Abstract
Signal intensity and feedback regulation are known to be major factors in the signaling events stemming from the T-cell receptor (TCR) and its various coreceptors, but the exact nature of these relationships remains in question. We present a mathematical model of the complex signaling network involved in T-cell activation with cross-talk between the Erk, calcium, PKCθ and mTOR signaling pathways. The model parameters are adjusted to fit new and published data on TCR trafficking, Zap70, calcium, Erk and IκBα signaling. The regulation of the early signaling events by phosphatases, CD45 and SHP1, and the TCR dynamics are critical to determining the behavior of the model. Additional model corroboration is provided through quantitative and qualitative agreement with experimental data collected under different stimulating and knockout conditions. The resulting model is analyzed to investigate how signal intensity and feedback regulation affect TCR- and coreceptor-mediated signal transduction and their downstream transcriptional profiles to predict the outcome for a variety of stimulatory and knockdown experiments. Analysis of the model shows that: (1) SHP1 negative feedback is necessary for preventing hyperactivity in TCR signaling; (2) CD45 is required for TCR signaling, but also partially suppresses it at high expression levels; and (3) elevated FOXP3 and reduced IL-2 signaling, an expression profile often associated with T regulatory cells (Tregs), is observed when the system is subjected to weak TCR and CD28 costimulation or a severe reduction in CD45 activity.
Keywords:
T-cell activation; CD28 costimulation; calcium; Erk; NFkB; mTOR; CD45; SHP1; mathematical model 1. Introduction
The actions of CD4+ T-cells are controlled by the stimulatory signals and cytokine milieu in their surrounding tissue environment [1,2]. Extracellular signals naturally drive CD4+ T-cells to activate and assume one of many important immunological roles, each characterized by a distinct profile of signaling events and cytokine secretion. These signals are processed by the intracellular signaling pathways to alter the transcription profiles that direct T-cell activation and differentiation. To explore this process from a quantitative perspective, a mathematical model is constructed from the known molecular interactions and regulations.
This model encompasses the signal transduction events relating to the process of directing gene expression that ultimately determine the actions or immunological role of the T-cell. This process begins at the engagement of the T-cell receptor (TCR) to cognate peptide-bound major histocompatibility complexes (pMHC) on the surface of antigen-presenting cells (APCs) [3,4,5]. TCR engagement triggers a number of early phosphorylation and dephosphorylation events. The CD45 phosphatase primes the CD4 coreceptor-associated 56-kD lymphocyte-specific tyrosine kinase (Lck), a member of the sarcoma (Src) family of kinases (SFK) [6,7], by removing the inhibitory phosphate group from the Y505 residue [8,9]. Primed Lck is activated by autophosphorylation at Y394, mediated by receptor clustering [9]. Activated Lck phosphorylates the immunoreceptor tyrosine-based activation motifs (ITAMs) on the TCR-associated CD3 ζ-chain. The zeta-chain-associated protein kinase 70 (Zap70) binds with high affinity to doubly phosphorylated ζ-chain ITAMs through its two Src homology 2 (SH2) domains and is activated by Lck-mediated phosphorylation of its Y319 and Y493 residues [10,11,12]. Zap70 facilitates the activation of phospholipase C-γ1 (PLCγ1), which hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP) to second messengers diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP) [13,14,15]. IP migrates to the cytoplasm to regulate intracellular calcium (Ca) flux from the endoplasmic reticulum [13,16,17], a key modulator of nuclear factor of activated T-cells (NFAT) [18]. While IP migrates to the cytoplasm, DAG operates within the plane of the cell membrane to activate two major signaling molecules: rat sarcoma (Ras) and protein kinase Cθ (PKCθ) [19]. Ras triggers the extracellular signal-regulated kinase (Erk) cascade that results in the activation of activator protein 1 (AP1) [20], while PKCθ activates AP1 and the nuclear factor κB (NFκB) pathway [21,22,23,24]. Upon entering the nucleus, transcription factors, such as NFAT, AP1, NFκB and others, interact to support cooperative regulation of interleukin-2 (IL-2) and other genes involved in T-cell activation [25,26].
A critical second signal driving T-cell activation and fate determination is achieved by incorporating CD28-mediated costimulation [27] and its downstream signaling events into the model. In spite of the ability of the TCR to upregulate factors necessary for full activation, TCR-antigen recognition ultimately leads to anergy or a state of hyporesponsiveness in the absence of a second signal [28]. Alternatively, TCR signaling with additional costimulation of CD28 on the surface of T-cells greatly enhances IL-2 production, T-cell proliferation and the prevention of anergy. This synergistic effect is believed to be mediated through PKCθ and Akt (also known as protein kinase B), which recruit and regulate several signaling molecules, including NFκB [23,29] and mammalian target for rapamycin (mTOR) [30]. The serine/threonine protein kinase mTOR is another important player in CD4+ T-cell activation. mTOR complex 1 (mTORC1) has many known functions, including the ability to control the expression of forkhead box P3 (FOXP3), a master transcriptional regulator in the development of T regulatory cells (Tregs) [30,31]. While much less is known about mTORC2, it was recently shown to promote PKC, Akt and NFκB signaling and to regulate T helper type 1 (Th1) and Th2 commitment [32].
A number of mathematical models exist that span the entirety or portions of these T-cell signaling events. The models presented in [33,34] span these events and are able to capture many dominant behaviors. However, these discrete-time logical models do not support a detailed analysis of the interactions in a continuous state and parameter space and the ability to predict responses to analog knockdown or mutation scenarios. Other models are very detailed, but only consider portions of the system, such as the MAPK [35] and NFκB [36,37] pathways. Some of these models consider every hypothesized molecular interaction or complex formation. This level of detail is prohibitive to complete for the entire signaling network, predominantly due to computational tractability, but also due to uncertainties in the existence or rates of these events. A few models set out to address the role of location as a regulatory event, analyzing T-cell signaling events within a spatial context [38,39]. These models tend to be limited in scope in order to offset the added computational expense of such an analysis. Our model differs from past approaches in that we consider a broad scope of early TCR and CD28 signaling leading to gene transcription in continuous time, consider only the major known phosphorylation, dephosphorylation, association and dissociation events and address location through compartments and not as a partial differential equation with diffusion.
Here, we present a mathematical model of T-cell activation that focuses on the early steps in TCR trafficking and signal amplification by kinases, phosphatases and other second messengers, as well as CD28 coreceptor signaling and gene transcription of IL-2 and FOXP3. Section 2 and Section 3 describe the formulation of the model and its fitting to training data. Model simulations and corroborating experimental evidence are provided in Section 4, demonstrating the model’s ability to reproduce many known experimental results. This section also describes predictions made by the model about the roles of phosphatases CD45 and SHP1 in T-cell activation. Finally, conclusions are summarized in Section 5.
2. Materials and Methods
This section provides an overview of the proposed T-cell activation model and details the methods and materials employed in its development. These include the computational methods and experimental datasets used for model parameter identification.
2.1. The Mathematical Model: Overview and Scope
The mathematical model reflects the current understanding of the intracellular signaling events leading to T-cell activation. The reaction scheme, as shown in Figure 1, encompasses the primary events that occur upon TCR engagement and CD28 costimulation. Appendix 1 describes each signaling element and assigns a symbol and initial value for use in constructing the mathematical model. Appendix 1 also lists the rate equations, parameter values and summarizes the reactions primarily in terms of protein binding, phosphate group transfers and kinase and phosphatase activation. The part of the model representing the TCR-mediated Erk pathway is based on the ordinary differential equation (ODE) model developed by Zheng [40] with updates in Perley et al. [41]. The Zheng model takes into account the major events of early T-cell signaling leading to the activation of the protein tyrosine kinases Zap70 and SFK, the downstream Erk pathway, as well as negative feedback regulation by phosphatase SHP1.
The Zheng model was extended to specifically incorporate TCR trafficking, tuning of CD45 and SHP1 phosphatase activity, refinement of Erk signaling with AP1 activation and the addition of the calcium, NFκB and mTOR pathways leading to IL-2 and FOXP3 transcription. These changes are further detailed in Section 3. Molecular association and complex dissociation are designed to obey the law of mass action, i.e., assuming that the rate of a reaction is proportional to the product of reactant concentrations. Most enzyme-catalyzed reactions utilize second-order kinetics derived from a simplification of the Michaelis–Menten enzyme kinetics by assuming that the substrate concentration is limiting. Such a simplification contributes to reducing the complexity of the nonlinearities while retaining the structure of the pathway connectivity. For reactions catalyzed by enzymes not explicitly included in this model, the expression further reduces to first-order kinetics, where the enzyme concentration becomes a constant. In addition, single molecular transitions are also governed by first-order kinetics. Alternatively, enzyme-catalyzed reactions in which cooperative binding between substrates plays a significant role utilize the Hill equation. Scenarios characterized by these equations include the cooperative transcriptional regulation of IL-2 and FOXP3 and the activation of certain enzymes. The model is also designed to mimic the cell response to stimulation by αCD3, αCD28, phorbol 12-myristate 13-acetate (PMA) and the calcium ionophore ionomycin, as well as mimic perturbations in kinase and phosphatase activity by various small molecule inhibitors. The complete model consists of 48 ODEs with 154 reaction parameters and simulated in MATLAB using the ode15s solver.
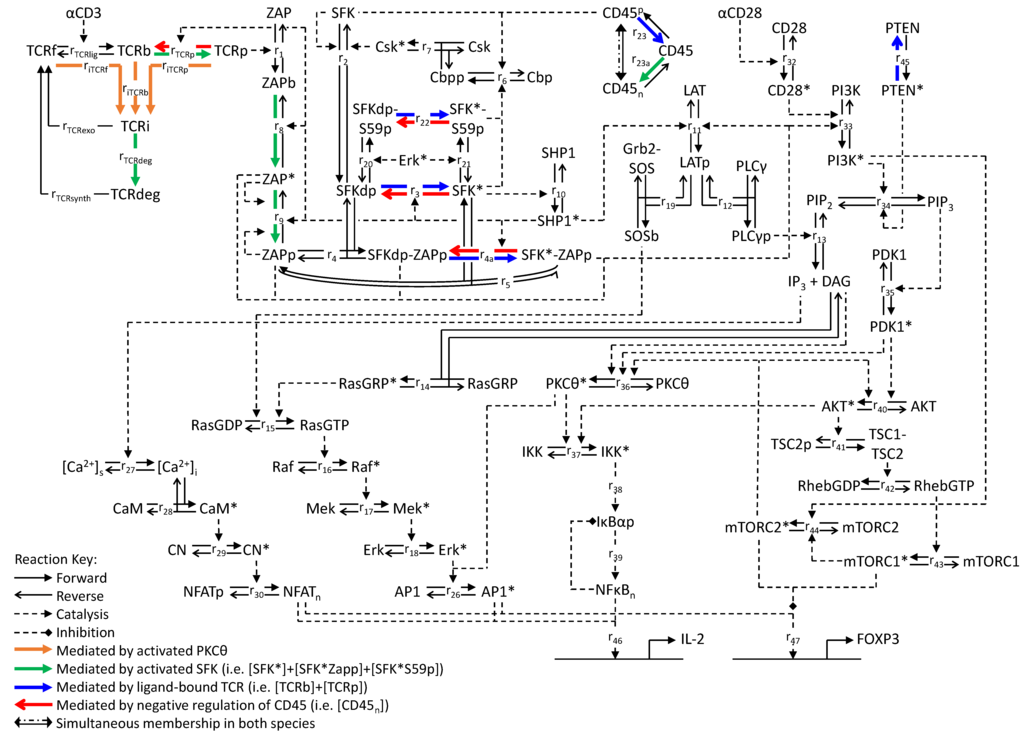
Figure 1.
Reaction network of the T-cell activation model. Solid arrows denote reactions for which the forward and reverse directions are indicated; dashed arrows connect reactions (either forward or reverse) with their catalysts. Colored arrows denote reactions that are catalyzed by specific species as indicated. Symbols and reactions are described in Appendix 1.
Figure 1.
Reaction network of the T-cell activation model. Solid arrows denote reactions for which the forward and reverse directions are indicated; dashed arrows connect reactions (either forward or reverse) with their catalysts. Colored arrows denote reactions that are catalyzed by specific species as indicated. Symbols and reactions are described in Appendix 1.
2.2. Global Sensitivity Analysis
Due to the size and complexity of the model, it is necessary to reduce the dimension of the uncertain parameter space to facilitate an effective and computationally tractable analysis of the model. Parameter sensitivity analysis provides a powerful tool for analyzing mathematical models of complex biological systems and can be used to facilitate parameter estimation by identifying and ranking the various contributors of parametric uncertainty in the model. Parameters with the lowest sensitivity ranks, i.e., parameters whose uncertainty causes the least amount of variability in the output space, can be neglected during the model fitting process and fixed at their initial guesses. For the purpose of this study, a variance-based sensitivity analysis is used to determine how the model output changes over the global parameter space:
where is the sensitivity index of the output to the parameter. The sensitivity indices are numerically computed using Sobol’s method by the efficient sparse-grid-based algorithm proposed by Buzzard [42,43]. Parameters having little effect on model outputs upon perturbation are determined to be insignificant and fixed at their nominal values. Significant parameters are retained for parameter identification.
2.3. Parameter Identification
The ability of the model simulated with a given parameter vector to reproduce available experimental data is quantified by the objective function:
where is the error at each sample time point, is the inverse of the measurement error covariance matrix at time , and are the simulated model outputs and the mean values of the available data at time , respectively, is a binary matrix that indicates which outputs are measured at time and is the total number of sampled time points. This cost function is computed in log space in order to compress the range of and smooth the function, making its approximation with sparse-grid interpolation more effective. The “1” in the cost function is added to avoid the singularity of log at zero.
Specific parameter values that are capable of producing data-consistent model simulations are extracted directly from the literature whenever possible. For those not found in the literature, they are either derived using known conditions or constraints of the system or estimated from experimental data using a combination of manual and automated calibration. First, the starting guess values are roughly estimated by back-of-the-envelope calculations and manual tuning to within a reasonable level of accuracy. This step is used to establish our parameter search space and its bounds, defined to be a hypercube spanning the nominal parameter vector by an order of magnitude both above and below. Next, Equation (2) is evaluated at a number of samples from the parameter hypercube, generated using Latin hypercube sampling in log space. Finally, starting from the parameter vector with the lowest sampled cost, MATLAB’s nonlinear constrained optimization solver, fmincon, is employed to identify the parameter vector that minimizes Equation (2) constrained within the hypercube. To increase the chance of identifying the global minimizer, multiple starting points were considered that span the region of low cost. This procedure of automated parameter identification is aided by the use of sparse-grid interpolation, a powerful tool for generating fast-evaluating approximations of mathematical models [44].
2.4. Experimental Datasets for Model Development
Experimental datasets for the purposes of model development are compiled from the published literature or generated from experiments performed in our lab specifically for this manuscript. For datasets compiled from the published literature, only those that are commensurate with our model in terms of cell type, stimuli, output species and time scale are considered. Table 1 summarizes the attributes of the compiled datasets used to train the model. These datasets consist of dynamics (i.e., time courses), dose response experiments (i.e., multiple doses of an input or stimulus) and input response experiments (i.e., single doses of different input or stimuli combinations). In order to compare the model and observed behaviors directly, simulations are generated to mimic the experimental conditions used in our experimental set-ups (see Appendix 3 and Appendix 4) or as indicated in the corresponding source articles. Model simulations are shown with the corresponding data and error bars whenever possible; however, for cases in which data could not be visualized on the same plot, the reader is referred to the original source.

Table 1.
