
A Chemical Engineering Perspective on the Origins of Life



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Background
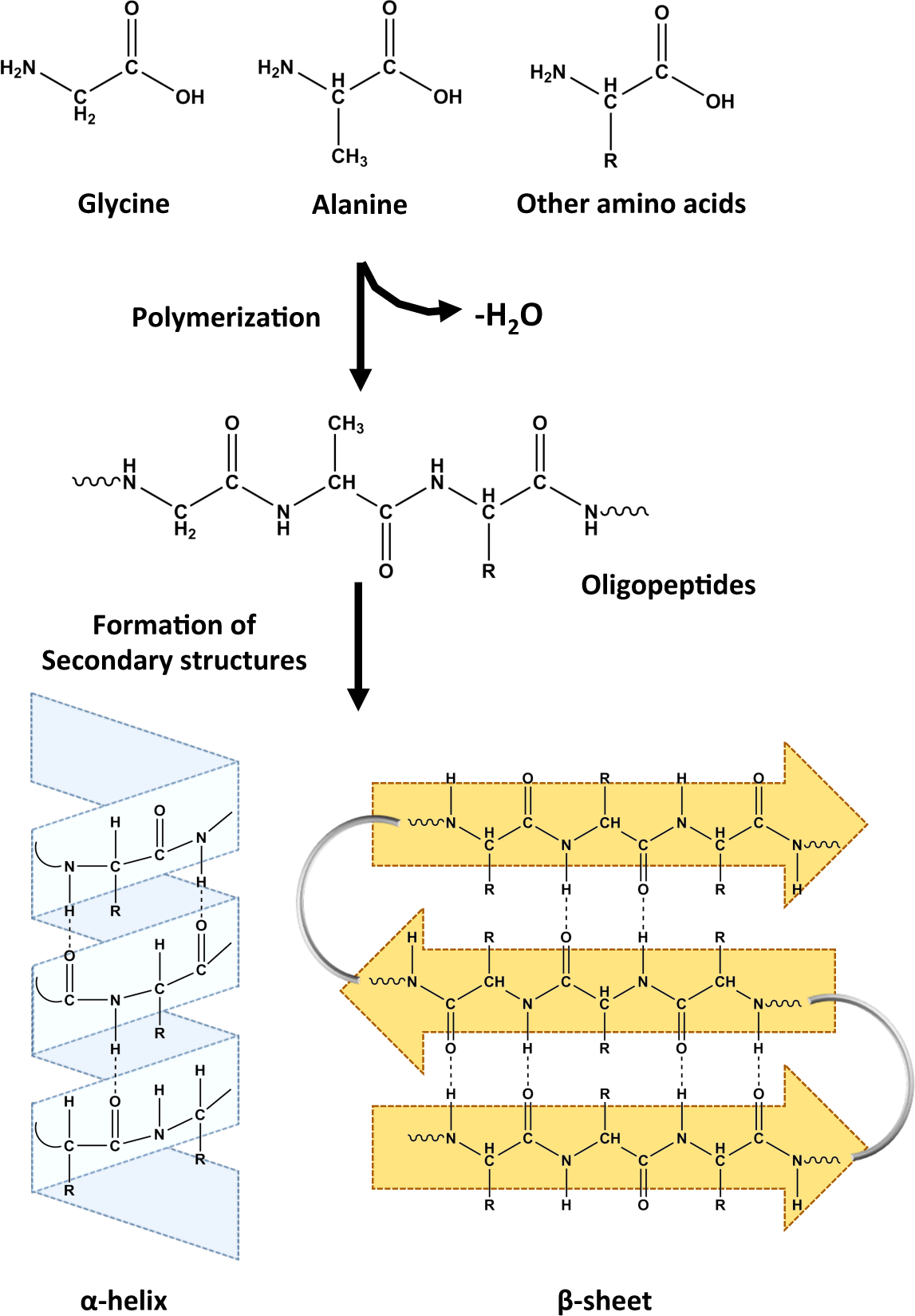
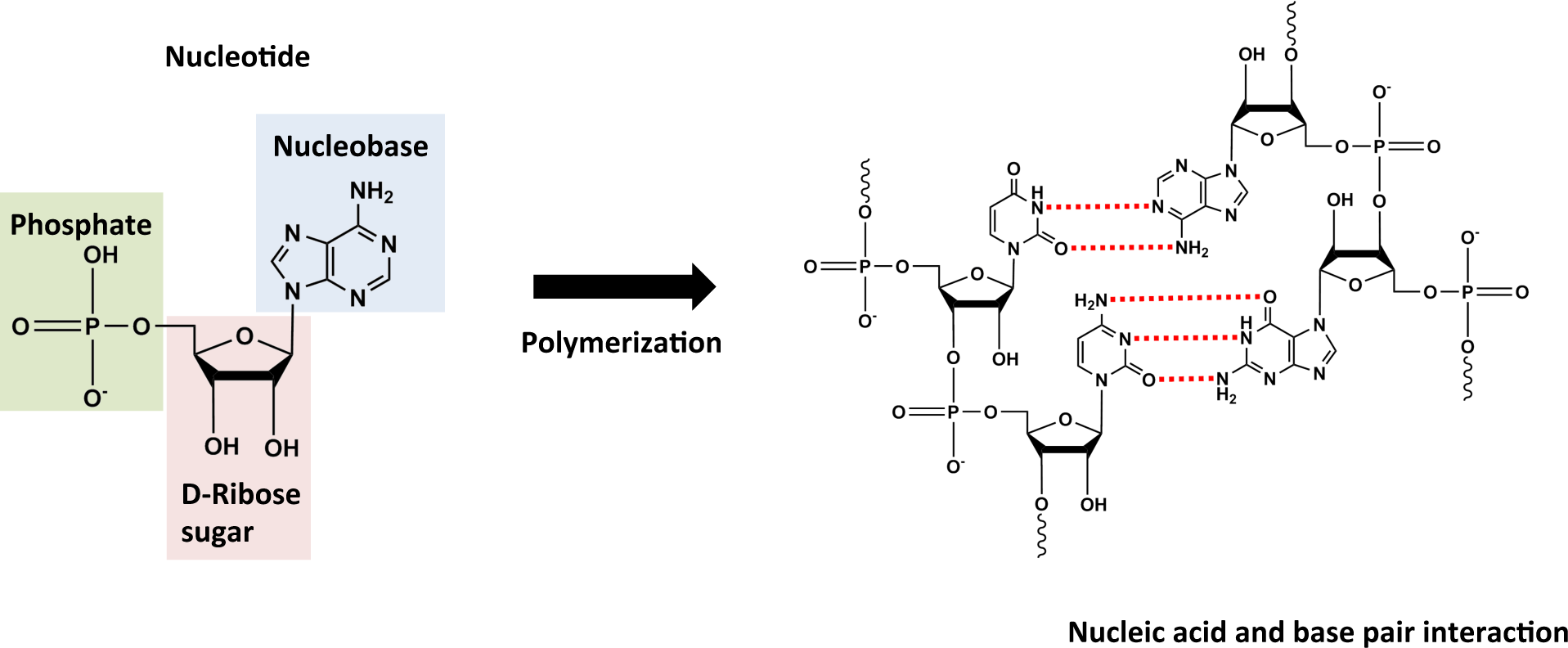
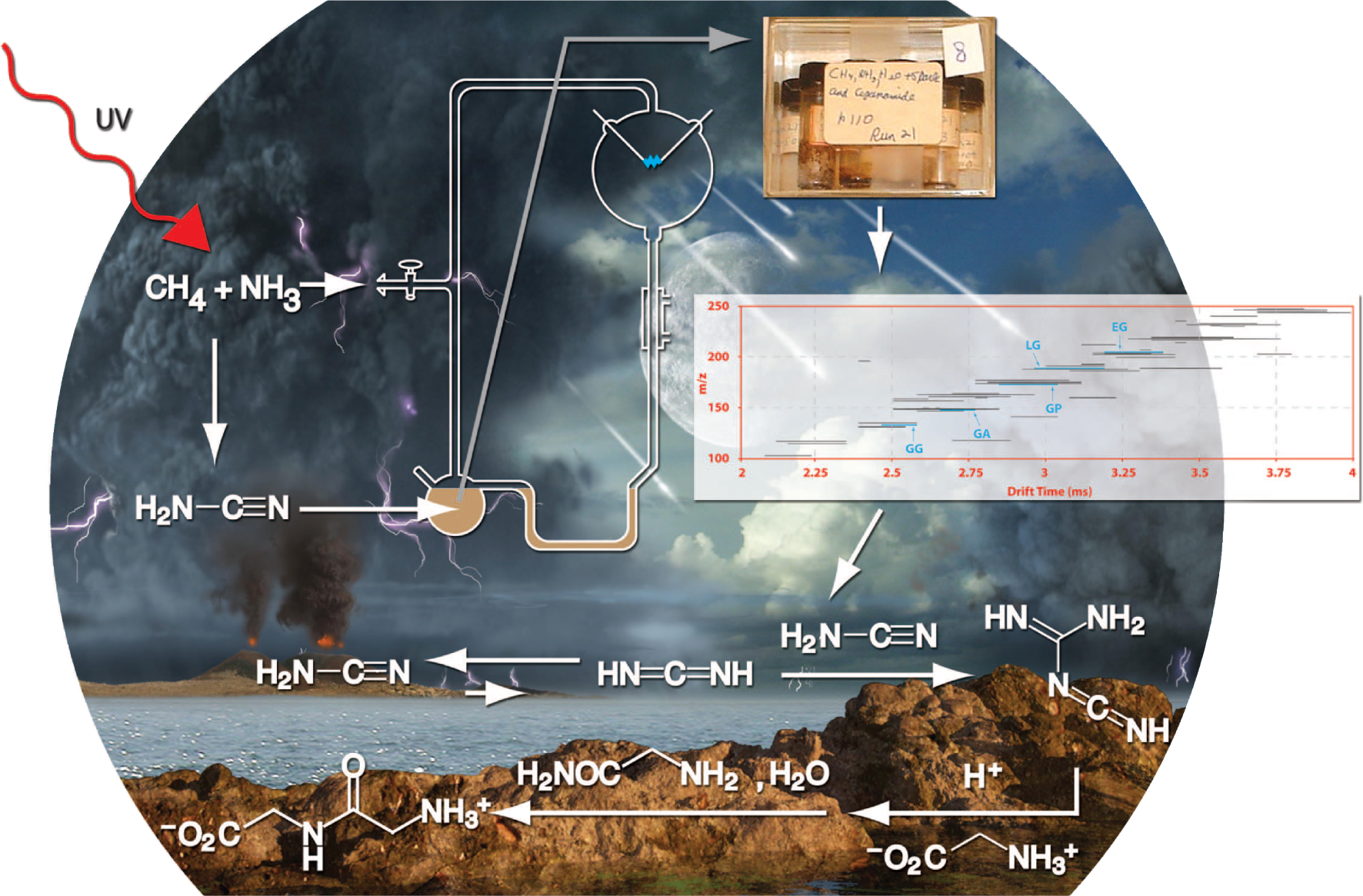
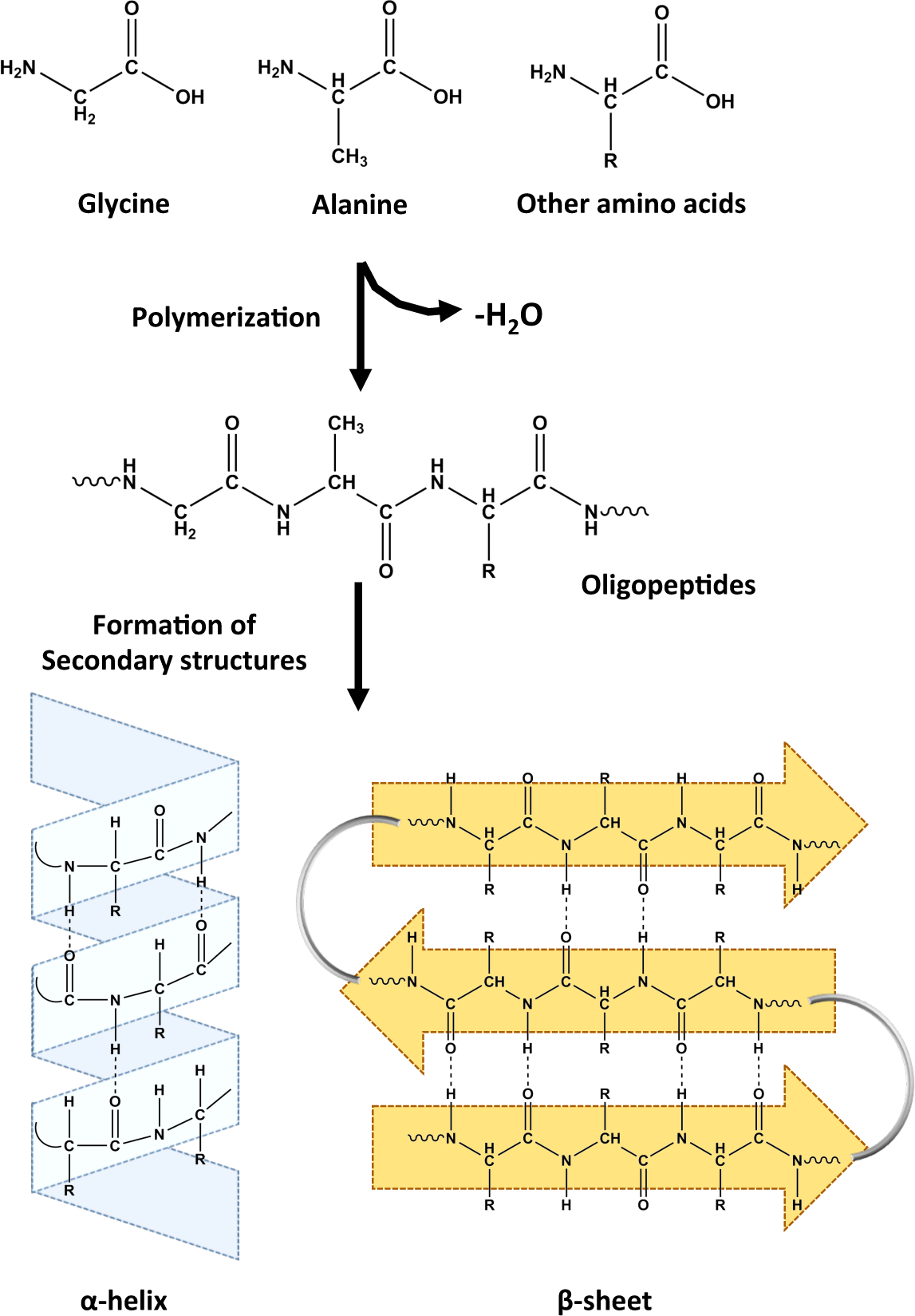
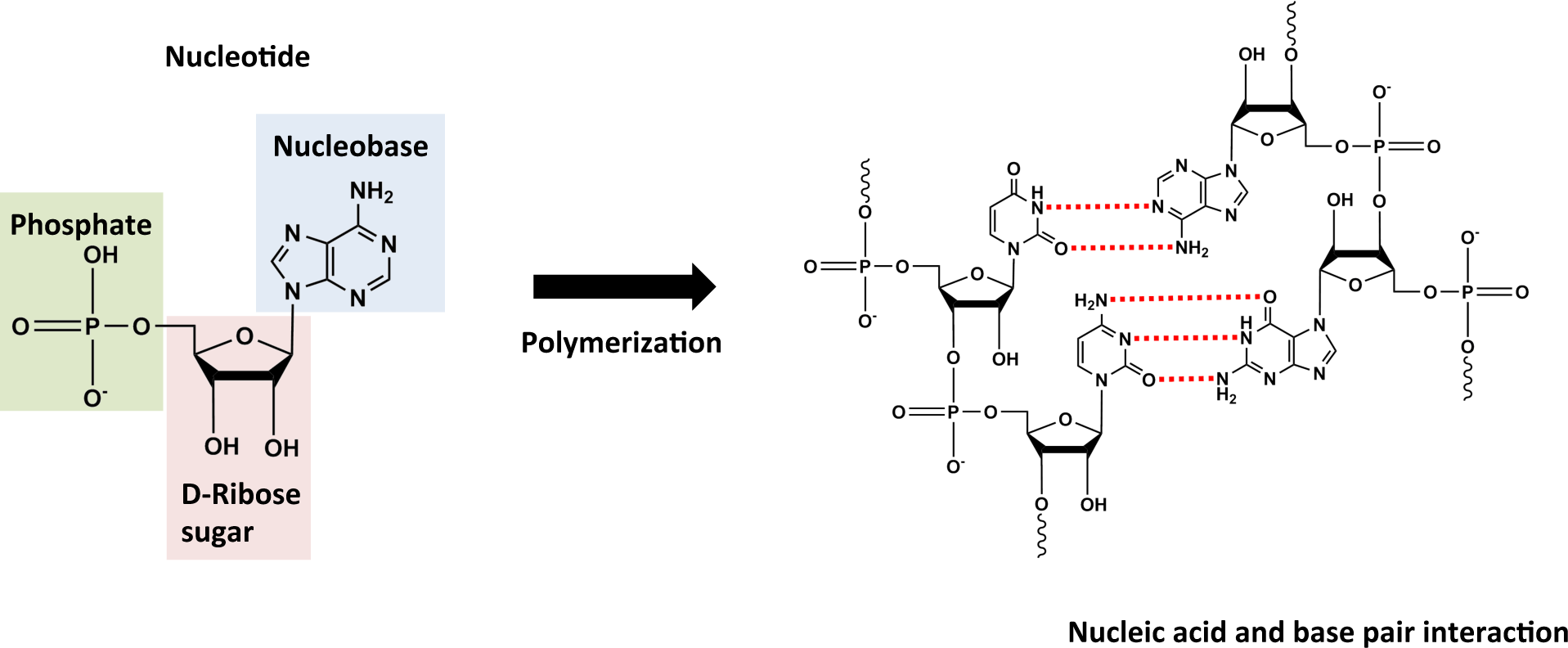
2.1. Monomers
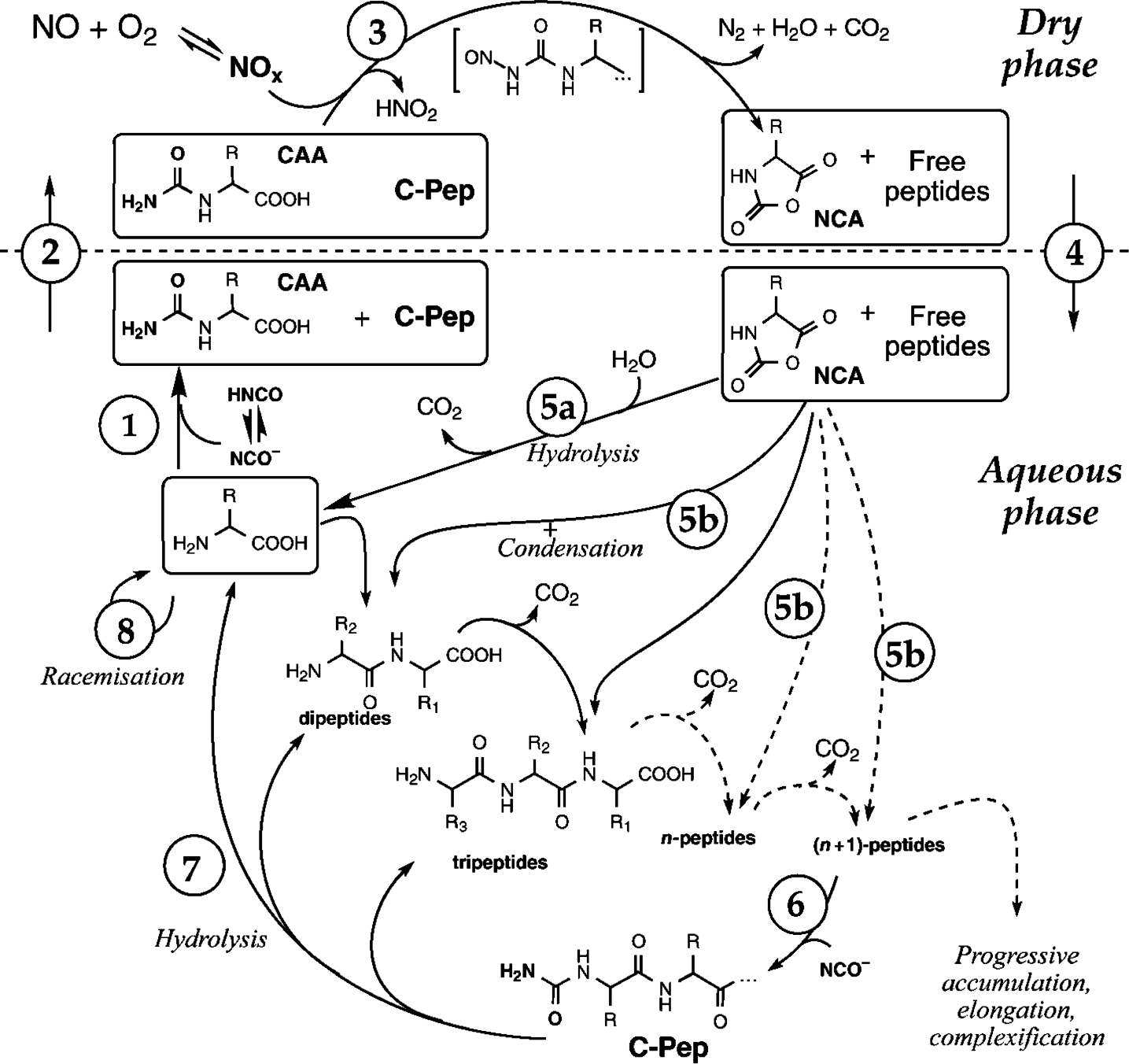
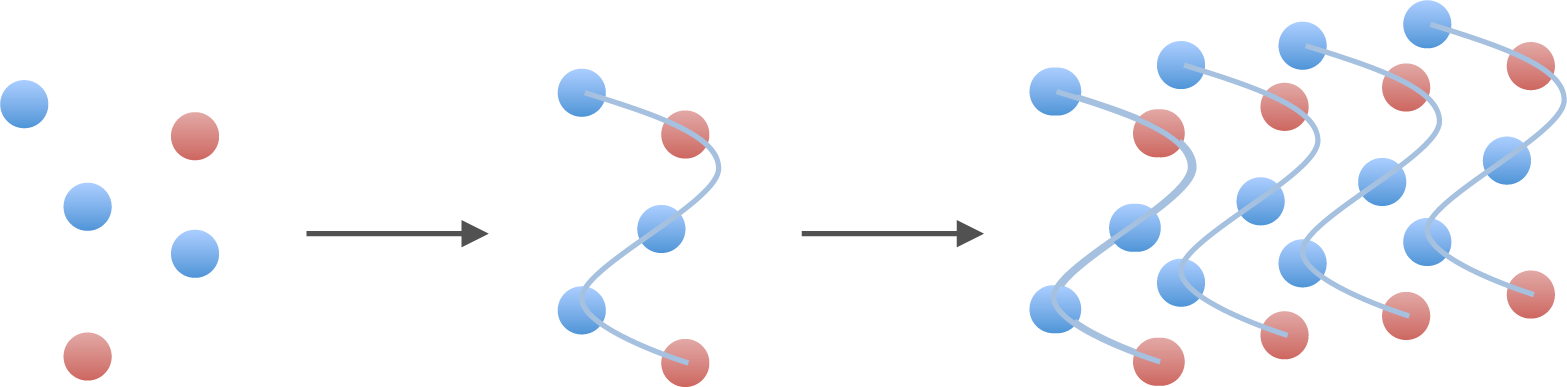
2.2. Polymers
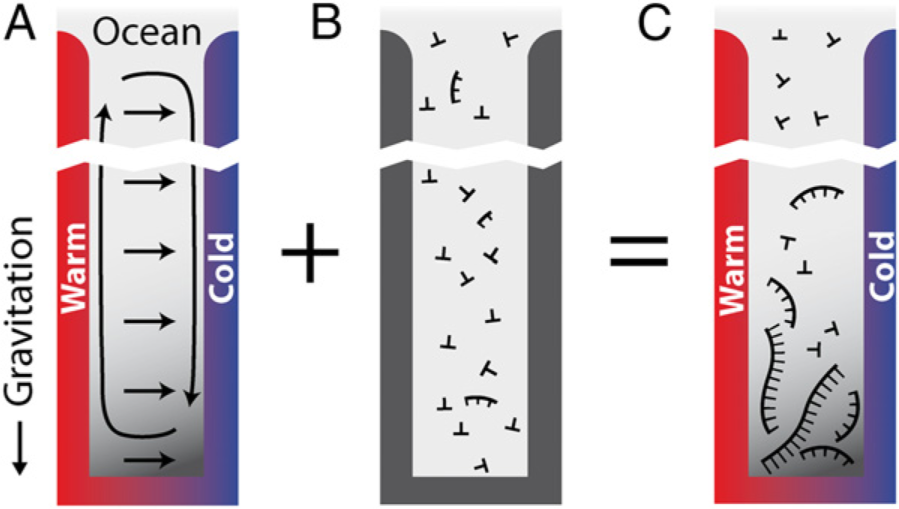
2.3. Assemblies
2.3.1. Selection
2.3.2. Evolution
