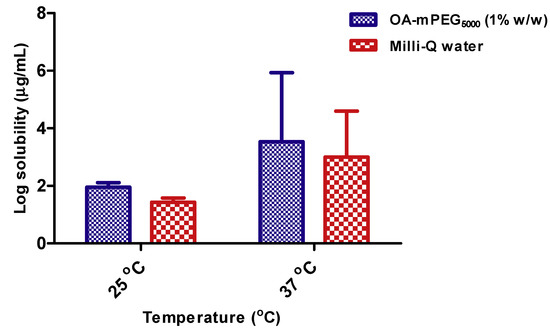
Figure 2.
Log solubility of furosemide in OA-mPEG5000 and milli-Q water at 25 °C and 37 °C (n = 3).
3.2. Dynamic Light Scattering
After confirming the solubility enhancement of furosemide by OA-mPEG5000, we further evaluated the polymersome formation process. For this purpose, the ratio of furosemide to OA-mPEG5000 used was 1:10. The experiments were performed in 12 replicates and in each case, the size of the polymersomes was below 200 nm. In some cases, bimodal distribution was observed. Size, PI and ZP for polymersomes ranged from 85–145.5 nm, 0.187–0.511 and −4.0–12.77 mV, respectively. Micelles smaller than 100 nm can easily evade the phagocyte scavenging system and thus improve the therapeutic efficacy [31,37]. The DLS results obtained suggest that OA-mPEG5000 could be a potential drug carrier for furosemide. However, a wide range in size, PI and ZP clearly indicates that there is a need for further investigations to obtain optimized polymersome formulation loaded with furosemide. No attempts or further formulation development were performed because this was a proof-of-concept study to show the applicability of OA-mPEG5000 as a solubility-enhancing excipient for poorly soluble drugs.
3.3. Entrapment Efficiency
%EE for all the formulations was >95% indicating a higher degree of encapsulation of furosemide in the polymersomes.
3.4. XRD
Properties such as solubility, dissolution rate, bioavailability, and stability are determined by studying the crystalline nature of drugs [34,38]. XRD studies were performed to confirm the transformation of crystalline furosemide to amorphous form. Results showed that pure furosemide was crystalline in nature with characteristic peaks at 6.18°, 13.28°, 21.20°, and 23.38° (Figure 3A), whereas XRD for furosemide polymersomes showed the absence of characteristic peaks of furosemide. This confirms that the formulation of polymersomes resulted in the transition of initially poorly soluble crystalline furosemide to amorphous form (Figure 3B). Amorphous forms of drug molecules exhibit enhanced solubility, dissolution rate and improved bioavailability compared to the crystalline form. Therefore, the formulation of furosemide polymersomes resulted in improvement of its solubility and complemented the solubility results.
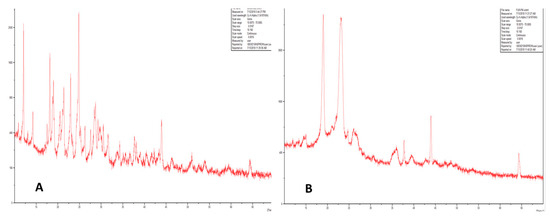
Figure 3.
XRD of (A) furosemide and (B) furosemide polymersomes.
3.5. In-Vitro Release Study
Figure 4 represents the release profile of furosemide from OA-mPEG5000 polymersomes. In the initial 1st h, 71% of the drug was released from polymersomes. After that, drug released slowed down and reached 83% after 3 h. These results indicated a burst release pattern OA-mPEG5000 polymersomes. A similar kind of burst release was observed for triclosan-loaded poly-ε-caprolactone nanoparticles where drug release after 1 h and 3 h was 76% and 97%, respectively. The authors attributed this burst release to the superficial drug entrapment [39]. After 3 h, furosemide release from polymersomes achieved a steady-state pattern.
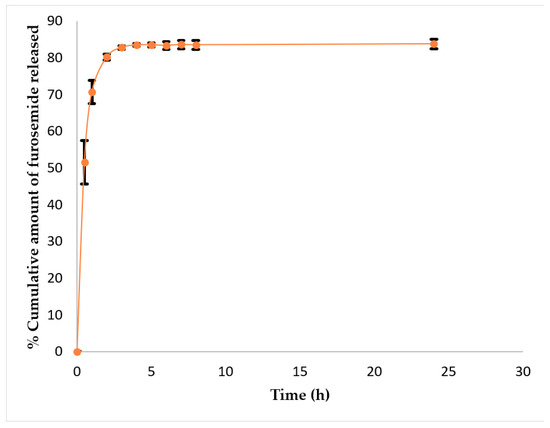
Figure 4.