Description of experimental datasets used for model development.
| Species | Variable(s) | Experiment Type | Cell Type | Figure | Source(s) |
|---|---|---|---|---|---|
| TCR | αCD3 | Dynamic dose response | Jurkat | 2 | [45,46] |
| Zap70 | αCD3 | Dose response | Jurkat | 4 | [40] |
| Erk | Sanguinarine, U0126 | Dynamic dose response | Jurkat | 5 | [41] |
| Ca | αCD3 | Dynamics | Jurkat | 6 | Appendix 3 |
| IκBα | αCD3+αCD28 | Dynamics | Jurkat | 7 | Appendix 4 |
| Akt, PKCθ | αCD3, αCD28 | Input response | CD4+ T-cells (murine) | 8 | [32] |
3. Model Development
The following subsections describe the process of formulating the proposed T-cell activation model as shown in Figure 1 from the original ODE model presented by Zheng [40]. These changes include the incorporation of TCR trafficking mechanisms, tuning of CD45 and SHP1 phosphatase activity, refinement of Erk signaling with AP1 activation and the addition of the calcium, NFκB and mTOR pathways leading to IL-2 and FOXP3 transcription. These changes are further detailed in the following subsections.
3.1. Modeling TCR Trafficking
The TCR expression level at the cell surface is the result of a dynamic equilibrium maintained by the membrane expression of newly synthesized TCR, receptor internalization, recycling to the cell surface and degradation [46,47]. As depicted in Figure 1, the TCR complex is represented in the model by five states: free TCR (), ligand-bound TCR (), phosphorylated ligand-bound TCR (), internalized TCR () and degraded TCR (). TCR trafficking is modeled by allowing TCR complexes from all surface states (, and ) to be internalized [38]. Internalized TCR subunits can be degraded or recycled back to the unbound state. Newly synthesized TCR contributes to the free receptor state with a rate proportional to degraded TCR.
Early studies demonstrated that the TCR is a constitutively cycling receptor [46,47]. Thus, at steady state, a certain amount of TCR is endocytosed, while at the same time, an equal amount of TCR is exocytosed. The associated model rate parameters are set by the following observations. Several studies seem to agree that the constitutive endocytic rate constant for the TCR in resting T-cells is ~0.01 min, meaning that ~1% of the cell surface-expressed TCR is internalized each minute [46].
TCR ligation induces downregulation and degradation of the TCR in a dose-dependent manner [45,46]. In theory, TCR downregulation can be accomplished by an increase in the endocytic rate constant, a decrease in the exocytic rate constant or a combination of both. However, most studies found that TCR downregulation is caused by an increase in the endocytic rate constant to ~0.038 min after TCR triggering [46], whereas the exocytic rate was constant. In addition, TCR degradation was also found to accelerate after TCR ligation. The TCR subunits in non-stimulated Jurkat cells were observed to degrade with rate constants of ~0.0011 min, resulting in a half-life of ~10.5 h. Triggering of the TCR by anti-TCR Abs resulted in a three-fold increase in the degradation rate constants to ~0.0033 min, resulting in a half-life of ~3.5 h [45]. This process was modeled by making the receptor internalization and degradation rate parameters functions related to T-cell activation. These mechanisms were mathematically encoded in the model using:
Equation (3) ensures a continuous transition between the constitutive endocytosis or degradation rates () and the ligand-induced rates () using a Hill equation, where X is the induction substrate concentration, K is the substrate concentration of the transition midpoint and n is the Hill coefficient denoting positive () or negative () cooperativity. Since the TCR cycling pathway is dependent on the CD3γ di-leucine-based motif and is activated by PKC [46,47], for receptor cycling, we set k = , X = , n = 2, and K is set to 5% of total PKCθ with = 0.01 min and = 0.038 min. Since the activation of Lck and Zap70 leads to recruitment of Cbl and ubiquitination (Ub) of the CD3 and ζ chains to induce degradation of the TCR in the lysosomes [46], for receptor degradation, we set k = , X = , n = 2, and K is set to 5% of total SFK with = 0.0011 min and = 0.0033 min.
At equilibrium without any stimulus, most studies also seem to agree that the pool of recycling TCR was distributed with approximately 75%–85% at the cell surface and 15%–25% inside the cells. Assuming the minimum constitutive rates for internalization and degradation with 80% surface TCR and 20% inside the cell, of which half are being degraded, we can compute the constitutive exocytosis rate as = 0.0789 min and the constitutive synthesis rate as = to achieve the observed dynamic equilibrium in resting T-cells.
The resulting simulated surface and degraded TCR dynamics are consistent with expectations at equilibrium and after receptor engagement with a ligand. The dose response curves in Figure 2A,B show good agreement with the behaviors reported by Menne et al. [47], Geisler et al. [46] and von Essen et al. [45].
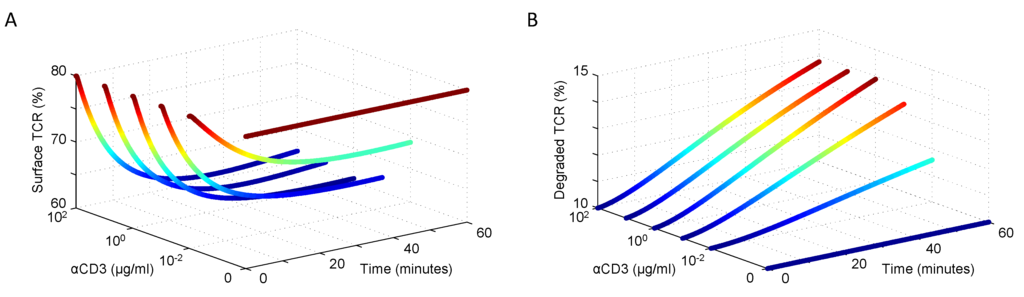
Figure 2.
Resting and ligand-induced (A) surface TCR expression and internalization and (B) degradation activity in response to varying doses of αCD3 stimulation (data shown in [45]).
Figure 2.
Resting and ligand-induced (A) surface TCR expression and internalization and (B) degradation activity in response to varying doses of αCD3 stimulation (data shown in [45]).
3.2. Tuning the Roles of CD45 and SHP1
The protein tyrosine phosphatases, CD45 and SHP1, are central players in signal amplification following antigen recognition by the TCR and the activation of many downstream second messenger and signaling molecules. CD45 is a leukocyte-specific transmembrane glycoprotein and a receptor-like protein tyrosine phosphatase (PTP) [8]. The SFK member Lck is the best characterized CD45 substrate in T-cells [7]. Lck exists in dynamic equilibrium with three main sub-populations: (1) open and activated ( in the model); (2) open and not activated (“primed”) (); (3) closed and not activated () [9]. Phosphorylation of SFKs at the negative regulatory site (Y505 in Lck) by the kinase Csk results in an intramolecular interaction with the SH2 domain, creating a folded inactive conformation [48]. Dephosphorylation at this site by CD45 opens up the molecule, creating a “primed” molecule. Clustering of these primed SFKs results in the transphosphorylation of the activation loop (Y394 in Lck), which displaces it from the catalytic site and creates an active kinase by allowing substrate access. Dephosphorylation at this site by CD45 or other PTPs, such as SHP1, downregulates SFK activity and returns them to the primed state [9]. Thus, CD45 functions as both a positive and negative regulator of the T-cell antigen receptor and in setting the threshold of activation.
The dual role of CD45 as both positive and negative regulator of T-cell activation is modulated in part by the distribution and movement of CD45 and its substrates proximal to the receptor complex inside and outside lipid rafts. Lck inside lipid rafts has been reported to be hyperphosphorylated and less active when CD45 is excluded [9]. The engineered inclusion of CD45 to the lipid domains also decreases TCR signaling, consistent with its ability to downregulate signaling by dephosphorylating the positive regulatory site at later time points [9]. These observations show that the localization of both CD45 and the SFKs can affect the phosphorylation state of SFKs. It is believed that upon receptor-mediated clustering, the SFKs are activated and often relocalize to lipid rafts, where the concentration of CD45 is much lower and the kinases can experience sustained signaling. The later recruitment of CD45 to these domains will then primarily dephosphorylate the activation site and downregulate activity [9].
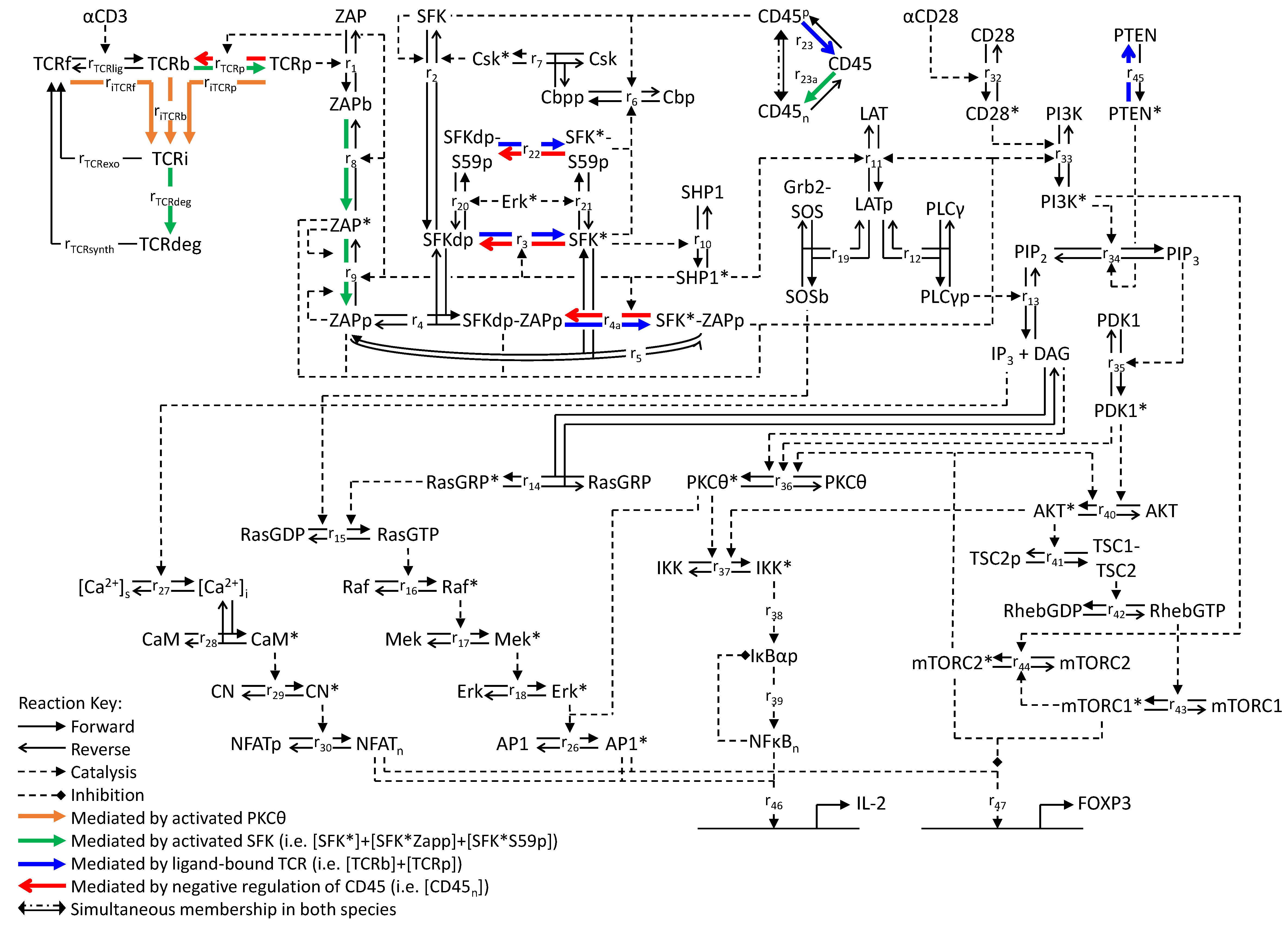
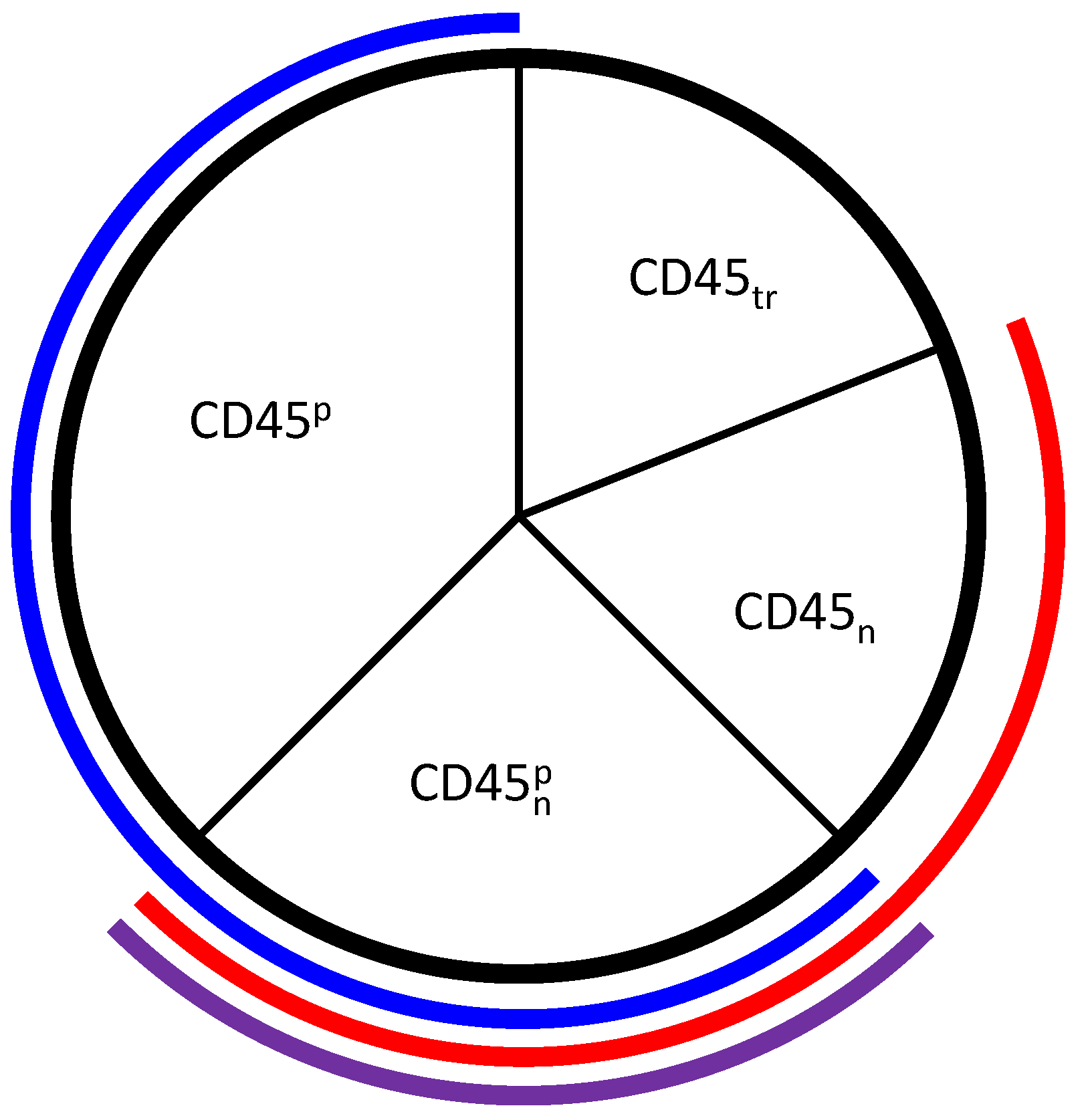
The model is augmented to include both the positive and negative regulatory roles of CD45, as well as spatial localization effects, on T-cell activation with two explicit states: and (see Figure 3). The first, , reflects its ability to activate substrates immediately following TCR triggering and promote signaling. Initially, this state exists at a high level with its major role being to dephosphorylate negative regulatory sites and “prime” SFKs (). Following TCR engagement, the receptor cluster formation (represented in simulation by ligand-bound TCR) separates CD45 and its substrate. This causes to enter an ineffective state, an implicitly modeled state called (), and decreases its ability to activate its substrate. The second state, , represents the negative regulatory function of CD45 on SFK. Initially, this state is minimally active with most existing in the inactive state, . As TCR signaling progresses (represented in simulation by activated SFK), CD45 is recruited back to the lipid rafts (), where its ability to dephosphorylate activated SFKs is the dominant role (, , and ). Since CD45 can possess both positive and negative roles simultaneously, the species and are not mutually exclusive; however, the intersection between these two sets, a CD45-related state possessing both roles (), is not explicitly modeled or tracked. , representing completely inactive CD45 that is translocated away from its substrates, is also not explicitly modeled.