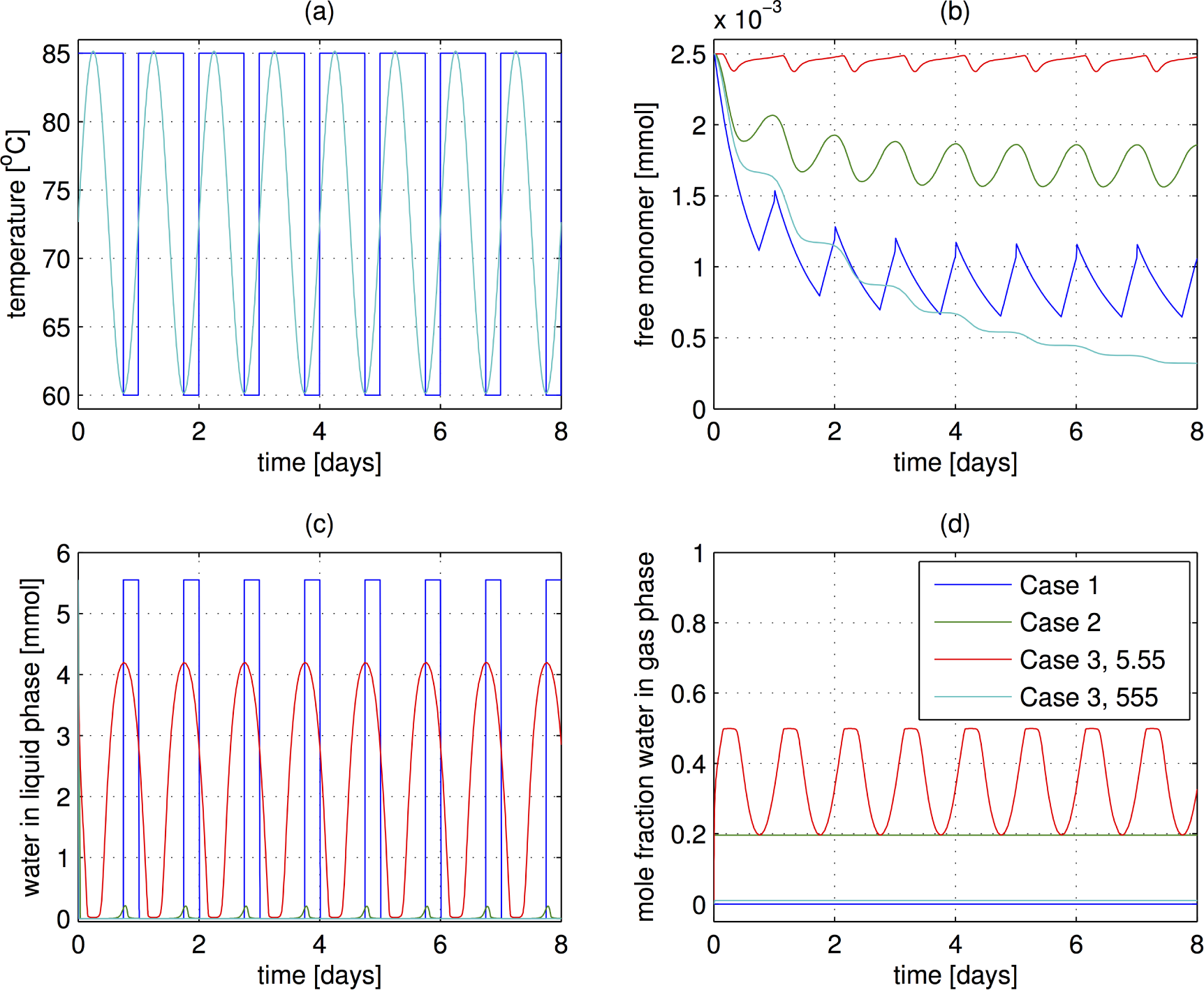
3. Case Study
3.1. Modeling
3.2. Environmental Scenarios
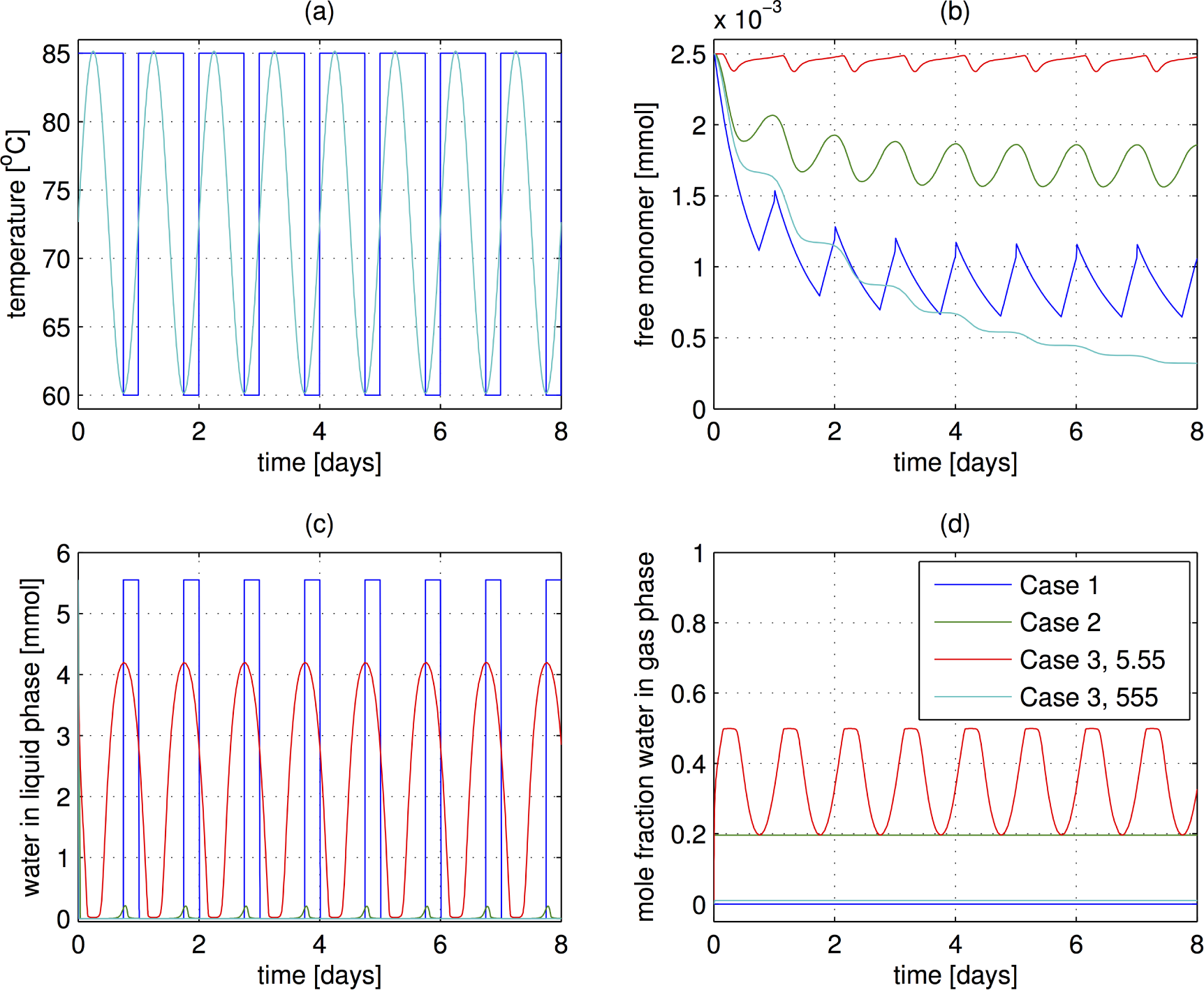
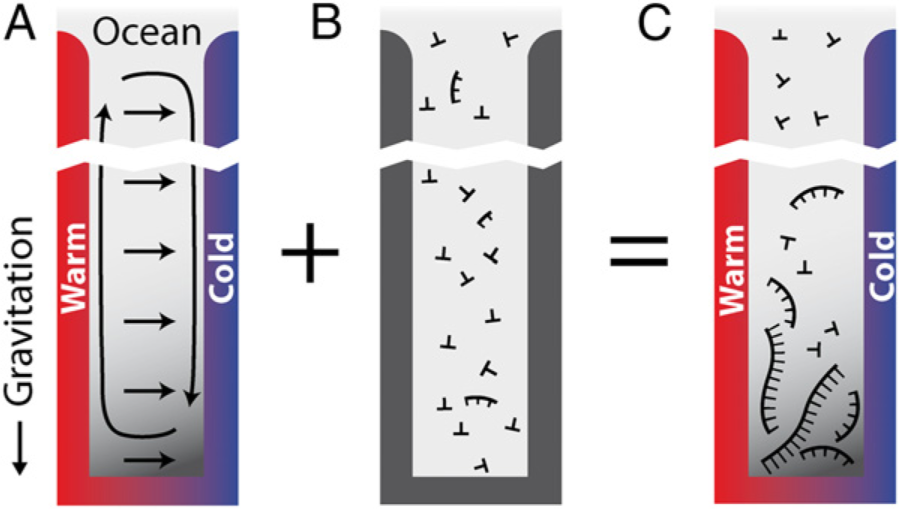
- The case of sudden temperature switches as implemented in Ref [35]. At a high temperature of 85 °C, the air is assumed to be completely dry, and at the low temperature of 60 °C the system is capped, allowing no transfer of water. The hot period lasts 18 h and the cold period is 6 h. Eight cycles are performed, corresponding to eight days. (Note: the day length on the early Earth was actually closer to 12 h, but that effect should not change these results substantially.)
- The temperature is varied sinusoidally with a 24 h period over 8 days. The temperature varies between 60 and 85 °C, the same levels as in Case 1. The system is open to mass transfer at all times, and the water content (mole fraction) in the air is constant, at the saturation level for 60 °C. This water level is motivated by an environmental scenario in which the air becomes saturated at night and dries out during the dry as the temperature heats up.
- The temperature is cycled as in the previous case, but the system is now closed to the mass transfer of water. The system does contain gas in the head space, and transfer between the liquid and gas phases can occur. However, the total water content is fixed. This case is reiminscent of reaction in the pore in a rock, another possible origins of life scenario. In this case, the amount of polymerization depends strongly on the amount of gas in the headspace. The total pressure therefore increases as the temperature rises.