Furosemide release profile from OA-mPEG5000 polymersomes. Data are represented as mean ± SD (n = 3).
4. Conclusions
In this study, we have explored the applicability of OA-mPEG5000 as a solubilizer and nanocarrier for furosemide, a BCS Class IV drug. The solubility of furosemide increased significantly in OA-mPEG5000 at 25 °C as well as 37 °C. The increase in solubility was greater than 3-fold, indicating the potential of mPEG-OA5000 in enhancing the solubility of furosemide. Further, the evaluation of OA-mPEG5000 in encapsulating furosemide into nanoparticles was evaluated by preparing polymersomes. It was noticed that OA-mPEG5000 formed furosemide-encapsulated polymersomes below 200 nm size. However, size variation ranged from 85–145.5 nm. Although encapsulation efficiency was >95% in polymersomes, a burst release of furosemide was observed with ~70% drug release in the 1st hour. These results suggest that although OA-mPEG5000 was able to form polymersomes with greater encapsulation of furosemide, there is a need for further investigations into formulation optimization to obtain furosemide-loaded polymersomes with size uniformity and controlled drug release pattern. Overall, it can be concluded that considering the increasing need of solubilizers for poorly soluble drugs by formulation scientists, OA-mPEG5000 can be a good addition to expand the available toolbox of solubilizers. In short, it can be concluded that OA-mPEG5000 could further be explored for its potential as a solubilizer and polymer for the preparation of polymersomes of poorly soluble BCS Class II and IV drugs.
Author Contributions
Conceptualization, R.S.K. and J.R.; methodology, R.S.K., P.K.B, S.H.S.B.; formal analysis, P.K.B., R.S.K.; writing—original draft preparation, R.S.K., P.K.B.; writing—review and editing, J.R., S.H.S.B.
Funding
This research received no external funding.
Acknowledgments
Authors are thankful to Julian Franco, Carlos Meraz, and Victor Rodriguez for help during DLS analysis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mittal, B. Chapter 2—Pharmacokinetics and Preformulation. In B.B.T.-H. to D.R.S.O.D.F. from C. to P.-A.; Mittal, B., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 17–37. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Saal, W.; Ross, A.; Wyttenbach, N.; Alsenz, J.; Kuentz, M. A Systematic Study of Molecular Interactions of Anionic Drugs with a Dimethylaminoethyl Methacrylate Copolymer Regarding Solubility Enhancement. Mol. Pharm. 2017, 14, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Savla, R.; Browne, J.; Plassat, V.; Wasan, K.M.; Wasan, E.K. Review and analysis of FDA approved drugs using lipid-based formulations. Drug Dev. Ind. Pharm. 2017, 43, 1743–1758. [Google Scholar] [CrossRef] [PubMed]
- McMurry, J.; Ballantine, D.S.; Hoeger, C.A.; Peterson, V.E.; Castellion, M. Fundamentals of General, Organic, and Biological Chemistry; Pearson Education: London, UK, 2010. [Google Scholar]
- Juluri, A.; Murthy, S.N. Transdermal iontophoretic delivery of a liquid lipophilic drug by complexation with an anionic cyclodextrin. J. Control. Release 2014, 189, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins: Basic science and product development. J. Pharm. Pharmacol. 2010, 62, 1607–1621. [Google Scholar] [CrossRef] [PubMed]
- USFDA, Guidance for Industry: Applications Covered by Section 505(b) (2), 1999. Available online: https://www.fda.gov/media/72419/download (accessed on 6 August 2019).