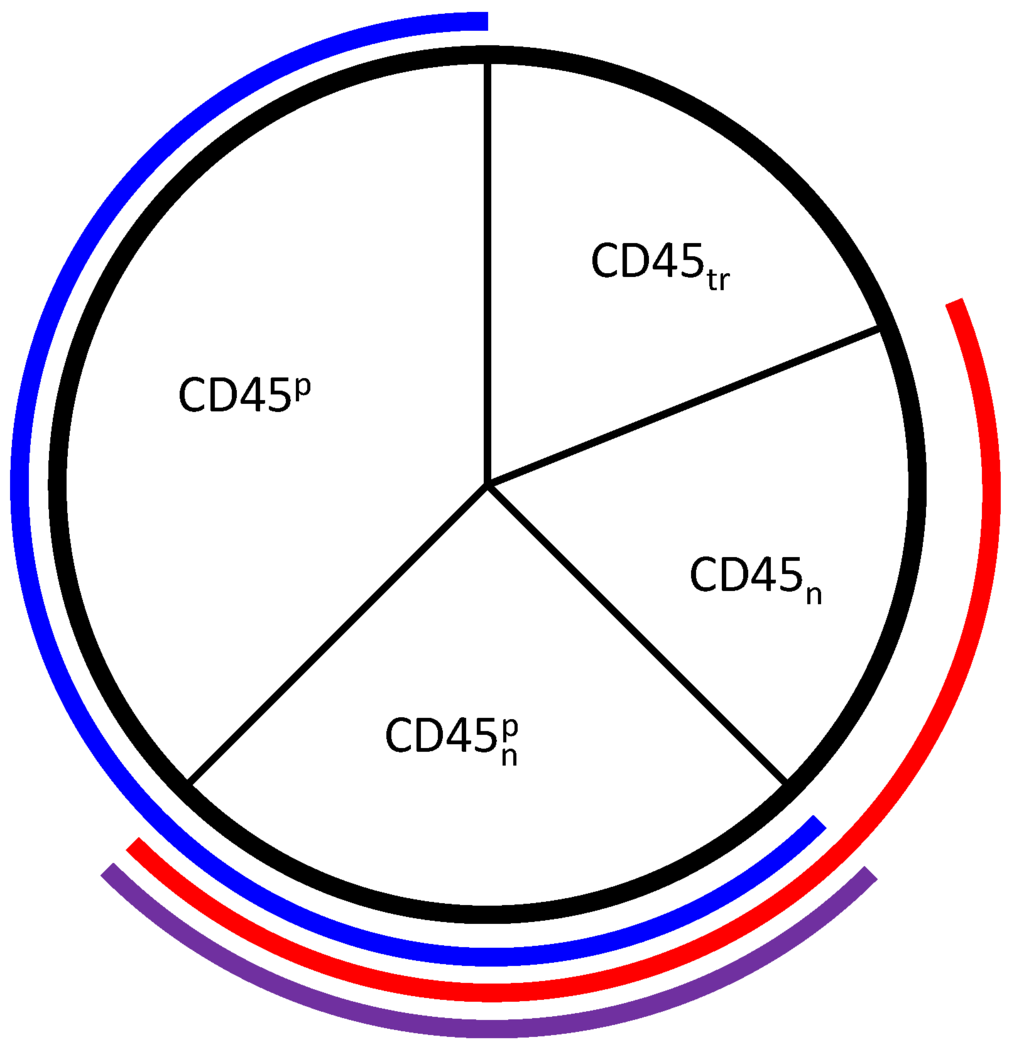
Figure 3.
Representation of the phosphatase CD45 in the model. The model states and are represented by the regions outlined in blue and red, respectively. , which is the intersection between the two modeled states outlined in purple, has both positive and negative roles, but is not explicitly modeled. represents completely inactive CD45 that is translocated away from its substrates and is also not explicitly modeled.
Figure 3.
Representation of the phosphatase CD45 in the model. The model states and are represented by the regions outlined in blue and red, respectively. , which is the intersection between the two modeled states outlined in purple, has both positive and negative roles, but is not explicitly modeled. represents completely inactive CD45 that is translocated away from its substrates and is also not explicitly modeled.
In contrast to the dual role of CD45, Src homology region 2 domain-containing phosphatase-1 (SHP1) functions primarily as a negative regulator of TCR signal transduction. SHP1 associates with and negatively regulates the Syk family kinase Zap70 upon T-cell activation ( and ), thereby suppressing TCR signaling [49]. SHP1 also forms a negative feedback loop that is composed of (1) SHP1 phosphorylation by activated Lck, (2) binding of phospho-SHP1 to Lck and (3) Lck inactivation by SHP1-mediated dephosphorylation () [50]. Erk, on the other hand, antagonizes SHP1 activity by modifying Lck (S59 phosphorylated by MAPK/Erk), which interferes with SHP1 recruitment and Lck inactivation [50,51]. These events are captured by partitioning the SFK states with S59 phosphorylation (i.e., and ) from those without ( and ).
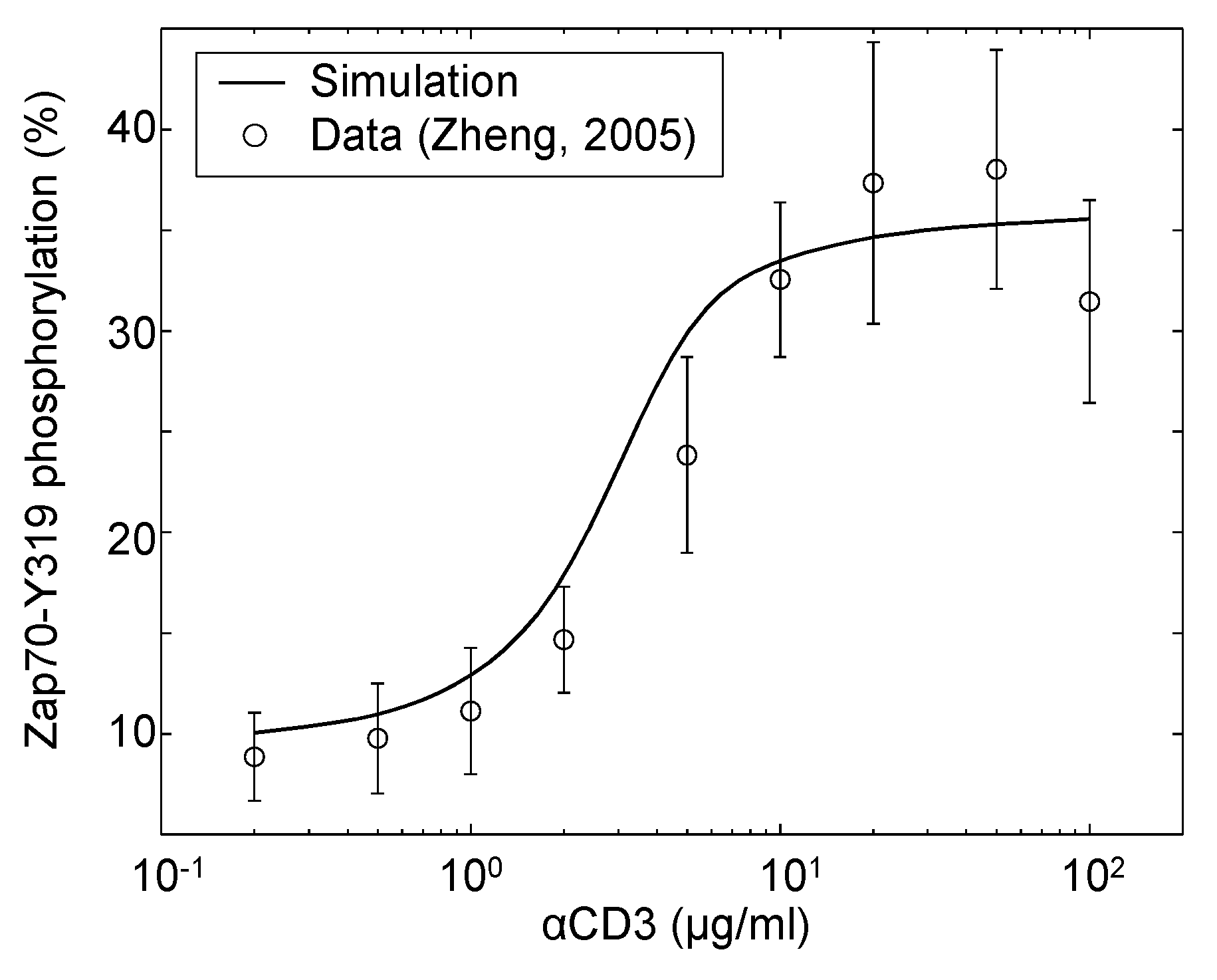
Parameters for this updated model structure, which include reactions - and -, are trained to recapitulate the behaviors and roles of CD45 and SHP1 in early TCR signaling. To effectively isolate the early TCR signaling module from downstream feedback, the Erk positive feedback loop is suppressed with the presence of the Mek1/2 inhibitor U0126. Figure 4 shows the changes in the Zap70-Y319 phosphorylation level with different doses of αCD3 stimulation in the presence of U0126. The results show that with increasing doses of stimulation, the response of Zap70 increases slowly initially, then rapidly between 2 and 5 μg/mL of αCD3 and eventually saturates and even decreases slightly at the highest stimulus level (100 μg/mL). The dose response curve demonstrates the existence of a threshold αCD3 stimulation concentration. Below the threshold level, CD45 is unable to activate enough SFK and indirectly Zap70 to overcome the negative regulation by SHP1 and CD45 itself to sustain signaling. Once the threshold is crossed, the negative feedback barrier caused by SHP1 is overcome, and Zap70 signaling increases rapidly and eventually saturates at a high stimulus level. The Zap70 dose response simulations show good agreement with the observations originally reported by Zheng [40].
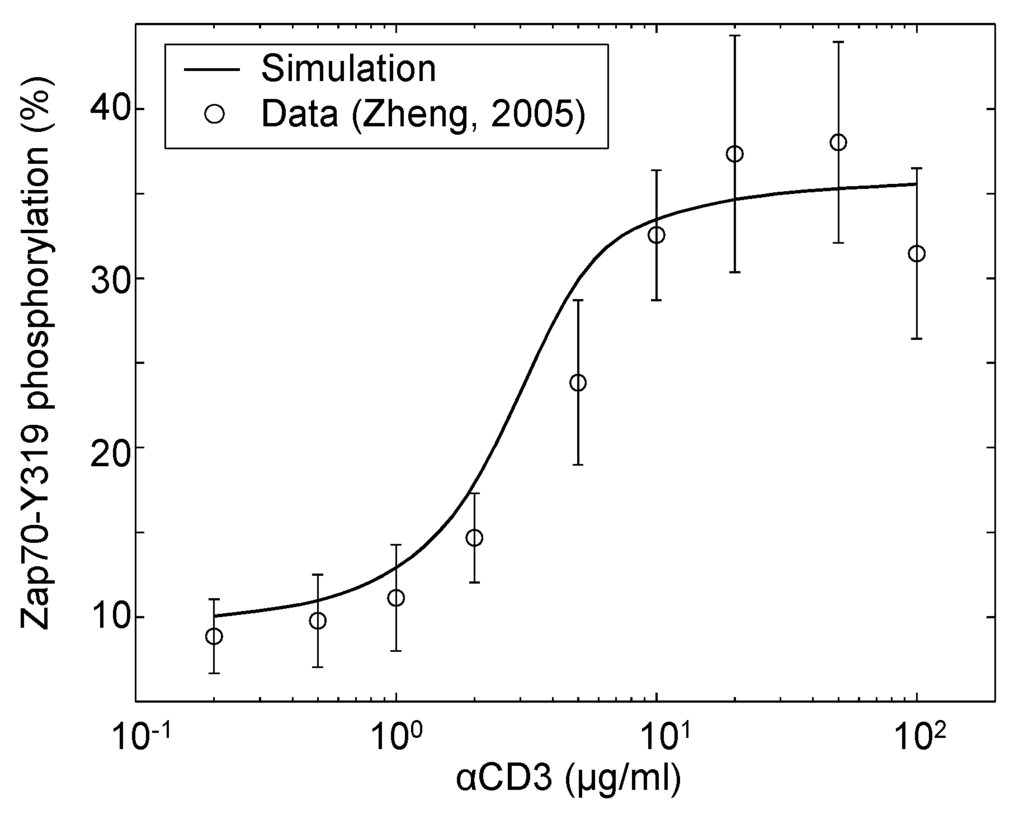
Figure 4.
Zap70-Y319 phosphorylation in response to various doses of αCD3 stimulation in the absence of Erk feedback. Jurkat cells were incubated in the presence of a Mek1/2 inhibitor (2 μg/mL U0126) and stimulated with αCD3 at the indicated concentrations. Samples were taken 5 min post-stimulation and analyzed by western blot. The data shown are the means and standard errors from at least three independent experiments (data source: [40]).
Figure 4.
Zap70-Y319 phosphorylation in response to various doses of αCD3 stimulation in the absence of Erk feedback. Jurkat cells were incubated in the presence of a Mek1/2 inhibitor (2 μg/mL U0126) and stimulated with αCD3 at the indicated concentrations. Samples were taken 5 min post-stimulation and analyzed by western blot. The data shown are the means and standard errors from at least three independent experiments (data source: [40]).
3.3. Tuning Erk Signaling
The Erk signaling cascade plays an important role in IL-2 activation via the transcription factor, AP1. Activated Zap70 mobilizes a number of adapter proteins to the receptor complex, including linker of activated T-cells (LAT) and growth factor receptor-bound protein 2 (Grb2) [52]. Son of Sevenless (SOS), a Ras guanine exchange factor (GEF), forms a complex with Grb2 and LAT to facilitate the activation of Ras in T-cells. LAT also facilitates the activation of Ras through the RAS guanyl nucleotide-releasing protein-1 (RasGRP1), which is itself activated by DAG [19]. Ras triggers the extracellular signal-regulated kinase (Erk) cascade that results in the activation of AP1 [20]. As mentioned in Section 3.2, Erk also promotes its own activation by phosphorylating Lck at S59 and antagonizing SHP1 activity [50,51].