3.3. Results
4. Future Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schopf, J.W. Cradle of Life: The Discovery of the Earth’s Earliest Fossils; Princeton University Press: Princeton, NJ, USA, 1999. [Google Scholar]
- Kasting, J.F. Earth’s early atmosphere. Science 1993, 259, 920–926. [Google Scholar]
- Cleaves, H.J.; Miller, S.L. Oceanic protection of prebiotic organic compounds from UV radiation. Proc. Natl. Acad. Sci. USA 1998, 95, 7260–7263. [Google Scholar]
- Valley, J.W.; Cavosie, A.J.; Ushikubo, T.; Reinhard, D.A.; Larson, D.J.; Clifton, P.H.; Kelly, T.F.; Wilde, S.A.; Moser, D.E.; Spicuzza, M.J. Hadean age for a post-magma-ocean zircon confirmed by atom-probe tomography. Nat. Geosci. 2014, 7, 219–223. [Google Scholar]
- Roth, A.S.G.; Bourdon, B.; Mojzsis, S.J.; Rudge, J.F.; Guitreau, M.; Blichert-Toft, J. Combined Sm-147, Sm-146-Nd-143,Nd-142 constraints on the longevity and residence time of early terrestrial crust. Geochem. Geophys. Geosyst. 2014, 15, 2329–2345. [Google Scholar]
- Woese, C. The universal ancestor. Proc. Natl. Acad. Sci. USA 1998, 95, 6854–6859. [Google Scholar]
- Orgel, L.E. Prebiotic chemistry and the origin of the RNA World. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 99–123. [Google Scholar]
- Woese, C. The Genetic Code; Harper & Row: New York, NY, USA, 1967; pp. 179–195. [Google Scholar]
- Orgel, L.E. Evolution of the genetic apparatus. J. Mol. Biol. 1968, 38, 381–393. [Google Scholar]
- Crick, F.H.C. The origin of the genetic code. J. Mol. Biol. 1968, 38, 367–379. [Google Scholar]
- Kruger, K.; Grabowski, P.J.; Zaug, A.J.; Sands, J.; Gottschling, D.E.; Cech, T.R. Self-splicing RNA: Autoexcision and autocyclization of the ribosomal RNA intervening sequence of tetrahymena. Cell 1982, 31, 147–157. [Google Scholar]
- Gilbert, W. Origin of life—The RNA World. Nature 1986, 319, 618–618. [Google Scholar]
- Joyce, G.F. The antiquity of RNA-based evolution. Nature 2002, 418, 214–221. [Google Scholar]
- Joyce, G.F.; Schwartz, A.W.; Miller, S.L.; Orgel, L.E. The case for an ancestral genetic system involving simple analogs of the nucleotides. Proc. Natl. Acad. Sci. USA 1987, 84, 4398–4402. [Google Scholar]
- Orgel, L.E. Some consequences of the RNA world hypothesis. Orig. Life Evol. Biosph. 2003, 33, 211–218. [Google Scholar]
- Orgel, L. Origin of life—A simpler nucleic acid. Science 2000, 290, 1306–1307. [Google Scholar]
- Higgs, P.G.; Lehman, N. The RNA World: Molecular cooperation at the origins of life. Nat. Genet. Rev. 2015, 16, 7–17. [Google Scholar]
- Orgel, L.E. Self-organizing biochemical cycles. Proc. Natl. Acad. Sci. USA 2000, 97, 12503–12507. [Google Scholar]
- Wachtershauser, G. Evolution of the first metabolic cycles. Proc. Natl. Acad. Sci. USA 1990, 87, 200–204. [Google Scholar]
- Danger, G.; Plasson, R.; Pascal, R. Pathways for the formation and evolution of peptides in prebiotic environments. Chem. Soc. Rev. 2012, 41, 5416–5429. [Google Scholar]
- Rode, B.; Son, H.; Suwannachot, Y.; Bujdak, J. The combination of salt induced peptide formation reaction and clay catalysis: A way to higher peptides under primitive Earth conditions. Orig. Life Evol. Biosph. 1999, 29, 273–286. [Google Scholar]
- Segre, D.; Ben-Eli, D.; Deamer, D.W.; Lancet, D. The lipid word. Orig. Life Evol. Biosph. 2001, 31, 119–145. [Google Scholar]
- Deamer, D.W. The first living systems: A bioenergetic perspective. Microbiol. Mol. Biol. Rev. 1997, 61, 239–261. [Google Scholar]
- Stern, R.; Jadrzejas, M.J. Carbohydrate polymers at the center of life’s origins: The importance of molecular processivity. Chem. Rev. 2008, 108, 5061–5085. [Google Scholar]
- Miller, S.L. A production of amino acids under possible primitive earth conditions. Science 1953, 117, 528–529. [Google Scholar]
- Bada, J.L.; Lazcano, A. Prebiotic Soup–Revisiting the Miller Experiment. Science 2003, 300, 745–746. [Google Scholar]
- Oro, J. Mechanism of synthesis of adenine from hydrogen cyanide under possible primitive Earth conditions. Nature 1961, 191, 1193–1194. [Google Scholar]
- Barks, H.L.; Buckley, R.; Grieves, G.A.; Di Mauro, E.; Hud, N.V.; Orlando, T.M. Guanine, adenine, and hypoxanthine production in UV-irradiated formamide solutions: Relaxation of the requirements for prebiotic purine nucleobase formation. Chembiochem 2010, 11, 1240–1243. [Google Scholar]
- Ruiz-Bermejo, M.; Menor-Salvan, C.; Osuna-Esteban, S.; Veintemillas-Verdaguer, S. Prebiotic microreactors: A synthesis of purines and dihydroxy compounds in aqueous aerosol. Orig. Life Evol. Biosph. 2007, 37, 123–142. [Google Scholar]
- Cleaves, H.J.; Chalmers, J.H.; Lazcano, A.; Miller, S.L.; Bada, J.L. A reassessment of prebiotic organic synthesis in neutral planetary atmospheres. Orig. Life Evol. Biosph. 2008, 38, 105–115. [Google Scholar]
- Benner, S.A.; Kim, H.J.; Carrigan, M.A. Asphalt, water, and the prebiotic synthesis of ribose, ribonucleosides, and RNA. Acc. Chem. Res. 2012, 45, 2025–2034. [Google Scholar]
- Pross, A. Seeking the chemical roots of Darwinism: Bridging between chemistry and biology. Chem.: Eur. J 2009, 15, 8374–8381. [Google Scholar]
- Goldenfeld, N.; Woese, C.W. Life is Physics: Evolutionas a Collective Phenomenon Far From Equilibrium. Annu. Rev. Condens. Matter Phys. 2011, 2, 375–399. [Google Scholar]
- Nelson, K.E.; Levy, M.; Miller, S.L. Peptide nucleic acids rather than RNA may have been the first genetic molecule. Proc. Natl. Acad. Sci. USA 2000, 97, 3868–3871. [Google Scholar]
- Mamajanov, I.; MacDonald, P.J.; Ying, J.; Duncanson, D.M.; Dowdy, G.R.; Walker, C.A.; Engelhart, A.E.; FernÃandez, F.M.; Grover, M.A.; Hud, N.V.; et al. Ester formation and hydrolysis during wet-dry cycles: Generation of far-from-equilibrium polymers in a model prebiotic reaction. Macromolecules 2014, 47, 1334–1343. [Google Scholar]
- Cairns-Smith, A.G. The chemistry of materials for artificial Darwinian systems. Int. Rev. Phys. Chem. 1988, 7, 209–250. [Google Scholar]
- Damer, B.; Deamer, B. Coupled Phases and Combinatorial Selection in Fluctuating Hydrothermal Pools: A Scenario to Guide Experimental Approaches to the Origin of Cellular Life. Life 2015, 5, 872–887. [Google Scholar]
- Darwin, F. The Life and Letters of Charles Darwin, Including an Autobiographical Chapter 3; John Murray: London, UK, 1887; p. 18. [Google Scholar]
- Strulson, C.A.; Molden, R.C.; Keating, C.D.; Bevilacqua, P.C. RNA catalysis through compartmentalization. Nat. Chem. 2012, 4, 941–946. [Google Scholar]
- Lahav, N.; White, D.; Chang, S. Peptide formation in the prebiotic era: Thermal condensation of glycine in fluctuating clay environments. Science 1978, 201, 67–69. [Google Scholar]
- Mast, C.B.; Schink, S.; Gerland, U.; Braun, D. Escalation of polymerization in a thermal gradient. Proc. Natl. Acad. Sci. USA 2013, 110, 8030–8035. [Google Scholar]
- Philip, G.K.; Freeland, S.J. Did evolution select a nonrandom “alphabet” of amino acids? Astrobiology 2011, 11, 235–240. [Google Scholar]
- Freeland, S.J.; Knight, R.D.; Landweber, L.F.; Hurst, L.D. Early fixation of an optimal genetic code. Mol. Biol. Evol. 2000, 17, 511–518. [Google Scholar]
- New, M.H.; Pohorille, A. An inherited efficiences model of non-genomic evolution. Simul. Pract. Theory. 2000, 8, 99–108. [Google Scholar]
- Eigen, M. The Hypercycle: A Principle of Natural Self-Organization; Springer: Berlin, Germany, 1979. [Google Scholar]
- Lincoln, T.A.; Joyce, G.F. Self-Sustained Replication of an RNA Enzyme. Science 2009, 323, 1229–1232. [Google Scholar]
- Sczepanski, J.T.; Joyce, G.F. A cross-chiral RNA polymerase ribozyme. Nature 2014, 515, 440–442. [Google Scholar]
- Ashkenasy, G.; Jagasia, R.; Yadav, M.; Ghadiri, M.R. Design of a directed molecular network. Proc. Natl. Acad. Sci. USA 2004, 101, 10872–10877. [Google Scholar]
- Wu, M.; Higgs, P. Origin of self-replicating biopolymers: Autocatalytic feedback can jumpstart the RNA world. J. Mol. Evol. 2009, 69, 541–554. [Google Scholar]
- Szathmary, E.; Maynard Smith, J. The major evolutionary transitions. Nature 1995, 374, 227–232. [Google Scholar]
- Schroedinger, E. What is Life? In Based on Lectures Delivered under the Auspices of the Dublin; Institute for Advanced Studies at Trinity College: Dublin, Germany, 1944. [Google Scholar]
- Defining Life. Astrobiology Magazine 2002.