- Ghadi, R.; Dand, N. BCS class IV drugs: Highly notorious candidates for formulation development. J. Control. Release 2017, 248, 71–95. [Google Scholar] [CrossRef] [PubMed]
- Kalhapure, R.S.; Akamanchi, K.G. Oleic acid based heterolipid synthesis, characterization and application in self-microemulsifying drug delivery system. Int. J. Pharm. 2012, 425, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Akinlade, B.; Elkordy, A.A.; Essa, E.A.; Elhagar, S. Liquisolid Systems to Improve the Dissolution of Furosemide. Sci. Pharm. 2010, 78, 325–344. [Google Scholar] [CrossRef]
- Chaulang, G.; Patel, P.; Hardikar, S.; Kelkar, M.; Bhosale, A.; Bhise, S. Formulation and Evaluation of Solid Dispersions of Furosemide in Sodium Starch Glycolate. Trop. J. Pharm. Res. 2009, 8, 43–51. [Google Scholar] [CrossRef]
- Gulsun, T.; Borna, S.E.; Vural, I.; Sahin, S. Preparation and characterization of furosemide nanosuspensions. J. Drug Deliv. Sci. Technol. 2018, 45, 93–100. [Google Scholar] [CrossRef]
- Toxnet, Furosemide, (n.d.). Available online: https://toxnet.nlm.nih.gov/cgi-bin/sis/search2/r?dbs+hsdb:@term+@rn+@rel+54-31-9 (accessed on 29 July 2019).
- Berthod, A.; Carda-Broch, S.; García-Álvarez-Coque, M.C. Hydrophobicity of Ionizable Compounds. A Theoretical Study and Measurements of Diuretic Octanol—Water Partition Coefficients by Countercurrent Chromatography. Anal. Chem. 1999, 71, 879–888. [Google Scholar] [CrossRef]
- Ruiz-Angel, M.J.; Carda-Broch, S.; Garcıa-Alvarez-coque, M.C.; Berthod, A. Micellar versus hydro-organic mobile phases for retention-hydrophobicity relationship studies with ionizable diuretics and an anionic surfactant. J. Chromatogr. A 2004, 1030, 279–288. [Google Scholar] [CrossRef]
- Shin, S.C.; Kim, J. Physicochemical characterization of solid dispersion of furosemide with TPGS. Int. J. Pharm. 2003, 251, 79–84. [Google Scholar] [CrossRef]
- Latere Dwan'Isa, J.P.; Rouxhet, L.; Preat, V.; Brewster, M.E.; Arien, A. Prediction of drug solubility in amphiphilic di-block copolymer micelles: The role of polymer-drug compatibility. Die Pharm. 2007, 62, 499–504. [Google Scholar] [CrossRef]
- Oh, S.W.; Han, S.Y. Loop Diuretics in Clinical Practice. Electrolytes Blood Press. 2015, 13, 17–21. [Google Scholar] [CrossRef]
- Nielsen, L.H.; Rades, T.; Müllertz, A. Characterization during storage and dissolution of solid dispersions containing furosemide and hydroxypropyl methylcellulose. J. Drug Deliv. Sci. Technol. 2013, 23, 409–415. [Google Scholar] [CrossRef]
- Miranda, J.A.; Garnero, C.; Zoppi, A.; Sterren, V.; Ayala, A.P.; Longhi, M.R. Characterization of systems with amino-acids and oligosaccharides as modifiers of biopharmaceutical properties of furosemide. J. Pharm. Biomed. Anal. 2018, 149, 143–150. [Google Scholar] [CrossRef]
- Popescu, C.; Manda, P.; Juluri, A.; Janga, K.Y.; Cidda, M.; Murthy, S.N. Enhanced Dissolution Efficiency of Zaleplon Solid Dispersions via Modified β-Cyclodextrin Molecular Inclusion Complexes. J. Pharma Pharm. Sci. 2015, 1, 12–21. [Google Scholar] [CrossRef]
- Juluri, A.; Popescu, C.; Zhou, L.; Murthy, R.N.; Gowda, V.K.; Kumar, C.; Pimparade, M.B.; Repka, M.A.; Murthy, S.N. Taste Masking of Griseofulvin and Caffeine Anhydrous Using Kleptose Linecaps DE17 by Hot Melt Extrusion. Aaps Pharmscitech 2016, 17, 99–105. [Google Scholar] [CrossRef]
- Marzan, A.L.; Tabassum, R.; Jahan, B.; Asif, M.H.; Reza, H.M.; Kazi, M.; AlShehri, S.M.; De Matas, M.; Shariare, M.H. Preparation and Characterization of Stable Nanosuspension for Dissolution Rate Enhancement of Furosemide: A Quality by Design (QbD) Approach. Curr. Drug Deliv. 2018, 15, 672–685. [Google Scholar] [CrossRef]