These reactions (-) are modeled using the first- and second-order mass action kinetic equations of the base model presented by Zheng [40]. The kinetic parameters in this module are fitted to Erk phosphorylation data presented by Perley et al. [41]. Figure 5A,B shows the changes in the Erk phosphorylation with different doses of the MKP inhibitor sanguinarine and Mek1/2 inhibitor U0126. For each experiment, Jurkat cells were stimulated with 10 μg/mL αCD3, followed by a dose of inhibitor once the indicated amount of time had past. Samples were taken before and after the inhibitor dose administrations to show how the system changes over time with varying degrees of inhibition. At the lowest concentrations (where inhibition is negligible), the transient nature of Erk phosphorylation can be seen by the decreasing level over time. As the inhibitor concentrations increase, a threshold is crossed, resulting in more forceful inhibition: 10 μg/mL for sanguinarine and 1 μg/mL for U0126. The phospho-Erk simulations show good agreement with the time course and dose response observations originally reported by Perley et al. [41].
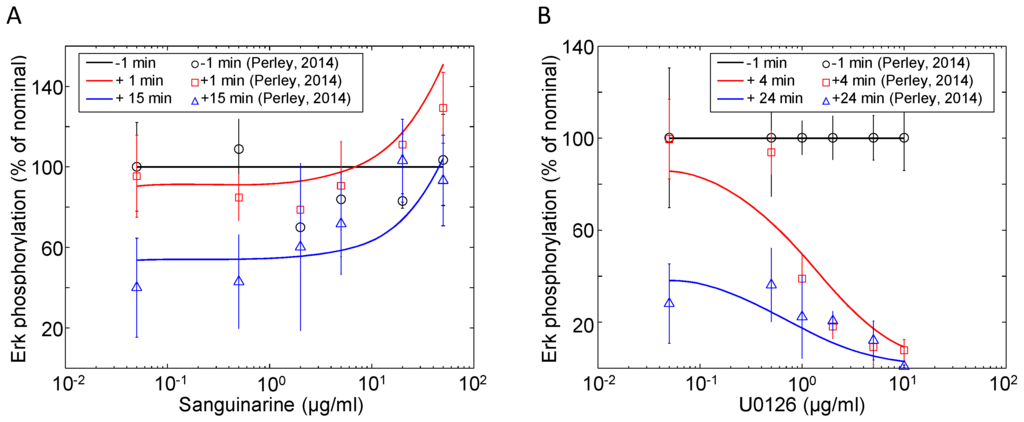
Figure 5.
Erk activation in response to various doses of (A) MKP inhibitor sanguinarine and (B) Mek1/2 inhibitor U0126. Doses of sanguinarine and U0126 were administered at 15 and 6 min post-stimulation (10 μg/mL αCD3), respectively. Samples of phospho-Erk were taken at indicated times relative to the inhibitor doses and measured via western blot. The data shown are the means and standard errors from at least three independent experiments (data source: [41]).
Figure 5.
Erk activation in response to various doses of (A) MKP inhibitor sanguinarine and (B) Mek1/2 inhibitor U0126. Doses of sanguinarine and U0126 were administered at 15 and 6 min post-stimulation (10 μg/mL αCD3), respectively. Samples of phospho-Erk were taken at indicated times relative to the inhibitor doses and measured via western blot. The data shown are the means and standard errors from at least three independent experiments (data source: [41]).
3.4. Modeling Calcium Signaling
Calcium is an important second messenger involved in a number of cellular processes, including the activation of NFAT and IL-2. PLCγ1, activated by Zap70 via LAT, hydrolyzes PIP to DAG and IP () [13,14,15]. IP migrates to the cytoplasm and interacts with IP receptors on the endoplasmic reticulum, inducing the release of stored calcium (Ca) () [13,16,17]. Calmodulin (CaM), a small calcium-sensing protein and signal transducer, binds released Ca ions () and undergoes a conformational change to activate the calcium-dependent serine-threonine phosphatase calcineurin (CN) () [53]. Activated CN dephosphorylates the nuclear factor of activated T-cell (NFAT) proteins, exposing their nuclear-localization signal (NLS) and inducing nuclear translocation () [18].
The model is modified to recapitulate calcium signaling with these reactions (, -), modeled using first- and second-order mass action kinetic expressions similar to that of the Erk signaling module. The kinetic parameters in this module are fitted to Ca flux data for which the experimental methodology is presented in Appendix 3. Figure 6 shows the experimental observations of intracellular calcium in Jurkat cells stimulated with 10 μg/mL αCD3 and the corresponding model simulation. Upon TCR stimulation, calcium ions are rapidly released from intracellular stores, peaking after 2 min before returning to a slightly elevated level. It is evident from the plot that the model readily captures the rapid transience of intracellular calcium flux.
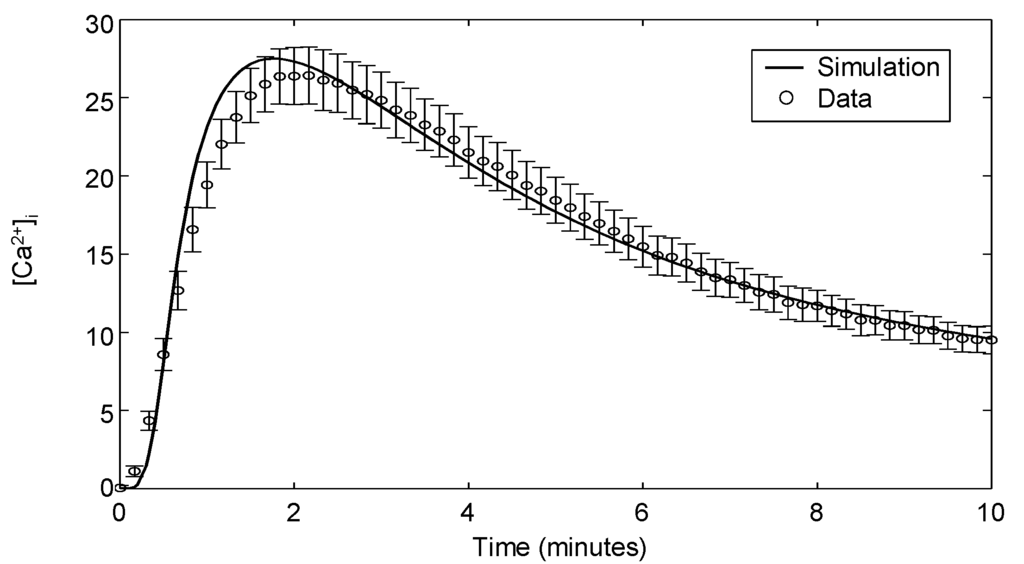
Figure 6.
Intracellular calcium release in response to stimulation with 10 μg/mL αCD3. The data shown are the means and standard errors from 12 independent experiments.
Figure 6.
Intracellular calcium release in response to stimulation with 10 μg/mL αCD3. The data shown are the means and standard errors from 12 independent experiments.
3.5. Modeling CD28 Costimulation
In spite of the ability of the TCR to upregulate many factors necessary for full activation, TCR-antigen recognition ultimately leads to anergy, or a state of hyporesponsiveness, in the absence of a second signal [28]. By contrast, signaling via CD28 on the surface of T-cells, in addition to TCR signaling, greatly enhances IL-2 production, T-cell proliferation and the prevention of anergy. As a result, CD28-mediated costimulation provides a critical second signal in T-cell activation and fate determination [27]. CD28 signaling is believed to be mediated by phosphoinositide 3-kinase (PIK) () [54]. PIK phosphorylates PIP to become phosphatidylinositol 3,4,5-trisphosphate (PIP), an activity that is directly antagonized by phosphatase and tensin homolog (PTEN) (). PIP serves as pleckstrin homology (PH) domain membrane anchors for 3-phosphoinositide-dependent kinase-1 (PDK1) (). PDK1 activation leads to the membrane recruitment of PKCθ () and protein kinase B (PKB, or Akt) () to regulate several signaling pathways, including NFκB [23,29] and mTOR [30].
The model is augmented to simulate the effects of CD28 costimulation on T-cell activation. As with the other signaling modules, these reactions (-) are modeled primarily using first- and second-order mass action kinetic expressions. The corresponding kinetic parameters are tuned after accounting for downstream signaling events, such as NFκB and mTOR signaling, which are described in the following sections.
3.6. Modeling NFκB Signaling
Several lines of evidence indicate that the NFκB pathway is perhaps the most relevant biochemical or transcriptional target for the costimulatory activity of CD28 [29]. TCR- and CD28-mediated induction of the NFκB signaling pathway intersect at the central regulator, PKCθ (via mechanisms described in Section 3.5). PKCθ induces NFκB signaling by activating IκB kinase (IKK) () [21], which phosphorylates the inhibitor IκB (at positions S32 and S36). This triggers the rapid polyubiquitination (at positions K21 and K22) and proteolysis of IκB in the 26S proteasome complex (). IκB degradation exposes the nuclear localization signal of NFκB, allowing its rapid translocation into the nucleus () [23,24].
Physiologically, the defining characteristic of IκBα is its ability to regulate rapid, but transient, induction of NFκB activity, owing to the participation of IκBα in an autoregulatory feedback loop. That is, the activation of NFκB causes the upregulation of transcription of IκBα, which, in turn, serves to shut off its own nuclear localization signal [36,37]. This upregulation occurs due to the presence of κB sites in the IκBα promoter. Thus, IκBα is thought to maintain the transient effect of inducing agents on the transcription of NFκB responsive genes [23]. It was demonstrated that in vivo degradation of IκBα is required for the appearance of NFκB in the nucleus. In addition, some investigators were able to demonstrate by co-immunoprecipitating rel proteins with IκBα, from stimulated cells treated with proteasome inhibitors to block the degradation of phosphorylated IκBα, that IκBα undergoes degradation mediated by the 26S proteasome after phosphorylation, but before dissociation [23].
We model the autoregulatory relationship between IκBα and NFκB ( and ) as a two-component negative feedback system:
The dynamic behavior of the negative feedback depends on the relative efficiency of the feedback regulation (α and β) regulating oscillation persistence versus self-regulation (γ and δ), causing oscillation damping. The output, , can range from persistent oscillations (high feedback efficiency and no damping, ) to gradual rising to a plateau level (low feedback efficiency and high damping). Figure 7 demonstrates the phosphorylation of IκBα by IKK, subsequent rapid degradation by the 26S proteasome and synthesis of IκBα promoted by nuclear NFκB. The corresponding NFκB signal is also depicted. Model parameters are fitted to phospho-IκBα data for which the experimental methodology is specified in Appendix 4. In our case, the fitted parameters correspond to damped oscillations (intermediate feedback efficiency and intermediate damping). Although we note that the sample size is small, the model simulation does show good agreement with the trend seen in the data.
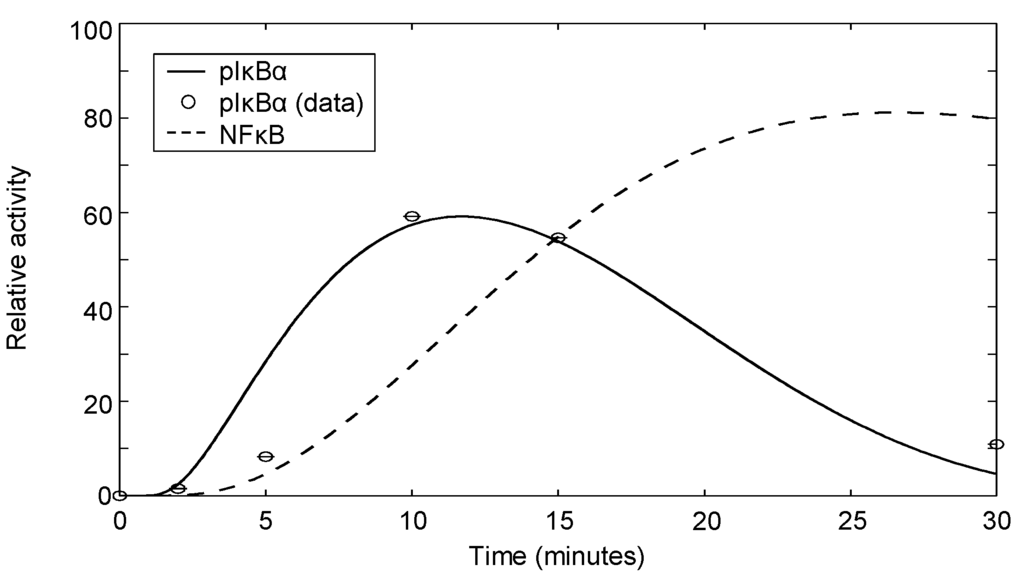
Figure 7.
IκBα phosphorylation following 2 μg/mL αCD3 and αCD28 costimulation. The data shown are quantified western blot data from one experiment.
Figure 7.
IκBα phosphorylation following 2 μg/mL αCD3 and αCD28 costimulation. The data shown are quantified western blot data from one experiment.
3.7. Modeling mTOR Signaling
Mammalian target of rapamycin (mTOR) is an evolutionary conserved serine/threonine protein kinase that is well known for its ability to control T-cell activation and differentiation. mTOR is necessary for TCR-induced signaling to drive differentiation into Th1, Th17 or Th2 effector types in vitro or in vivo under polarizing conditions. On the other hand, mTOR-deficiency drives CD4+ T-cells to the generation of FOXP3+ T-cells, even under normal activating conditions [30]. The ability of mTOR to regulate the expression of FOXP3 makes it a key player in the development of Tregs [30,31].
TCR and CD28 costimulation leads to the membrane recruitment of Akt (via mechanisms described in Section 3.5), where it is phosphorylated (at position T308) by PDK1 (). Activated Akt phosphorylates tuberous sclerosis complex 2 (TSC2) in an inhibitory manner, yielding a separation of the TSC1/TSC2 complex (). This causes Ras homolog enriched in brain (Rheb) to lose its GTPase-activating protein (GAP) activity (). The resulting accumulation of Rheb-GTP promotes mTOR complex 1 (mTORC1) function (), which acts to downregulate FOXP3 expression () [30]. By contrast, mTORC2 appears to be involved in cross-talk between Akt, FOXP3, PKCθ and NFκB. mTORC2 was shown to promote phosphorylation of Akt () and PKC (), Akt activity, nuclear NFκB and to regulate Th1 and Th2 commitment in response to T-cell activation [32].
The model is augmented with the described reactions (-), which consist primarily of first- and second-order mass action kinetic expressions. Hill equations are employed in cases, such as PTEN deactivation by TCR triggering (), TSC2 phosphorylation () and IL-2 () and FOXP3 () transcription, to better describe apparent cooperativity between transcriptions factors [26,31]. Kinetic parameters are chosen to reproduce qualitative observations of Akt and PKC phosphorylation in response to αCD3 and αCD28 costimulation originally reported by Lee et al. [32], the results of which are shown in Figure 8. In the model, as with the data, while αCD3 and αCD28 are each capable of inducing Akt and PKCθ phosphorylation, both are required to induce full activation of these pathways.
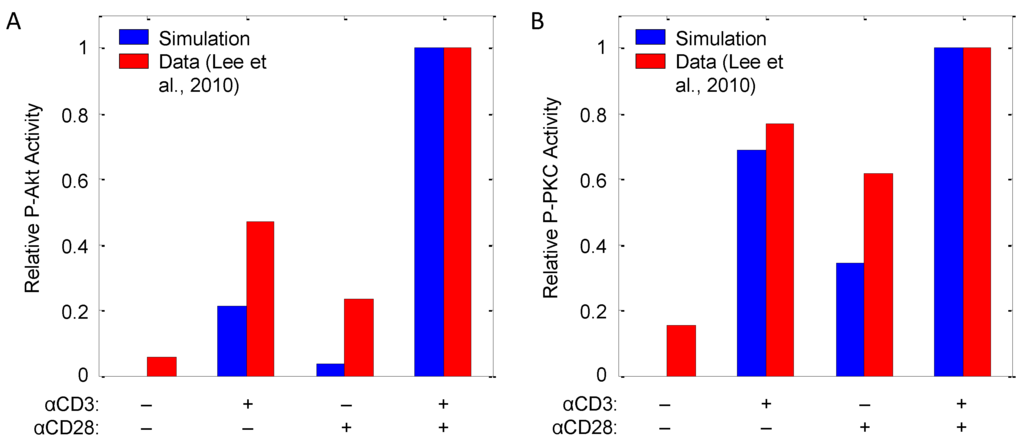
Figure 8.