- Cleland, C.E.; Chyba, C.F. Defining “Life”. Orig. Life Evol. Biosph. 2002, 32, 387–393. [Google Scholar]
- Joyce, G.F. RNA evolution and the origins of life. Nature 1989, 338, 217–224. [Google Scholar]
- Ruiz-Mirazo, K.; Briones, C.; de la Escosura, A. Prebiotic Systems Chemistry: New Perspectives for the Origins of Life. Chem. Rev. 2014, 114, 285–366. [Google Scholar]
- Eschenmoser, A. Chemical Etiology of Nucleic Acid Structure. Science 1999, 284, 2188–2124. [Google Scholar]
- Krishnamurthy, R. On the emergence of RNA. Isr. J. Chem. 2015. [Google Scholar] [CrossRef]
- Hennet, R.J.C.; Holm, N.G.; Engel, M.H. Abiotic synthesis of amino-acids under hydrothermal conditions and the origin of life—A perpetual phenomenon. Naturwissenschaften 1992, 79, 361–365. [Google Scholar]
- Martin, W.; Baross, J.; Kelley, D.; Russell, M.J. Hydrothermal vents and the origin of life. Nat. Rev. Microbiol. 2008, 6, 805–814. [Google Scholar]
- Plankensteiner, K.; Reiner, H.; Schranz, B.; Rode, B.M. Prebiotic formation of amino acids in a neutral atmosphere by electric discharge. Angew. Chem.-Int. Ed. 2004, 43, 1886–1888. [Google Scholar]
- Dworkin, J.P.; Deamer, D.W.; Sandford, S.A.; Allamandola, L.J. Self-assembling amphiphilic molecules: Synthesis in simulated interstellar/precometary ices. Proc. Natl. Acad. Sci. USA 2001, 98, 815–819. [Google Scholar]
- Hayatsu, R.; Studier, M.H.; Anders, E. Origin of organic matter in early solar system. 4. Amino acids—Confirmation of catalytic synthesis by mass spectrometry. Geochim. Cosmochim. Acta. 1971, 35, 939. [Google Scholar]
- Kobayashi, K.; Tsuchiya, M.; Oshima, T.; Yanagawa, H. Abiotic synthesis of amino-acids and imidazole by proton irradiation of simulated primitive earth atmospheres. Orig. Life Evol. Biosph. 1990, 20, 99–109. [Google Scholar]
- McCollom, T.M.; Ritter, G.; Simoneit, B.R.T. Lipid synthesis under hydrothermal conditions by Fischer-Tropsch-type reactions. Orig. Life Evol. Biosph. 1999, 29, 153–166. [Google Scholar]
- Nooner, D.W.; Gibert, J.M.; Gelpi, E.; Oro, J. Closed system Fischer-Tropsch synthesis over meteoritic iron, iron-ore and nickel-iron alloy. Geochim. Cosmochim. Acta. 1976, 40, 915–924. [Google Scholar]
- Parker, E.T.; Zhou, M.S.; Burton, A.S.; Glavin, D.P.; Dworkin, J.P.; Krishnamurthy, R.; Fernandez, F.M.; Bada, J.L. A plausible simultaneous synthesis of amino acids and simple peptides on the primordial earth. Angew Chem.–Int. Ed. 2014, 53, 8132–8136. [Google Scholar]
- Larralde, R.; Robertson, M.P.; Miller, S.L. Rate of decomposition of ribose and other sugars—Implications for chemical evolution. Proc. Natl. Acad. Sci. USA 1995, 92, 8158–8160. [Google Scholar]
- Levy, M.; Miller, S.L. The stability of the RNA bases: Implications for the origin of life. Proc. Natl. Acad. Sci. USA 1998, 95, 7933–7938. [Google Scholar]
- Simonov, A.N.; Pestunova, O.P.; Matvienko, L.G.; Parmon, V.N. The nature of autocatalysis in the Butlerov reaction. Kinet. Catal. 2007, 48, 245–254. [Google Scholar]
- Weber, A.L. The sugar model: Catalysis by amines and amino acid products. Orig. Life Evol. Biosph. 2001, 31, 71–86. [Google Scholar]
- Miller, S.L.; Urey, H.C. Organic compound synthesis on the primitive Earth. Science 1959, 130, 245–251. [Google Scholar]
- Kolb, V.M.; Dworkin, S.L.; Miller, S.L. Alternative bases in the RNA World: The prebiotic synthesis of urazole and its ribosides. J. Mol. Evol. 1994, 38, 549–557. [Google Scholar]
- Fuller, W.D.; Orgel, L.E.; Sanchez, R.A. Studies in prebiotic synthesis .7. Solid-state synthesis of purine nucleosides. J. Mol. Evol. 1972, 1, 249. [Google Scholar]
- Bean, H.D.; Sheng, Y.H.; Collins, J.P.; Anet, F.A.L.; Leszczynski, J.; Hud, N.V. Formation of a beta-pyrimidine nucleoside by a free pyrimidine base and ribose in a plausible prebiotic reaction. J. Am. Chem. Soc. 2007, 129, 9556–9557. [Google Scholar]
- Powner, M.W.; Gerland, B.; Sutherland, J.D. Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature 2009, 459, 239–242. [Google Scholar]
- Powner, M.W.; Sutherland, J.D. Prebiotic chemistry: A new modus operandi. Philos. Trans. R. Soc. B-Biol. Sci. 2011, 366, 2870–2877. [Google Scholar]
- Pasek, M.A.; Harnmeijer, J.P.; Buick, R.; Gull, M.; Atlas, Z. Evidence for reactive reduced phosphorus species in the early Archean ocean. Proc. Natl. Acad. Sci. USA 2013, 110, 10089–10094. [Google Scholar]
- Pizzarello, S. The chemistry of life’s origin: A carbonaceous meteorite perspective. Acc. Chem. Res. 2006, 39, 231–237. [Google Scholar]
- Abramov, O.; Mojzsis, S.M. Microbial habitability of the Hadean Earth during the late heavy bombardment. Nature 2009, 459, 419–422. [Google Scholar]
- Rufo, C.M.; Moroz, Y.S.; Moroz, O.V.; Stohr, J.; Smith, T.A.; Hu, X.Z.; DeGrado, W.F.; Korendovych, I.V. Short peptides self-assemble to produce catalytic amyloids. Nat. Chem. 2014, 6, 303–309. [Google Scholar]
- Adamala, K.; Engelhart, A.E.; Szostak, J.W. Generation of functional RNAs from inactive oligonucleotide complexes by non-enzymatic primer extension. J. Am. Chem. Soc. 2015, 137, 483–489. [Google Scholar]
- Martin, R.B. Free energies and equilibria of peptide bond hydrolysis and formation. Biopolymers 1998, 45, 351–353. [Google Scholar]
- Dickson, K.S.; Burns, C.M.; Richardson, J.P. Determination of the free-energy change for repair of a DNA phosphodiester bond. J. Biol. Chem. 2000, 275, 15828–15831. [Google Scholar]
- Liu, Z.; Beaufils, D.; Rossi, J.C.; Pascal, R. Evolutionary Importance of the Intramolecular Pathways of Hydrolysis of Phosphate Ester Mixed Anhydrides with Amino Acids and Peptides. Sci. Rep. 2014, 4, 7440. [Google Scholar]
- Imai, E.I.; Honda, H.; Hatori, K.; Brack, A.; Matsuno, K. Elongation of oligopeptides in a simulated submarine hydrothermal system. Science 1999, 283, 831–833. [Google Scholar]
- Ogasawara, H.; Yoshida, A.; Imai, E.I.; Honda, H.; Hatori, K.; Matsuno, K. Synthesizing oligomers from monomeric nucleotides in simulated hydrothermal environments. Orig. Life Evol. Biosph. 2000, 30, 519–526. [Google Scholar]
- Orgel, L. The origin of polynucleotide-directed protein synthesis. J. Mol. Evol. 1989, 29, 465–474. [Google Scholar]
- Cleaves, H.J.; Aubrey, A.D.; Bada, J.L. An evaluation of the critical parameters for abiotic peptide synthesis in submarine hydrothermal systems. Orig. Life Evol. Biosph. 2009, 39, 109–126. [Google Scholar]
- Commeyras, A.; Collet, H.; Boiteau, L.; Taillades, J.; Vandenabeele-Trambouze, O.; Cottet, H.; Biron, J.P.; Plasson, R.; Mion, L.; Lagrille, O.; et al. Prebiotic synthesis of sequential peptides on the Hadean beach by a molecular engine working with nitrogen oxides as energy sources. Polym. Int. 2002, 51, 661–665. [Google Scholar]
- Griffith, E.C.; Vaida, V. In situ observation of peptide bond formation at the water-air interface. Proc. Natl. Acad. Sci. USA 2012, 109, 15697–15701. [Google Scholar]