- Morales, D.; Gutiérrez, J.M.; Garcia-Celma, M.J.; Solans, Y.C. Preparation and characterization of solid lipid nanoparticles of furosemide using quality by design. Part. Sci. Technol. 2018, 36, 695–709. [Google Scholar] [CrossRef]
- Nielsen, L.H.; Melero, A.; Keller, S.S.; Jacobsen, J.; Garrigues, T.; Rades, T.; Müllertz, A.; Boisen, A. Polymeric microcontainers improve oral bioavailability of furosemide. Int. J. Pharm. 2016, 504, 98–109. [Google Scholar] [CrossRef]
- Karashima, M.; Kimoto, K.; Yamamoto, K.; Kojima, T.; Ikeda, Y. A novel solubilization technique for poorly soluble drugs through the integration of nanocrystal and cocrystal technologies. Eur. J. Pharm. Biopharm. 2016, 107, 142–150. [Google Scholar] [CrossRef]
- Bader, H.; Ringsdorf, H.; Schmidt, B. Watersoluble polymers in medicine. Die Angew. Makromol. Chem. 1984, 123, 457–485. [Google Scholar] [CrossRef]
- Khadka, P.; Ro, J.; Kim, H.; Kim, I.; Kim, J.T.; Kim, H.; Cho, J.M.; Yun, G.; Lee, J. Pharmaceutical particle technologies: An approach to improve drug solubility, dissolution and bioavailability. Asian J. Pharm. Sci. 2014, 9, 304–316. [Google Scholar] [CrossRef]
- Latere Dwan'Isa, J.P.; Rouxhet, L.; Brewster, M.E.; Préat, V.; Ariën, A. Spontaneously self-assembled micelles from poly(ethylene glycol)-b-poly(ε-caprolactone-co-trimethylene carbonate) for drug solubilization. Die Pharm. 2008, 63, 235–240. [Google Scholar] [CrossRef]
- Crothers, M.; Attwood, D.; Collett, J.H.; Yang, Z.; Booth, C.; Taboada, P.; Mosquera, V.; Ricardo, N.M.P.S.; Martini, L.G.A. Micellization and Gelation of Diblock Copolymers of Ethylene Oxide and Styrene Oxide in Aqueous Solution. Langmuir 2002, 18, 8685–8691. [Google Scholar] [CrossRef]
- Omolo, C.A.; Kalhapure, R.S.; Jadhav, M.; Rambharose, S.; Mocktar, C.; Ndesendo, V.M.; Govender, T. Pegylated oleic acid: A promising amphiphilic polymer for nano-antibiotic delivery. Eur. J. Pharm. Biopharm. 2017, 112, 96–108. [Google Scholar] [CrossRef]
- Bolla, P.K.; Kalhapure, R.S.; Rodriguez, V.A.; Ramos, D.V.; Dahl, A.; Renukuntla, J. Preparation of solid lipid nanoparticles of furosemide-silver complex and evaluation of antibacterial activity. J. Drug Deliv. Sci. Technol. 2019, 49, 6–13. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, C.; Xia, X.; Liu, Y. Self-assembled lecithin/chitosan nanoparticles for oral insulin delivery: Preparation and functional evaluation. Int. J. Nanomed. 2016, 11, 761–769. [Google Scholar] [CrossRef]
- Kalhapure, R.S.; Bolla, P.; Dominguez, D.C.; Dahal, A.; Boddu, S.H.S.; Renukuntla, J. FSE–Ag complex NS: preparation and evaluation of antibacterial activity. IET Nanobiotechnol. 2018, 12, 836–840. [Google Scholar]
- Kwon, G.S.; Okano, T. Polymeric micelles as new drug carriers. Adv. Drug Deliv. Rev. 1996, 21, 107–116. [Google Scholar] [CrossRef]
- Rodriguez, V.A.; Bolla, P.K.; Kalhapure, R.S.; Boddu, S.H.S.; Neupane, R.; Franco, J.; Renukuntla, J. Preparation and Characterization of Furosemide-Silver Complex Loaded Chitosan Nanoparticles. Processes 2019, 7, 206. [Google Scholar] [CrossRef]
- Aminu, N.; Baboota, S.; Pramod, K.; Singh, M.; Dang, S.; Ansari, S.H.; Sahni, J.K.; Ali, J. Development and evaluation of triclosan loaded poly-ε-caprolactone nanoparticulate system for the treatment of periodontal infections. J. Nanopart. Res. 2013, 15, 2075. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).