T-cell signaling in response to doses of αCD3 and αCD28 as measured by (A) P-Akt and (B) P-PKCθ. CD4+ T-cells were stimulated (40 min) with 0.5 mg/mL plate-bound αCD3, 2.5 mg/mL of soluble αCD28 or both. Bar graphs quantify phosphorylation of Akt and PKC, with each sample normalized to the level of unphosphorylated protein in one experiment representative of three replicates (data source: [32]).
Figure 8.
T-cell signaling in response to doses of αCD3 and αCD28 as measured by (A) P-Akt and (B) P-PKCθ. CD4+ T-cells were stimulated (40 min) with 0.5 mg/mL plate-bound αCD3, 2.5 mg/mL of soluble αCD28 or both. Bar graphs quantify phosphorylation of Akt and PKC, with each sample normalized to the level of unphosphorylated protein in one experiment representative of three replicates (data source: [32]).
4. Results and Discussion
The following results and discussion subsections are divided into two main groups. First, the complete and calibrated model, as described in Section 3, is evaluated against additional data not used in the model tuning process. This experimental evidence and corroboration of the model’s predictive accuracy is described in Section 4.1. The sections that follow then describe predictions made using the model. As mentioned in the introduction, these will mainly focus on the roles of signal strength and feedback loops on the regulation of T-cell activation.
4.1. Model Corroboration
4.1.1. Experimental Datasets for Model Corroboration
Experimental datasets for the purposes of model corroboration are compiled from the published literature. Only those that are commensurate with our model in terms of cell type, stimuli, output species and time scale are considered. Table 2 summarizes the attributes of the compiled dataset. Each dataset is used to analyze the model’s ability to recapitulate the dynamics of the biological system as indicated. These datasets consist of dynamics (i.e., time courses), dose responses (i.e., multiple doses of an input or stimulus), input responses (i.e., single doses of different input or stimuli combinations) and knockdown or knockout (i.e., activity of a particular species is reduced or eliminated) experiments. In order to compare the model and observed behaviors directly, simulations are generated to mimic the experimental conditions used in the published experimental set-ups. Model simulations are shown with the corresponding data and error bars whenever possible.
Table 2.
Description of experimental datasets used for model corroboration.
| Species | Variable(s) | Experiment Type | Cell Type | Figure | Source |
|---|---|---|---|---|---|
| Ca | αCD3, αCD28, PMA, ionomycin | Dynamic input response | Jurkat | 9 | [27] |
| NFAT | Cyclosporin A | Dose response | Jurkat | 9 | [53] |
| NFAT, AP1, NFκB | αCD3, αCD28, PMA | Input response | Jurkat | 10 | [27] |
| Lck, Erk | CD45 | Knockdown | Murine DPthymocytes | 11 | [55] |
| Ca | CD45 | Knockout | Murine DP thymocytes | 11 | [56] |
| IL-2 | SHP1, PMA, aTCR, ionomycin | Input response, Knockdown | Jurkat | 12 | [49] |
4.1.2. Corroboration of Signaling Events and Transcription Factor Activation
In order to corroborate the model, we perform several in silico experiments and compare the simulations to published experimental results (described in Table 2). Figure 9A shows the changes in the intracellular calcium release in response to different combinations of stimuli. As shown in the figure, only αCD3 and ionomycin are capable of inducing calcium signaling, with ionomycin (administered with PMA) being the stronger of the two inducers. For the most part, the model simulations are in good qualitative agreement with the observations reported by Smeets et al. [27]; however, there are two noticeable discrepancies. First, these data show that the model slightly overestimates the system’s calcium sensitivity to ionomycin stimulation relative to that of αCD3. Since the model is shown to fit calcium signaling quite well (see Figure 6), more information on the effects of ionomycin would likely resolve this issue. Second, the model’s predicted calcium response demonstrates a slight sensitivity to PMA, a trend that is not observed in the data. This model behavior is likely caused by the presence of the positive feedback loop mediated by Erk. PMA is considered a DAG substitute, activating the substrates of DAG, such as RasGRP, which leads to the activation of Erk. Erk, in turn, promotes signaling by preventing SFK from activating the SHP1 negative feedback loop. In the model, the effect of elevated SFK facilitates further signaling through LAT, PLCγ, IP and eventually calcium. This may be evidence that the model, particularly the positive feedback loop, may be more sensitive than these experimental data suggest.
Figure 9B shows changes in nuclear translocation of NFAT, a downstream target of calcium signaling, with varying doses of the calcineurin inhibitor cyclosporin A (CsA). Model simulations show excellent agreement with the observations reported by Clipstone et al. [53] in terms of half-maximal inhibitory concentration (IC) at ~3.5 ng/mL CsA. However, the model does appear to be slightly less sensitive to changes in inhibitor concentrations, as the slopes of the two curves are slightly different.
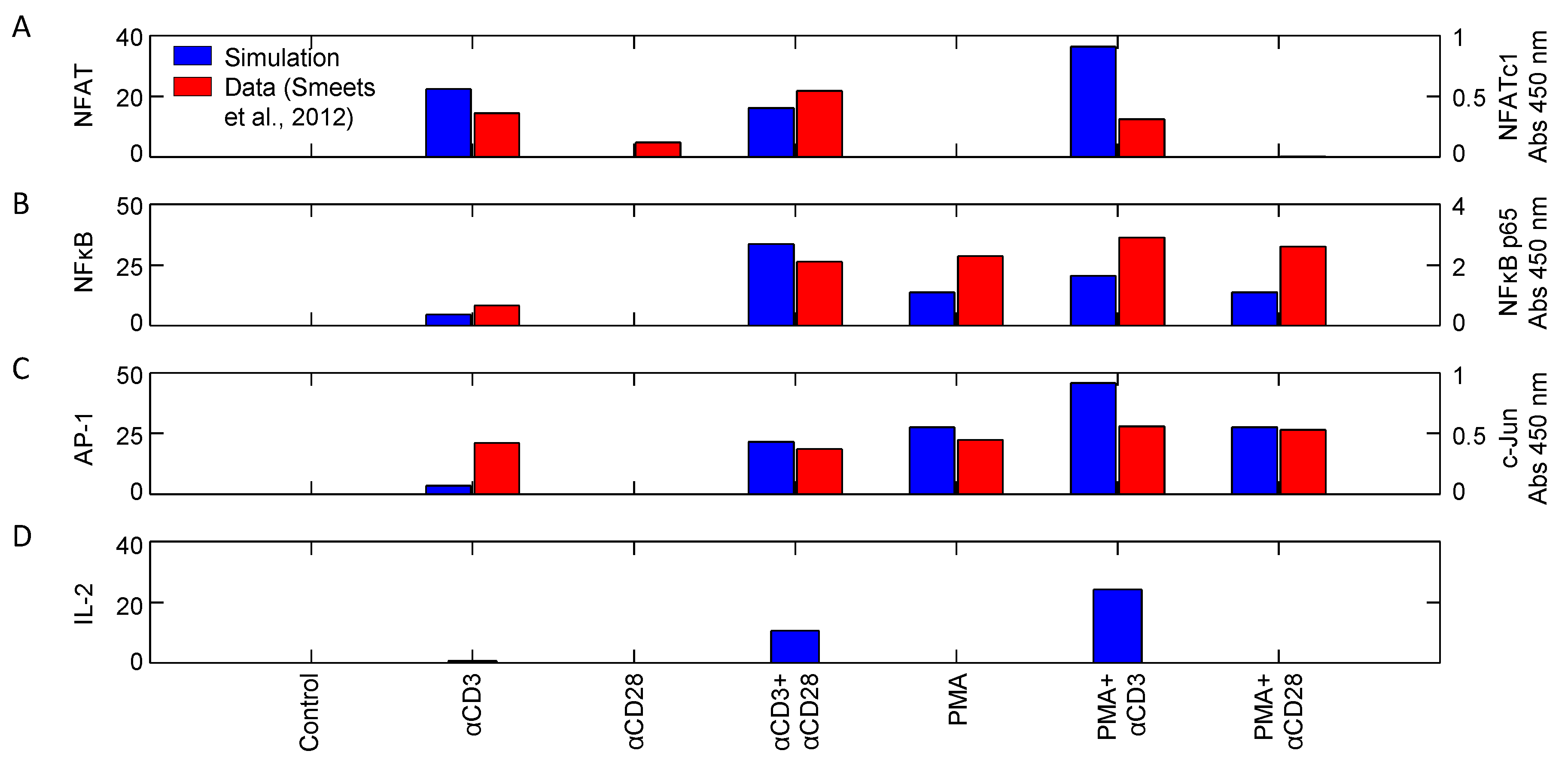
Figure 10 shows the activity of transcription factors NFAT, NFκB and AP1, as well as IL-2 synthesis in response to different combinations of stimuli. While there are a few obvious quantitative discrepancies between the model simulations and the data presented by Smeets et al., there is good qualitative agreement among them in the input/output relationships. Nuclear localization of NFAT is only achieved when stimulating with combinations involving αCD3 (Figure 10A). This is because NFAT translocation requires strong calcium signaling, which αCD28 and PMA stimulation alone could not induce, consistent with [17] and Figure 9A. On the other hand, both αCD3 and PMA are sufficient to induce nuclear localization of NFκB and AP1 (Figure 10B,C). This is promoted by αCD28, but a combination of αCD3 and PMA have the greatest influence on translocation. IL-2 transcription in response to these same stimuli is shown in Figure 10D. The results indicate that TCR signaling is necessary for IL-2 transcription, but not sufficient. Coupling with CD28 coreceptor signaling causes a moderate increase in IL-2 transcription, but coupling with PMA results in the greatest observed increase, consistent with [17,27]. These experimental results corroborate the model and suggest that it is capable of accurately recapitulating the transcription profiles and, indirectly, the upstream signaling pathways, for a wide variety of stimuli.
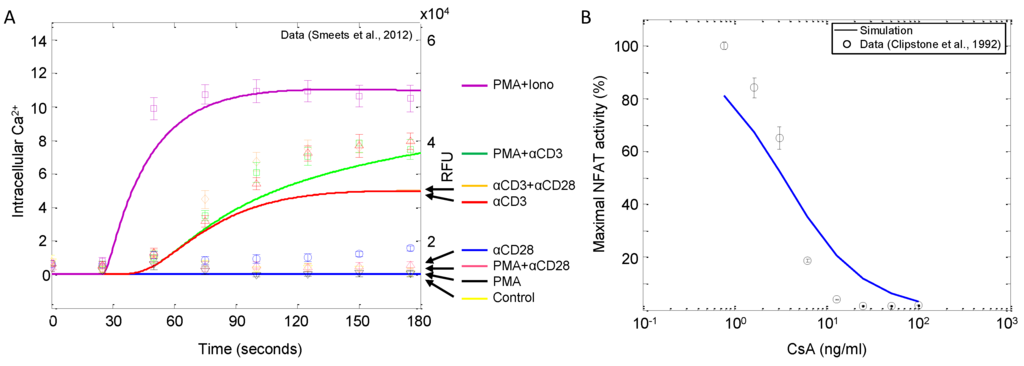
Figure 9.
Intracellular calcium and NFAT signaling in response to various combinations of stimuli. (A) Jurkat cells were stimulated as indicated (αCD3, αCD28, ionomycin: 1 μg/mL; PMA: 10 ng/mL), and intracellular Ca release was monitored over time (data sampled from [27]). Solid lines represent corresponding model simulations. (B) NFAT activity in response to 10 μg/mL αCD3 stimulation and varying doses of calcineurin inhibitor cyclosporin A (CsA). The data are measured at 30 min post-stimulation (data sampled from [53]).
Figure 9.
Intracellular calcium and NFAT signaling in response to various combinations of stimuli. (A) Jurkat cells were stimulated as indicated (αCD3, αCD28, ionomycin: 1 μg/mL; PMA: 10 ng/mL), and intracellular Ca release was monitored over time (data sampled from [27]). Solid lines represent corresponding model simulations. (B) NFAT activity in response to 10 μg/mL αCD3 stimulation and varying doses of calcineurin inhibitor cyclosporin A (CsA). The data are measured at 30 min post-stimulation (data sampled from [53]).
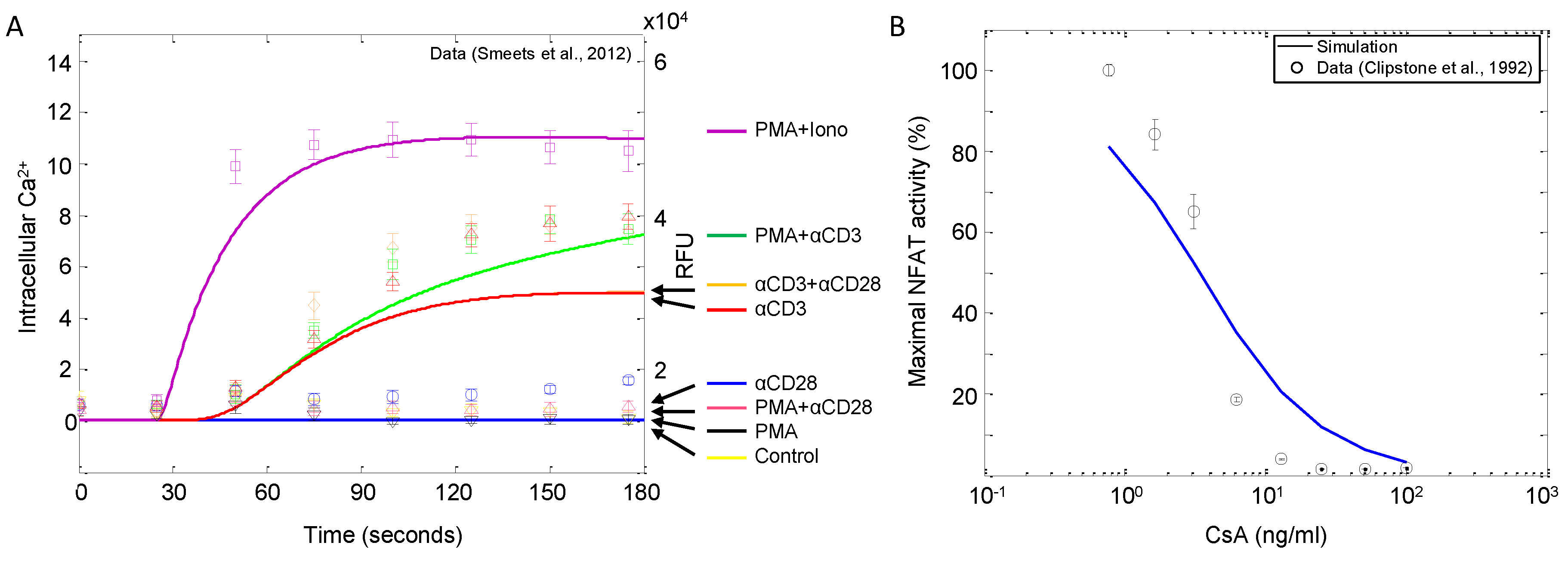
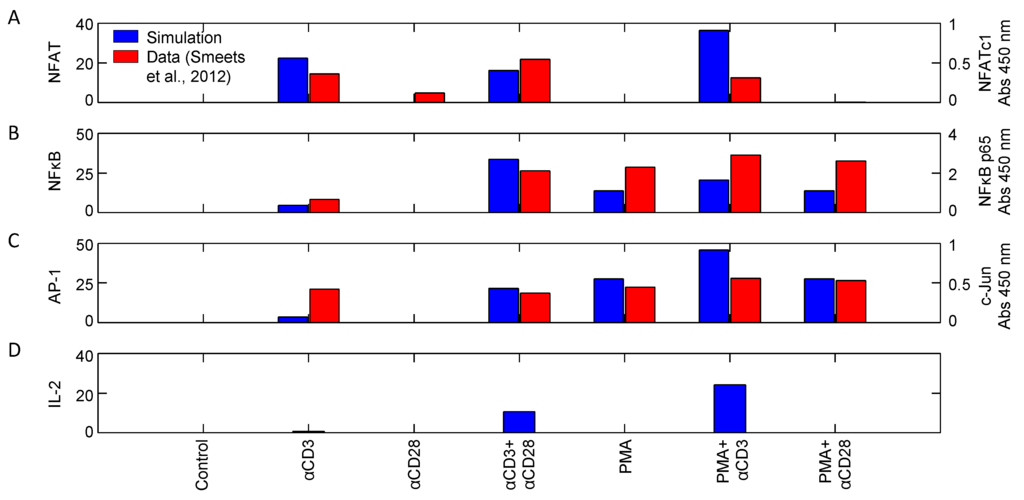
Figure 10.