- Ferris, J.P.; Hill, A.R.; Liu, R.; Orgel, L.E. Synthesis of long prebiotic oligomers on mineral surfaces. Nature 1996, 381, 59–61. [Google Scholar]
- Leman, L.; Orgel, L.; Ghadiri, M.R. Carbonyl sulfide-mediated prebiotic formation of peptides. Science 2004, 306, 283–286. [Google Scholar]
- Budjak, J.; Rode, B.M. Peptide bond formation on the surface of activated alumina: Peptide chain elongation. Catal. Lett. 2003, 91, 149–154. [Google Scholar]
- Huang, W.; Ferris, J.P. One-step, regioselective synthesis of up to 50-mers of RNA oligomers by montmorillonite catalysis. J. Am. Chem. Soc. 2006, 128, 8914–8919. [Google Scholar]
- Costanzo, G.; Pino, S.; Ciciriello, F.; Di Mauro, E. Generation of long RNA chains in water. J. Biol. Chem. 2009, 284, 33206–33216. [Google Scholar]
- Monnard, P.A.; Kanavarioti, A.; Deamer, D.W. Eutectic phase polymerization of activated ribonucleotide mixtures yields quasi-equimolar incorporation of purine and pyrimidine nucleobases. J. Am. Chem. Soc. 2003, 125, 13734–13740. [Google Scholar]
- Milner-White, E.J. The relevance of peptides that bind FeS clusters, phosphate groups, cations or anions for prebiotic evolution. In Origins of Life: The Primal Self-Organization; Springer: Berlin, Germany, 2011; pp. 155–166. [Google Scholar]
- Lambert, J.F. Adsorption and polymerization of amino acids on mineral surfaces: A review. Orig. Life Evol. Biosph. 2008, 38, 211–242. [Google Scholar]
- Hud, N.V. Mineral surfaces: A mixed blessing for the RNA world? Astrobiology 2009, 9, 253–255. [Google Scholar]
- Plankensteiner, K.; Reiner, H.; Rode, B.M. Stereoselective differentiation in the salt-induced peptide formation reaction and its relevance for the origin of life. Peptides 2005, 26, 535–541. [Google Scholar]
- Zepik, H.H.; Rajamani, S.; Maurel, M.C.; Deamer, D. Oligomerization of thioglutamic acid: Encapsulated reactions and lipid catalysis. Orig. Life Evol. Biosph. 2007, 37, 495–505. [Google Scholar]
- Rajamani, S.; Vlassov, A.; Benner, S.; Coombs, A.; Olasagasti, F.; Deamer, D. Lipid-assisted synthesis of RNA-like polymers from mononucleotides. Orig. Life Evol. Biosph. 2008, 38, 57–74. [Google Scholar]
- Gull, M.; Zhou, M.; Fernandez, F.M.; Pasek, M.A. Prebiotic phosphate ester syntheses in a deep eutectic solvent. J. Mol. Evol. 2014, 78, 109–117. [Google Scholar]
- Fahnestock, S.; Rich, A. Ribosome-catalyzed polyester formation. Science 1971, 173, 340–343. [Google Scholar]
- Weber, A.L. Thermal synthesis and hydrolysis of polyglyceric acid. Orig. Life Evol. Biosph. 1989, 19, 7–19. [Google Scholar]
- Nielsen, P. Peptide nucleic acids and the origin of life. Chem. Biodivers. 2007, 4, 1996–2002. [Google Scholar]
- Bean, H.D.; Anet, F.A.L.; Gould, I.R.; Hud, N.V. Glyoxylate as a backbone linkage for a prebiotic ancestor of RNA. Orig. Life Evol. Biosph. 2006, 36, 39–63. [Google Scholar]
- Pizarello, S.; Davidowski, S.K.; Holland, G.P.; Williams, L.B. Processing of meteoritic organic materials as a possible analog of early molecular evolution in planetary environments. Proc. Natl. Acad. Sci. USA 2013, 110, 15614–15619. [Google Scholar]
- Reches, M.; Gazit, E. Designed aromatic homo-dipeptides: formation of ordered nanostructures and potential nanotechnological applications. Phys. Biol. 2006, 3(6), S10–S19. [Google Scholar]
- Szostak, J.W. The eightfold path to non-enzymatic RNA replication. J. Syst. Chem. 2012, 3(6), 1–14. [Google Scholar]
- Hagenbuch, P.; Kervio, E.; Hochgesand, A.; Plutowski, U.; Richert, C. Chemical primer extension: Efficiently determining single nucleotides in DNA. Angew. Chem.-Int. Ed. 2005, 44, 6588–6592. [Google Scholar]
- Von Kiedrowski, G. A Self-Replicating Hexadeoxynucleotide. Angew. Chem. Int. Ed. 1986, 25, 932–935. [Google Scholar]
- Li, T.; Nicolaou, K. Chemical self-replication of palindromic duplex DNA. Nature 1994, 369, 218–221. [Google Scholar]
- Zielinski, W.S.; Orgel, L.E. Autocatalytic synthesis of a tetranucleotide analogue. Nature 1987, 327, 346–347. [Google Scholar]
- Sievers, D.; von Kiedrowski, G. Self-replication of complementary nuceotide-based oligomers. Nature 1994, 369, 221–224. [Google Scholar]
- Freier, S.M.; Kerzek, R.; Jaeger, J.A.; Sugimoto, N.; Caruthers, M.H.; Neilson, T.; Turner, D.H. Improved free-energy parameters for predictions of RNA duplex stability. Proc. Natl. Acad. Sci. USA 1986, 83, 9373–9377. [Google Scholar]
- SantaLucia, J.; Hicks, D. The thermodynamics of DNA structural motifs. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 415–440. [Google Scholar]
- Grossmann, T.N.; Strohback, A.; Seitz, O. Achieving Turnover in DNA-Templated Reactions. ChemBioChem 2008, 9, 2185–2192. [Google Scholar]
- Fernando, C.; von Kiedrowski, G.; Szathmary, E. A Stochastic Model of Nonenzymatic Nucleic Acid Replication: “Elongators” Sequester Replicators. J. Mol. Evol. 2007, 64, 572–585. [Google Scholar]
- Zhan, Z.Y.; Lynn, D.G. Chemical Amplification through Template-Directed Synthesis. J. Am. Chem. Soc. 1997, 119, 12420–12421. [Google Scholar]
- Kausar, A.; McKay, R.D.; Lam, J.; Bhogal, R.S.; Tang, A.Y.; Gibbs-Davis, J.M. Tuning DNA stability to achieve turnover in template for an enzymatic ligation reaction. Angew. Chem. Int. Ed. 2011, 50, 8922–8926. [Google Scholar]
- Dose, C.; Ficht, S.; Seitz, O. Reducing product inhibition in DNA-template-controlled ligation reactions. Angew. Chem. Int. Ed. 2006, 45, 5369–5373. [Google Scholar]
- Luther, A.; Brandsch, R.; Kiedrowski, G.V. Surface-promoted replication and exponential amplification of DNA analogues. Nature 1998, 396, 245–248. [Google Scholar]
- Deck, C.; Jauker, M.; Richert, C. Efficient enzyme-free copying of all four nucleobases templated by immobilized RNA. Nat. Chem. 2011, 3(6), 603–608. [Google Scholar]
- Kreysing, M.; Keil, L.; Lanzmich, S.; Braun, D. Heat flux across an open pore enables the continuous replication and selection of oligonucleotides toward increasing length. Nat. Chem. 2015, 7, 203–208. [Google Scholar]
- Engelhart, A.E.; Powner, M.W.; Szostak, J.W. Functional RNAs exhibit tolerance for non-heritable 2′–5′ versus 3′–5′ backbone heterogeneity. Nat. Chem. 2013, 5, 390–394. [Google Scholar]
- Eigen, M. Self-organization of matter and evolution of biological macromolecules. Naturwissenschaften 1971, 58, 465–523. [Google Scholar]
- Podlech, J. Origin of organic molecules and biomolecular homochirality. Cell. Mol. Life Sci. 2001, 58, 44–60. [Google Scholar]
- Hein, J.E.; Blackmond, D.G. On the origin of single chirality of amino acids and sugars in biogenesis. Acc. Chem. Res. 2012, 45, 2045–2054. [Google Scholar]
- Cronin, J.R.; Pizzarello, S. Enantiomeric excesses in meteoritic amino acids. Science 1997, 275, 951–955. [Google Scholar]