Activity of transcription factors (A) NFAT, (B) NFκB and (C) AP1, as well as (D) synthesis of IL-2 in response to various combinations of stimuli (αCD3, αCD28: 1 μg/mL; PMA: 10 ng/mL). Activity was measured 15 min post-stimulation (data sampled from [27]). Left and right axes correspond to model simulations and measured relative absorbance values, respectively.
Figure 10.
Activity of transcription factors (A) NFAT, (B) NFκB and (C) AP1, as well as (D) synthesis of IL-2 in response to various combinations of stimuli (αCD3, αCD28: 1 μg/mL; PMA: 10 ng/mL). Activity was measured 15 min post-stimulation (data sampled from [27]). Left and right axes correspond to model simulations and measured relative absorbance values, respectively.
4.1.3. Corroboration of CD45 Activity
In this model, CD45 exists initially as fully active (proximal to the SFKs), then is deactivated (translocates away from the SFKs) by the formation of the TCR complex. At later time points, CD45 is reintroduced to the receptor cluster with an increased negative regulatory role. This results in dephosphorylating active SFK and phosphorylated ligand-bound TCR, thus terminating the TCR signal. Figure 11A depicts Lck-Y505 phosphorylation as a function of CD45 activity after 15 min of stimulation. At normal expression levels (i.e., 100% of WT), Y505 phosphorylation is reduced by approximately 75%. Phosphorylation at Y505 is inversely related to CD45 activity, which is corroborated by the observations of McNeill et al. [55]. The model is able to approximate the data from 100% WT activity down through 5% WT activity very well. Only when CD45 is completely suppressed do the model and data diverge, although the trend is still present.
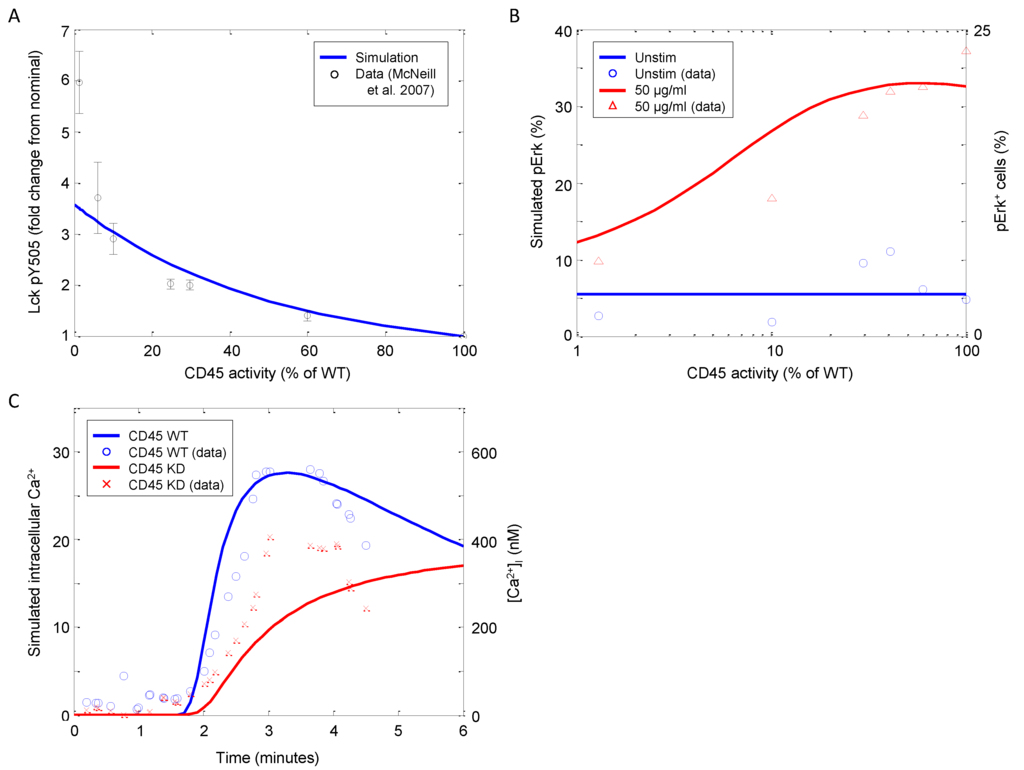
Figure 11.
Downstream TCR signaling in response to CD45 knockdown. (A) Model simulations of Lck phosphorylation at the negative regulatory residue Y505 as a function of CD45 activity (data sampled from [55]). (B) Model simulations of Erk phosphorylation 3 min after stimulation with 0 or 50 μg/mL αCD3 as a function of CD45 activity (data sampled from [55]). (C) Model simulations of intracellular calcium release over time in wild-type (WT) and CD45 knockdown mutant (5% of WT activity) stimulated with 10 μg/mL αCD3 at 90 s as a function of CD45 activity (data sampled from [56]).
Figure 11.
Downstream TCR signaling in response to CD45 knockdown. (A) Model simulations of Lck phosphorylation at the negative regulatory residue Y505 as a function of CD45 activity (data sampled from [55]). (B) Model simulations of Erk phosphorylation 3 min after stimulation with 0 or 50 μg/mL αCD3 as a function of CD45 activity (data sampled from [55]). (C) Model simulations of intracellular calcium release over time in wild-type (WT) and CD45 knockdown mutant (5% of WT activity) stimulated with 10 μg/mL αCD3 at 90 s as a function of CD45 activity (data sampled from [56]).
In CD45-null thymocytes, the p56Lck and p59Fyn tyrosine kinases are hyperphosphorylated, and p56Lck is found in its inactive conformation [56]. Both basal and TCR-stimulated tyrosine phosphorylation of TCRζ and CD3ϵ are also much reduced. These defects are associated with the failure of Zap70 kinase recruitment to the TCRζ chain; however, TCR-induced signaling is not entirely ablated. Figure 11B shows Erk phosphorylation as a function of αCD3 stimulation and CD45 activity. It is clearly evident that pErk increases with CD45 activity; however, above ~50%, there is qualitative switch, and pErk begins to decrease as CD45 expression approaches wild-type levels, which is also corroborated by observations by McNeill et al. [55].
Furthermore, significant inositol phosphate and calcium signals are observed in CD45-null thymocytes. In our simulation, the CD45 defect is approximated with a knockdown to 5% of WT activity as complete CD45 knockout suppressed all signaling. Although greatly reduced from the nominal system, calcium signaling does not appear to be insignificant in CD45-defective CD4+ T-cells (Figure 11C). The molecular analysis presented by Stone et al. suggests that the threshold for TCR signal transduction is greatly increased in CD45-null T-cells, thus explaining the profound defects in thymic development [56].
4.1.4. Corroboration of SHP1 Activity
SHP1 functions as a negative regulator by deactivating Zap70 and SFK upon T-cell activation [49]. SHP1 also forms a negative feedback loop that is composed of SHP1 phosphorylation by activated Lck, binding of phospho-SHP1 to Lck and Lck inactivation by SHP1-mediated dephosphorylation [50]. To evaluate the ability of the model to capture the role of SHP1 feedback on T-cell activation, we simulate the SHP1 knockdown experiment presented by [49]. Figure 12 shows the stimulation of the IL-2 reporter with different combinations of stimuli and two different levels of SHP1 activity. Model-simulated results and data are depicted by bars and dots, respectively. In the wild-type system, neither PMA nor αCD3 alone are sufficient to upregulate IL-2 transcription. However, the combination of the two is able to generate a substantial signal. Pairing PMA with ionomycin produces the maximally-observed signal. In the SHP1-knockdown system, IL-2 synthesis is drastically increased as a result of αCD3 and PMA + αCD3 stimulation, thus demonstrating the lack of inhibition. PMA + ionomycin stimulation is not affected by SHP1 knockdown because SHP1 functions upstream of their substrates: RasGRP1, PKCθ and calcium. The experimental results show mostly good agreement with the model, suggesting that the model is able to adequately capture the reported behavior of SHP1.
4.2. Weak CD28 Costimulation Predicted to Elevate FOXP3 Transcription
TCR signal intensity and costimulation is known to differentially affect the activation of T-cells. We study this by performing a two-way dose response experiment, measuring the effects of αCD3 and αCD28 on IL-2 and FOXP3 transcription. Figure 13A,B shows IL-2 and FOXP3 transcription, respectively, following 30 min of stimulation by various combinations of αCD3 and αCD28. At trivial doses of both stimuli, neither species is active, indicating that the cell is at rest. At high doses of both stimuli, IL-2 is the dominant species, indicating that the cell is fully active. However, when costimulation through CD28 is relatively weak, the balance of dominance shifts toward FOXP3, while leaving IL-2 at a much reduced level. Indeed, this behavior is corroborated by the observations by Kretschmer et al. [57] that weak TCR signals and limited costimulation have been linked to FOXP3 induction and the development of the regulatory phenotype in CD4+ T-cells.
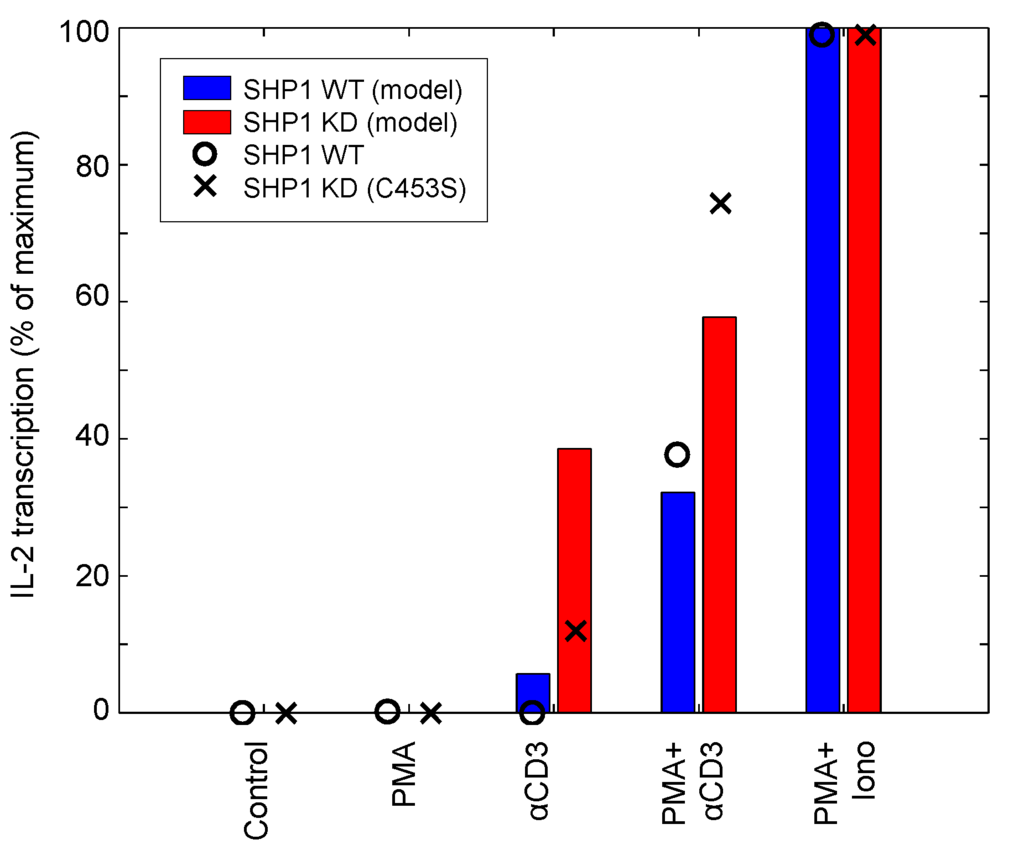
Figure 12.
IL-2 reporter stimulation in response to SHP1 knockdown. The model simulates the wild-type and SHP1 knockdown mutant (C453S mutation, resulting in catalytically inactive SHP1) stimulated with 10 μg/mL αCD3. IL-2 reporter stimulation was measured 2 h post-stimulation (data sampled from [49]).
Figure 12.
IL-2 reporter stimulation in response to SHP1 knockdown. The model simulates the wild-type and SHP1 knockdown mutant (C453S mutation, resulting in catalytically inactive SHP1) stimulated with 10 μg/mL αCD3. IL-2 reporter stimulation was measured 2 h post-stimulation (data sampled from [49]).
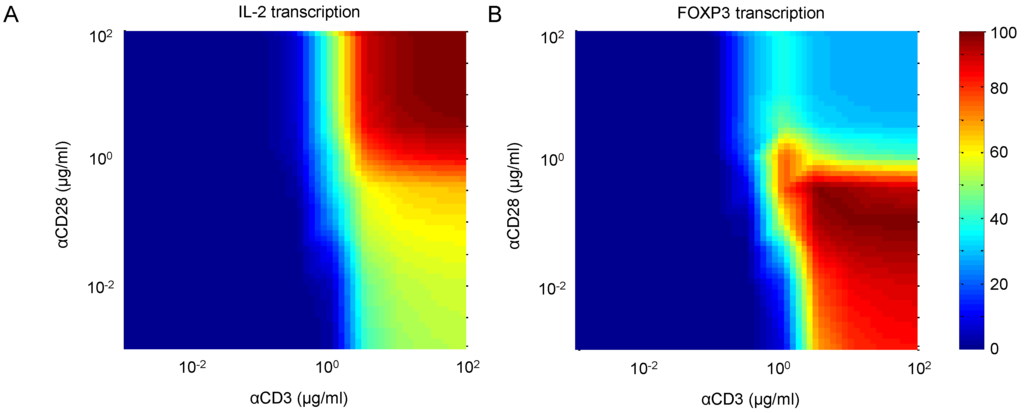
Figure 13.
(A) IL-2 and (B) FOXP3 transcription in response to stimulation by combinations of αCD3 and αCD28. Results show outputs 30 min after stimulation.
Figure 13.
(A) IL-2 and (B) FOXP3 transcription in response to stimulation by combinations of αCD3 and αCD28. Results show outputs 30 min after stimulation.
4.3. Reduced CD45 Activity Predicted to Elevate FOXP3 Transcription
We investigate the roles of CD45 and SHP1 on T-cell activation by simulating a two-way knockdown response experiment and measuring the effects on a small number of important outputs. Figure 14A–F show the model response to various CD45- and SHP1-knockdown scenarios. SHP1 clearly has the effect of downregulating most species playing a role in T-cell activation. As SHP1 activity is reduced (i.e., moving top to bottom on the right axis in each plot), calcium, Erk, PKCθ and IL-2 are all upregulated. This is due to the loss of their transient behaviors and constitutive activation at elevated levels. For CD45, on the other hand, the analysis demonstrates a dual role as both activator and inhibitor of T-cell activation. As CD45 activity is reduced (i.e., moving right to left on the top axis in each plot), TCR signaling increases until CD45 activity reaches ~1%–10% of the wild-type, then decreases to complete inactivation as CD45 is completely suppressed. This result indicates that CD45 is required for TCR signaling, without which the pool of SFKs would remain in the closed and inactive state; however, the highest level of activation is not actually achieved for wild-type CD45 expression, but rather at a reduced rate between approximately 1%–10% of the wild-type with intact SHP1 activity. Stronger TCR signaling can be achieved at full CD45 activity by downregulating SHP1.
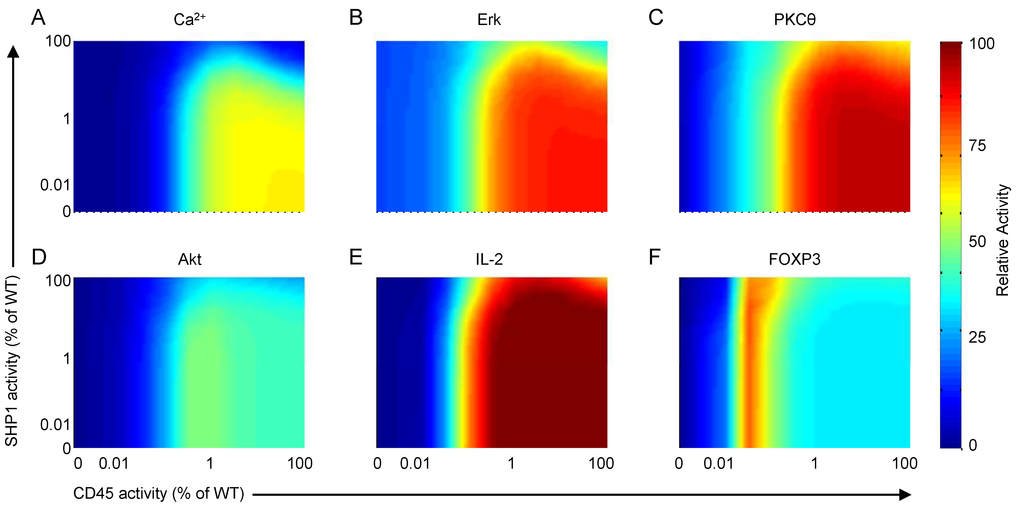
Figure 14.
Model response to various CD45- and SHP1-knockdown scenarios. (A) Intracellular calcium; (B) Erk; (C) PKCθ; (D) Akt; (E) IL-2; and (F) FOXP3 transcription factor activation in systems with various levels of CD45 and SHP1 downregulation. Note that downregulation is shown as percentages of wild-type activity (i.e., 100% corresponds to normal function, 0% corresponds to full knockout). Results show outputs 30 min after stimulation by 10 μg/mL αCD3 and αCD28.
Figure 14.
Model response to various CD45- and SHP1-knockdown scenarios. (A) Intracellular calcium; (B) Erk; (C) PKCθ; (D) Akt; (E) IL-2; and (F) FOXP3 transcription factor activation in systems with various levels of CD45 and SHP1 downregulation. Note that downregulation is shown as percentages of wild-type activity (i.e., 100% corresponds to normal function, 0% corresponds to full knockout). Results show outputs 30 min after stimulation by 10 μg/mL αCD3 and αCD28.
It is evident that the system is quite sensitive to CD45 activity, particularly at the lower end of expression. By contrast, SHP1 is much more effective closer to wild-type levels; however, its activity is heavily dependent on the presence or absence of CD45. The results suggest that there exists a distinct threshold of CD45 activity (~1% of WT) that may be critical to determining the outcome of TCR-mediated activation. Below this threshold, the system is presumably unable to activate with every state remaining at baseline. However, at the threshold and above, the signaling molecules relating to Th development become active and the system is rescued. Recently, McNeill et al. reported that only 3% of normal CD45 activity is sufficient to reconstitute CD45-deficient mice with normal numbers of mature T-cells, biased toward CD4+ T-cell lineage commitment [55].
CD45 also appears to have an effect on the activation of FOXP3. At total CD45 suppression, the cell remains unresponsive, even at full stimulation. For wild-type activity, TCR signaling promotes mTORC1 activation, thereby also suppressing FOXP3. However, a reduction in CD45 activity to around the aforementioned threshold actually induces a substantial upregulation in FOXP3 expression while IL-2 remains low. This may be due to the transcriptional regulators of FOXP3 becoming sufficiently induced, while signaling remains too low to promote mTOR-mediated inhibition. This result suggests that CD45 downregulation may lead to weak IL-2 induction and increased FOXP3 expression, which is often observed in Treg lineage commitment.
5. Conclusions
Signal intensity and feedback regulation are known to be major determinants of the signaling profile for the TCR and its various coreceptors. While the exact nature of these relationships remains under investigation, it is believed to involve a complex signaling network with cross-talk between the calcium, Erk, PKCθ and mTOR signaling pathways.
In this manuscript, we present a mathematical model encompassing the signal transduction events relating to the process of TCR-mediated cell activation and gene expression. The model is able to reproduce key behaviors in: (1) ligand-induced TCR trafficking, synthesis and degradation; (2) early kinase and phosphatase interactions between SFK, Zap70, CD45 and SHP1; (3) CD28 costimulation; and (4) downstream signal transduction pathways leading to IL-2 and FOXP3 synthesis, including calcium, Erk, PKCθ and mTOR.
In addition to corroborating many experimentally-observed behaviors, the model is able to provide insight into the positive and negative regulatory roles of the phosphatases CD45 and SHP1 during T-cell activation. Analysis of the model demonstrates that: (1) SHP1 negative feedback is necessary for preventing hyperactivity in TCR signaling; (2) CD45 is required for TCR signaling, but also partially suppresses it at high activity levels; and (3) elevated FOXP3 and reduced IL-2 signaling, an expression profile often observed in developing Tregs, can be achieved either by weak TCR and CD28 stimulation or a severe reduction in CD45 activity. However, we do note that further investigation and experimental evidence is required in order to corroborate these predictions.
While the proposed model is a demonstrably powerful tool for predicting many events involved in CD4+ T-cell activation, there are certain caveats that need to be considered to accurately define the model’s scope and capability. First, as a finite mathematical approximation, this model is inherently an abstraction of the biological reality. As such, the model does not attempt to explain every possible mechanism involved in the processes in question. For example, the Foxo family protein, Foxo1, is thought to bind to the FOXP3 locus and induce FOXP3 gene transcription. Foxo1 activity is subject to modulation by Akt kinase signaling, and Tregs have dampened Akt signaling in response to TCR stimulation compared with conventional T-cells. However, it is not within the scope of this work to test and corroborate every one of these mechanisms. Second, the purpose of the model is to simulate the dynamics of a small number of key species involved in T-cell activation or, more specifically, and clinically relevant, activation of human primary CD4+ T lymphocytes. Due to practical limitations on the availability of experimental data, however, the model is partially calibrated with datasets from alternate sources, including Jurkat cells. While Jurkat cells are an immortalized cell line of human T lymphocytes, they share many similarities with their primary counterparts and are very useful in studying T-cell signaling and IL-2 production. However, we do recognize that this constitutes an amalgamation of data from a variety of sources and that further investigation using primary T-cells is highly desirable.
As demonstrated in this manuscript, the mathematical model captures the key events in TCR and CD28 co-mediated signal transduction events leading to IL-2 and FOXP3 activation. As such, the model enables researchers to study these processes with a combination of broad scope, using a large-scale highly-connected network and with quantitative detail and accuracy not allowed by comparable models. In the future, we plan to use this mathematical model to design informative and hypothesis-driven experiments to refine our understanding of the dynamical nature of CD4+ T-cell activation. Our goal is to use such a model as a foundation for the design of strategies to drive and control T-cell activation and differentiation for therapeutic gain.
Acknowledgments
This research was funded by a grant awarded by the National Science Foundation, No. DMS-09002677. The authors would also like to thank current and former members of the Rundell research group for their helpful discussions and expertise.
Author Contributions
Jeffrey Perley performed the literature review, developed the model and contributed to the analysis. Judith Mikolajczak performed the experiments and collected the data used to develop and tune the model. Marietta Harrison and Gregery Buzzard contributed to designing and supervising the experimental and mathematical modeling aspects, respectively, and Ann Rundell oversaw all aspects of the research.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix 1: Model Equations
Table A1 presents a comprehensive list of all species illustrated in Figure 1 that were chosen to participate in the model. The list also includes the rate equation (further defined in Table A2), biological meaning, the initial and total quantities for each state in units of molecules and the source (if applicable) providing these values or used to compute them. Stars () denote active forms.
Table A1.
Summary of model states.
| State | Rate Equation | Biological Meaning | Initial | Total | Source(s) |
|---|---|---|---|---|---|
| TCR | Ligand-bound TCR | 0 | [40] | ||
| TCR | Phosphorylated TCR-ζ chain | 0 | [40] | ||
| TCR | Internalized TCR | [40,46] | |||
| TCR | Degraded TCR | [40,46] | |||
| Zapb | Protein tyrosine kinase Zap70 bound to the phosphorylated TCR-ζ chain | 0 | [40] | ||
| Zap* | Activated Zap70 (phosphorylated at Y493 in the activation loop) | 0 | [40] | ||
| Zapp | Doubly phosphorylated Zap70 (at Y493 and Y319) | [40] | |||
| SFKdp | Src family kinases (including Lck and Fyn) with dephosphorylated inhibitory site (Y505 on Lck) | 100 | [40] | ||
| SFKdp-Zapp | Dephosphorylated SFK bound to pY319 of Zap70 | 0 | [40] | ||
| SFKdpS59p | Dephosphorylated SFK phosphorylated at serine-59 by activated Erk | 0 | [40] | ||
| SFK* | Free fully activated SFK | 0 | [40] | ||
| SFK*-Zapp | Fully activated SFK bound to pY319 of Zap70 | 0 | [40] | ||
| SFK*S59p | Fully activated SFK phosphorylated at serine-59 by activated Erk | 0 | [40] | ||
| CD45 | Positive regulatory role of transmembrane tyrosine phosphatase CD45 | [40] | |||
| CD45 * | Negative regulatory role of CD45 | 0 | [40] | ||
| Cbpp | Phosphorylated transmembrane scaffold protein Cbp (also known as PAG) | 50 | [40] | ||
| Csk* | Membrane-localized protein tyrosine kinase Csk recruited by Cbpp | [40] | |||
| SHP1* | Tyrosine phosphatase SHP1 recruited to the membrane and activated | 0 | [40] | ||
| LATp | Phosphorylated transmembrane protein LAT at tyrosine residues | 0 | [40] | ||
| SOSb | LATp-bound scaffold protein Grb2 and guanine nucleotide exchange factor SOS | 0 | [40] | ||
| PLCγp | Activated phospholipase Cγ and bound to LATp | 0 | [40] | ||
| DAG | Diacylglycerol | 0 | [40] | ||
| IP | Inositol 1,4,5-triphosphate | 0 | [40] | ||
| RasGRP* | Activated Ras guanine nucleotide releasing protein (RasGRP) | 0 | [40] | ||
| RasGTP | Guanine triphosphate (GTP)-bound Ras protein | 0 | [40] | ||
| Raf* | Phosphorylated and activated mitogen-activated protein (MAP) kinase kinase kinase Raf | 0 | [40] | ||
| Mek* | Phosphorylated and activated MAP kinase kinase Mek | 0 | [40] | ||
| Erk* | Phosphorylated and activated MAP kinase (MAPK) Erk | [40] | |||
| AP1* | Activated transcription factor activator protein 1 | 0 | Derived | ||
| Ca | Cytoplasmic calcium ions released from intracellular stores (endoplasmic reticulum) | [58] | |||
| CaM* | Calcium-binding protein calmodulin bound to calcium | 0 | Derived | ||
| CN* | Activated calcium-dependent serine-threonine phosphatase calcineurin | 0 | Derived | ||
| NFATn | Dephosphorylated NFAT with unobstructed nuclear localization signal | 0 | Derived | ||
| CD28* | Ligand-bound and activated CD28 coreceptor | 0 | Derived | ||
| PI3K* | Activated phosphoinositide 3-kinase related kinase (PI3K) | 0 | Derived | ||
| PIP | Phosphatidylinositol 3,4,5-trisphosphate | 0 | Derived | ||
| PDK1* | Activated 3-phosphoinositide-dependent kinase-1 (PDK1) | 0 | Derived | ||
| PKCθ* | Active protein kinase C-θ | 0 | Derived | ||
| IKK* | Activated IκB kinase | 0 | Derived | ||
| IκBαp | Phosphorylated IκB marked for proteasomal degradation | 0 | Derived | ||
| NFκBn | Nuclear NFκB | 0 | Derived | ||
| AKT* | Activated serine-threonine kinase Akt, also known as protein kinase B (PKB) | 0 | Derived | ||
| TSC1-TSC2 | GTPase-activating protein (GAP) consisting of tuberous sclerosis complex 1 (TSC1) and TSC2 | Derived | |||
| RhebGTP | GTP-bound Ras homolog enriched in brain (Rheb) GTPase | Derived | |||
| mTORC1* | Activated mammalian target of rapamycin (mTOR) complex 1 | 0 | Derived | ||
| mTORC2* | Activated mTORC2 | 0 | Derived | ||
| PTEN* | Activated phosphatase and tensin homolog (PTEN) | Derived | |||
| IL2 | Interleukin-2, a cytokine marking T-cell activation | 0 | Derived | ||
| FOXP3 | Forkhead box P3, regulator of regulatory T-cell development and function | 0 | Derived |
Table A2 presents a comprehensive list of all reaction equations used to model the biochemical reactions of this system (illustrated in Figure 1). These expressions form the basis for the rate equations of the ODE-based model presented in Table A1. The list includes the reaction identifier, mathematical equation and biological meaning for each reaction.
Table A2.