- Bailey, J.; Chrysostomou, A.; Hough, J.H.; Gledhill, T.M.; McCall, A.; Clark, S.; Menard, F.; Tamura, M. Circular polarization in star-formation regions: Implications for biomolecular homochirality. Science 1998, 281, 672–674. [Google Scholar]
- Hazen, R.M.; Filley, T.R.; Goodfriend, G.A. Selective adsorption of L- and D-amino acids on calcite: Implications for biochemical homochirality. Proc. Natl. Acad. Sci. USA 2001, 98, 5487–5490. [Google Scholar]
- Glavin, D.P.; Dworkin, J.P. Enrichment of the amino acid L-isovaline by aqueous alteration on CI and CM meteorite parent bodies. Proc. Natl. Acad. Sci. USA 2009, 106, 5487–5492. [Google Scholar]
- Goldberg, S.I. Enantiomeric Enrichment on the Prebiotic Earth. Orig. Life Evol. Biosph. 2007, 37, 55–60. [Google Scholar]
- Blair, N.E.; Bonner, W.A. A model for the enantiomeric enrichment of polypeptides on the primitive Earth. Orig. Life. 1981, 11, 331–335. [Google Scholar]
- Hein, J.E.; Tse, E.; Blackmond, D.G. A route to enantiopure RNA precursors from nearly racemic starting materials. Nat. Chem. 2011, 3(6), 704–706. [Google Scholar]
- Breslow, R.; Cheng, Z.L. L-amino acids catalyze the formation of an excess of D-glyceraldehyde, and thus of other D sugars, under credible prebiotic conditions. Proc. Natl. Acad. Sci. USA 2010, 107, 5723–5725. [Google Scholar]
- Hud, N.V.; Cafferty, B.J.; Krishnamurthy, R.; Williams, L.D. The origin of RNA and ’my grandfather’s axe’. Chem. Biol. 2013, 20, 466–474. [Google Scholar]
- Lehn, J.M. Perspectives in supramolecular chemistry—From molecular recognition towards molecular information processing and self-organization. Angew. Chem. Int. Ed. 1990, 29, 1304–1319. [Google Scholar]
- Whitesides, G.M.; Mathias, J.P.; Seto, C.T. Molecular self-assembly and nanochemistry: A chemical strategy for the synthesis of nanostructures. Science 1991, 254, 1312–1319. [Google Scholar]
- Cafferty, B.J.; Gallego, I.; Chen, M.C.; Farley, K.I.; Eritja, R.; Hud, N.V. Efficient self-assembly in water of long noncovalent polymers by nucleobase analogs. J. Am. Chem. Soc. 2013, 135, 2447–2450. [Google Scholar]
- Menor-Salvan, C.; Ruiz-Bermejo, M.; Guzman, M.I.; Osuna-Esteban, S.; Veintemillas-Verdaguer, S. Synthesis of pyrimidines and triazines in ice: Implications for the prebiotic chemistry of nucleobases. Chem.-A Eur. J 2009, 15, 4411–4418. [Google Scholar]
- Chen, M.C.; Cafferty, B.J.; Mamajanov, I.; Gallego, I.; Khanam, J.; Krishnamurthy, R.; Hud, N.V. Spontaneous prebiotic formation of a β-ribofuranoside that self-assembles with a complementary heterocycle. J. Am. Chem. Soc. 2014, 136, 5640–5646. [Google Scholar]
- Athavale, S.S.; Spicer, B.; Chen, I.A. Experimental fitness landscapes to understand the molecular evolution of RNA-based life. Curr. Opin. Chem. Biol. 2014, 22, 35–39. [Google Scholar]
- Maynard Smith, J.; Szathmary, E. The Major Transitions in Evolution; Oxford: New York, NY, USA, 1995. [Google Scholar]
- Wattis, J.A.D.; Coveney, P.V. The origin of the RNA world: A kinetic model. J. Phys. Chem. B 1999, 103, 4231–4250. [Google Scholar]
- Coveney, P.V.; Swadling, J.B.; Wattis, J.A.D.; Greenwell, H.C. Theory, modelling and simulation in origins of life studies. Chem. Soc. Rev. 2012, 41, 5430–5446. [Google Scholar]
- Walker, S.I.; Grover, M.A.; Hud, N.V. Universal sequence replication, reversible polymerization and early functional biopolymers: A model for the initiation of prebiotic sequence evolution. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Boerlijst, M.; Hogeweg, P. Spiral wave structure in pre-biotic evolution: Hypercycles stable against parasites. Phys. D 1991, 48, 17–28. [Google Scholar]
- Novak, V.J.A. Present state of the coacervate in coacervate theory—Origin and evolution of the cell structure. Orig. Life Evol. Biosph. 1984, 14, 513–522. [Google Scholar]
- Monnard, P.A.; Deamer, D.W. Membrane self-assembly processes: Steps toward the first cellular life. Anat. Rec. 2002, 268, 196–207. [Google Scholar]
- Koga, S.; Williams, D.S.; Perriman, A.W.; Mann, S. Peptide-nucleotide microdroplets as a step towards a membrane-free protocell model. Nat. Chem. 2011, 3(6), 720–724. [Google Scholar]
- Monnard, P.A.; Szostak, J.W. Metal-ion catalyzed polymerization in the eutectic phase in water-ice: A possible approach to template-directed RNA polymerization. J. Inorg. Biochem. 2008, 102, 1104–1111. [Google Scholar]
- Chen, I.A.; Roberts, R.W.; Szostak, J.W. The emergence of competition between model protocells. Science 2004, 305, 1474–1476. [Google Scholar]
- Budin, I.; Szostak, J.W. Physical effects underlying the transition from primitive to modern cell membranes. Proc. Natl. Acad. Sci. USA 2011, 108, 5249–5254. [Google Scholar]
- Harshe, Y.M.; Storti, G.; Morbidelli, M.; Gelosa, S.; Moscatelli, D. Polycondensation kinetics of lactic acid. Macromol. React. Eng. 2007, 1, 611–621. [Google Scholar]
- Smith, J.M.; Van Ness, H.C.; Abbott, M.M. Introduction to Chemical Engineering Thermodynamics, 7th ed; McGraw-Hill: New York NY, USA, 2005; p. 682. [Google Scholar]
- Frederix, P.W.J.M.; Scott, G.G.; Abul-Haija, Y.M.; Kalafatovic, D.; Pappas, C.G.; Javid, N.; Hunt, N.T.; Ulijn, R.W.; Tuttle, T. Exploring the sequence space for (tri-)peptide self-assembly to design and discover new hydrogels. Nat. Chem. 2015, 7, 30–37. [Google Scholar]
- Otto, S. Dynamic combinatorial chemistry: A new method for selection and preparation of synthetic receptors. Curr. Opin. Drug Discov. Dev. 2003, 6, 509–520. [Google Scholar]
- Lehn, J.M. Toward self-organization and complex matter. Science 2002, 295, 2400–2403. [Google Scholar]
- Ludlow, R.F.; Otto, S. Systems Chemistry. Chem. Soc. Rev. 2008, 37, 101–108. [Google Scholar]
- Otto, S. Dynamic molecular networks: From synthetic receptors to self-replicators. Acc. Chem. Res. 2012, 45, 2200–2210. [Google Scholar]
- Hernandez, A.F.; Wagner, M.J.; Grover, M.A. Model identification of a template-directed peptide network for optimization in a continuous reactor. Chem. Commun. 2014, 50, 3849–3851. [Google Scholar]


© 2015 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grover, M.A.; He, C.Y.; Hsieh, M.-C.; Yu, S.-S. A Chemical Engineering Perspective on the Origins of Life. Processes 2015, 3, 309-338. https://doi.org/10.3390/pr3020309
Grover MA, He CY, Hsieh M-C, Yu S-S. A Chemical Engineering Perspective on the Origins of Life. Processes. 2015; 3(2):309-338. https://doi.org/10.3390/pr3020309
Chicago/Turabian StyleGrover, Martha A., Christine Y. He, Ming-Chien Hsieh, and Sheng-Sheng Yu. 2015. "A Chemical Engineering Perspective on the Origins of Life" Processes 3, no. 2: 309-338. https://doi.org/10.3390/pr3020309
APA StyleGrover, M. A., He, C. Y., Hsieh, M.-C., & Yu, S.-S. (2015). A Chemical Engineering Perspective on the Origins of Life. Processes, 3(2), 309-338. https://doi.org/10.3390/pr3020309