Summary of model equations.
| Reaction | Equation | Biological Meaning | |
|---|---|---|---|
| R | = | Association/dissociation of ligand and TCR complex | |
| = | SFK-mediated phosphorylation and SHP1/CD45-mediated dephosphorylation of ligand-bound TCR complex | ||
| = | Internalization of free TCR | ||
| = | Internalization of ligand-bound TCR | ||
| = | Internalization of phosphorylated TCR | ||
| = | Exocytosis of internalized TCR | ||
| = | Degradation of internalized TCR | ||
| = | Synthesis of new TCR | ||
| = | Association/dissociation of phosphorylated TCR complex and Zap70 | ||
| = | SFK dephosphorylation by CD45 and re-phosphorylation by Csk* at the inhibitory site (Y505 in Lck, Y528 in Fyn) | ||
| = | SFK phosphorylation at the activation loop (Y394 in Lck, Y417 in Fyn) by autophosphorylation (or by another kinase) and dephosphorylation by SHP1 | ||
| = | Association/dissociation of Zapp and SFKdp | ||
| = | TCR-mediated phosphorylation of Zapp-bound SFKdp | ||
| = | Dissociation/association of activated SFK and Zapp | ||
| = | Cbp phosphorylation by activated SFK (or other kinases) and dephosphorylation by CD45 | ||
| = | Cbpp-mediated activation of Csk | ||
| = | Zap70 phosphorylation at the activation loop (Y493) by activated SFK (SFK* and SFK*-Zapp) and dephosphorylation by PTPs including SHP1 | ||
| = | Additional Zap70 phosphorylation at Y319 by activated SFK and Zap70 and dephosphorylation by PTPs including SHP1 | ||
| = | SHP1 activation (by SFK*) and deactivation | ||
| = | LAT phosphorylation by activated Zap70 and dephosphorylation SHP1 | ||
| = | PLCγ phosphorylation by LATp | ||
| = | PLCγ-mediated hydrolysis of PIP to IP and DAG | ||
| = | Degradation of DAG | ||
| = | Degradation of IP | ||
| = | DAG-mediated activation of RasGRP | ||
| = | DAG-mediated activation of PKCθ | ||
| = | RasGRP- and Grb2SOS-mediated activation of Ras | ||
| = | Ras-mediated activation of Raf | ||
| = | Rafp-mediated activation of Mek | ||
| = | Mekp-mediated activation of Erk | ||
| = | LATp-mediated association and activation of the Grb2-SOS complex | ||
| = | Erkp-mediated phosphorylation of SFKdp at serine-59 | ||
| = | Erkp-mediated phosphorylation of SFKact at serine-59 | ||
| = | TCR-mediated activation and SHP1-mediated deactivation of SFK-s59p | ||
| = | Positive regulatory role of CD45 and translocation caused by receptor cluster formation | ||
| = | Negative regulatory role of CD45 and recruitment to receptor cluster | ||
| = | Erkp- and PKCθ-mediated activation of AP1 | ||
| = | IP -induced calcium release into the cytoplasm | ||
| = | Association/dissociation of calcium and calmodulin | ||
| = | Calmodulin-mediated activation of calcineurin | ||
| = | Calcineurin-mediated dephosphorylation and nuclear translocation of NFAT | ||
| = | Association/dissociation of ligand and CD28 | ||
| = | PI3K activation by CD28 and Zap70 and deactivation | ||
| = | PI3K-mediated phosphorylation of PIP and PTEN-mediated dephosphorylation of PIP | ||
| = | PIP -mediated activation of PDK1 | ||
| = | Activation of PKCθ mediated by PDK1, DAG, and mTORC2 | ||
| = | PKCθ- and AKT-mediated activation of IKK | ||
| = | IKK-mediated phosphorylation and 26S proteasome-mediated degradation of IκBα; NFκB-induced synthesis of new IκBα | ||
| = | Activation and nuclear translocation of NFκB | ||
| = | PDK1- and mTORC2-mediated phosphorylation of AKT | ||
| = | AKT-mediated phosphorylation, dissociation, and deactivation of TSC | ||
| = | GAP activity of TSC on Rheb | ||
| = | RhebGTP-mediated activation of mTORC1 | ||
| = | PI3K-mediated activation and mTORC1-mediated inhibition of mTORC2 | ||
| = | TCR-mediated inhibition and FOXP3-mediated activation of PTEN | ||
| = | AP1, NFAT, and NFkB regulate transcription of IL-2; FOXP3-mediated inhibition of IL-2 | ||
| = | AP1 and NFAT regulate transcription of FOXP3; mTOR-mediated inhibition of FOXP3 |
Table A3 presents a comprehensive list of all reaction parameters included in the model. The list also includes the biological meaning, value, 95% confidence interval (if applicable), units and source (if applicable) for each parameter. For parameters not provided by external sources, they are either estimated from data or explicitly derived to satisfy a condition of the model, for example, to ensure equilibrium when the system should be at rest. Confidence intervals are estimated for the model calibration data (Table 1) using the likelihood ratio method [59].
Table A3.
Summary of model parameters.
| Parameter | Biological Meaning | Value | 95% CI | Units | Source |
|---|---|---|---|---|---|
| Association rate of ligand and TCR complex | 0.0900 | [0.0558, 0.1231] | (mol·min) | Fitted | |
| Dissociation rate of ligand and TCR complex | [, ] | min | Fitted | ||
| Phosphorylation rate of ligand-bound TCR mediated by SFK* and SFK*-S59p | 0.3000 | [0.0475, 3.8961] | (mol·min) | [40] | |
| Phosphorylation rate of ligand-bound TCR mediated by SFK*-Zapp | 1.13 | (mol·min) | [40] | ||
| Dephosphorylation rate of ligand-bound TCR mediated by activated SHP1 | 0.0022 | [0.0015, 0.0221] | (mol·min) | [40] | |
| Constitutive dephosphorylation rate of ligand-bound TCR | 16.1100 | [1.0731, 61.7493] | min | [40] | |
| Dephosphorylation rate of ligand-bound TCR mediated by activated CD45 | 0.0300 | [, 0.1574] | (mol·min) | Fitted | |
| Resting TCR internalization rate | 0.0100 | min | [46] | ||
| Maximum induced TCR internalization rate | 0.0380 | min | [46] | ||
| TCR internalization Hill coefficient | 2 | unitless | Derived | ||
| Enzyme quantity producing half-maximum TCR internalization rate | 0.05·[PKCθ] | mol | Derived | ||
| Constitutive TCR exocytosis rate | 0.0789 | min | Derived | ||
| Resting TCR degradation rate | 0.0011 | min | [45] | ||
| Maximum induced TCR degradation rate | 0.0033 | min | [45] | ||
| TCR degradation Hill coefficient | 2 | unitless | Derived | ||
| Enzyme quantity producing half-maximum TCR internalization rate | 0.05·[SFK] | mol | Derived | ||
| TCR synthesis rate | min | Derived | |||
| Association rate of Zap70 to phosphorylated TCRζ-chain | [, ] | (mol·min) | Fitted | ||
| Dissociation rate of Zap70 to phosphorylated TCRζ-chain | 1.2600 | [0.4913, 41.7225] | min | Fitted | |
| Dephosphorylation rate of SFK at the inhibitory site by CD45 | [, ] | (mol·min) | Fitted | ||
| Phosphorylation rate of SFK at the inhibitory site by Csk | 0.1199 | (mol·min) | Derived | ||
| Phosphorylation rate of SFKdp at the activation site mediated by TCRb and TCRp | 13.7700 | [1.3177, 75.4525] | (mol·min) | Fitted | |
| Dephosphorylation rate of SFK* at the activation site by activated SHP1 | (mol·min) | [40] | |||
| Constitutive dephosphorylation rate of SFK* at the activation site | min | [40] | |||
| Dephosphorylation rate of SFK* at the activation site by activated CD45 | (mol·min) | Derived | |||
| Association rate of SFKdp to Zapp | 0.0217 | [, 2.1690] | (mol·min) | Fitted | |
| Dissociation rate of SFKdp to Zapp | 0.0025 | [, 0.0151] | min | Fitted | |
| Phosphorylation rate of Zapp-bound SFKdp at the activation site mediated by TCRb and TCRp | (mol·min) | [40] | |||
| Dephosphorylation rate of Zapp-bound SFK* at the activation site by activated SHP1 | 0.0068 | [0.0012, 0.0641] | (mol·min) | Fitted | |
| Constitutive dephosphorylation rate of Zapp-bound SFK* at the activation site | 13.4040 | [1.3409, 84.0744] | min | Fitted | |
| Dephosphorylation rate of Zapp-bound SFK* at the activation site by activated CD45 | 0.0300 | [0.0019, 0.3542] | (mol·min) | Fitted | |
| Dissociation rate of SFK* to Zapp | 70.7880 | [68.7880, 102.3198] | min | Fitted | |
| Association rate of SFK* to Zapp | (mol·min) | [40] | |||
| Phosphorylation rate of Cbp by SFK* | [, ] | (mol·min) | Fitted | ||
| Constitutive phosphorylation rate of Cbp | 0.0207 | [0.0127, 0.1200] | min | Fitted | |
| Dephosphorylation rate of Cbpp by CD45 | (mol·min) | Derived | |||
| Association rate of Csk to Cbpp | [, 0.0070] | (mol·min) | Fitted | ||
| Dissociation rate of Csk to Cbpp | 0.6635 | min | Derived | ||
| Phosphorylation rate of bound Zap by SFK* and SFK*S59p | 0.0021 | [0.0015, 0.0022] | (mol·min) | Fitted | |
| Phosphorylation rate of bound Zap by SFK*-Zapp | 1.13 | (mol·min) | [40] | ||
| Dephosphorylation rate of Zap* by activated SHP1 | (mol·min) | [40] | |||
| Constitutive dephosphorylation rate of Zap* | min | [40] | |||
| Phosphorylation rate of Zap* by free and bound Zapp | [, ] | (mol·min) | Fitted | ||
| Phosphorylation rate of Zap* by SFK* and SFK*S59p | (mol·min) | [40] | |||
| Phosphorylation rate of Zap* by SFK*-Zapp | 1.13 | (mol·min) | [40] | ||
| Dephosphorylation rate of Zapp by activated SHP1 | (mol·min) | [40] | |||
| Constitutive dephosphorylation rate of Zapp | min | [40] | |||
| Activation rate of SHP1 by SFK* | [, ] | (mol·min) | Fitted | ||
| Deactivation rate of SHP1 | 0.3660 | [0.3450, 0.3861] | min | Fitted | |
| Phosphorylation rate of LAT by activated Zap | [, ] | (mol·min) | Fitted | ||
| Dephosphorylation rate of LAT by activated SHP1 | 0.0020 | [0.0017, 0.0025] | (mol·min) | Fitted | |
| Constitutive dephosphorylation rate of SHP1 | 90 | [87.9332, 130.0896] | min | Fitted | |
| Association rate of PLCγ and LATp (PLCγ is immediately phosphorylated) | 0.0030 | [0.0021, 0.0033] | (mol·min) | Fitted | |
| Lumped rate of dephosphorylation of PLCγ and its dissociation from LATp | 300 | [295.1992, 302.0020] | min | Fitted | |
| PIP hydrolysis rate catalyzed by PLCγp | [, ] | (mol·min) | Fitted | ||
| DAG degradation rate | 2.4311 | [1.3990, 2.9228] | min | Fitted | |
| IP degradation rate | min | Derived | |||
| Activation rate of RasGRP by DAG | 0.0030 | [0.0025, 0.0052] | (mol·min) | Fitted | |
| Inactivation rate of RasGRP | 30 | [17.2632, 36.0679] | min | Fitted | |
| Activation rate of PKCθ by DAG | [, ] | (mol·min) | Fitted | ||
| Rate of Ras guanine nucleotide exchange catalyzed by activated RasGRP | [, ] | (mol·min) | Fitted | ||
| Rate of Ras guanine nucleotide exchange catalyzed by recruited SOS | [, ] | (mol·min) | Fitted | ||
| Constitutive rate of RasGTP hydrolysis to RasGDP | 30 | [24.9529, 43.3632] | min | Derived | |
| Activation rate of Raf by RasGTP | [, ] | (mol·min) | Fitted | ||
| Constitutive rate of Raf inactivation by phosphatase | 30 | [24.9529, 43.3632] | min | Derived | |
| Activation rate of Mek by activated Raf | 0.0030 | [0.0025, 0.0036] | (mol·min) | Fitted | |
| Constitutive rate of Mek inactivation by phosphatase | 30 | [24.9529, 36.0679] | min | Derived | |
| Activation rate of Erk by activated Mek | [, ] | (mol·min) | Fitted | ||
| Constitutive rate of Erk inactivation by phosphatase | 30 | [24.9529, 36.0679] | min | Derived | |
| Association rate of Grb-SOS complex to LATp | [, ] | (mol·min) | Fitted | ||
| Dissociation rate of Grb-SOS complex from LATp | 30 | [3.3632, 100] | min | Derived | |
| Phosphorylation rate of SFKdp at S59 by activated Erk | [, 0.0011] | (mol·min) | Fitted | ||
| Constitutive dephosphorylation rate of SFKdp at S59 | 30 | [4.5543, 80.0101] | min | Fitted | |
| Phosphorylation rate of SFK* at S59 by activated Erk | (mol·min) | [40] | |||
| Constitutive dephosphorylation rate of SFK* at S59 | min | [40] | |||
| Phosphorylation rate of SFKdpS59p at the activation site mediated by TCRb and TCRp | (mol·min) | Derived | |||
| Dephosphorylation rate of SFK*S59p at the activation site by activated SHP1 | (mol·min) | [40] | |||
| Constitutive dephosphorylation rate of SFK*S59p at the activation site | min | [40] | |||
| Dephosphorylation rate of SFK*S59p at the activation site by activated CD45 | (mol·min) | Derived | |||
| Translocation rate of CD45 mediated by receptor complex | [, ] | (mol·min) | Fitted | ||
| Constitutive return rate of CD45 | 0.0030 | [, 0.1009] | min | Fitted | |
| Activation rate of CD45 negative regulator by SFK* | [, ] | (mol·min) | Fitted | ||
| Constitutive deactivation rate of CD45 negative regulator | 0.0030 | [, 0.0961] | min | Fitted | |
| Activation rate of AP1 by Erk* and PKCθ* | [, ] | (molmin) | Fitted | ||
| Constitutive deactivation rate of AP1* | 30 | [23.1193, 43.8583] | min | Fitted | |
| Release rate of calcium stored in the endoplasmic reticulum by IP | 0.0300 | [0.0212, 0.0380] | (mol·min) | Fitted | |
| Constitutive calcium uptake rate | 30 | [19.0031, 54.1565] | min | Fitted | |
| Association rate of calmodulin to calcium | [, ] | (mol·min) | Fitted | ||
| Dissociation rate of calmodulin from calcium | 0.3000 | [0.0182, 3.2856] | min | Fitted | |
| Activation rate of calcineurin by calmodulin* | [, ] | (mol·min) | Fitted | ||
| Constitutive deactivation rate of calcineurin* | 0.3000 | [0.0943, 19.1092] | min | Fitted | |
| Activation rate of NFAT (NFAT immediately translocates to nucleus) | [, ] | (mol·min) | Fitted | ||
| Constitutive deactivation rate of NFAT | 0.3000 | [0.0210, 0.3433] | min | Fitted | |
| Association rate of ligand to CD28 coreceptor | 0.0300 | [0.0199, 0.0360] | (mol·min) | Fitted | |
| Dissociation rate of ligand from CD28 coreceptor | [, ] | min | Fitted | ||
| Activation rate of PI3K by ligand-bound CD28 and activated Zap | [, ] | (molmin) | Fitted | ||
| Constitutive deactivation rate of PI3K | 30 | [15.3560, 37.5731] | min | Fitted | |
| Phosphorylation rate of PIP by PI3K* | 3 | [0.3923, 5.7161] | (mol·min) | Fitted | |
| Dephosphorylation rate of PIP by PTEN* | 30 | [17.4422, 65.3412] | (mol·min) | Fitted | |
| Constitutive dephosphorylation rate of PIP | [, ] | min | Fitted | ||
| Activation rate of PDK1 by PIP | [, ] | (mol·min) | Fitted | ||
| Constitutive deactivation rate of PDK1* | 30 | [5.4422, 40.4623] | min | Fitted | |
| Activation rate of PKCθ by PDK1* | [, ] | (mol·min) | Fitted | ||
| Activation rate of PKCθ by mTORC2* | [, ] | (mol·min) | Fitted | ||
| Constitutive deactivation rate of PKCθ* | 0.3000 | [0.1754, 1.3217] | min | Fitted | |
| Activation rate of IKK by PKCθ* | 0.0015 | [, 0.0065] | (mol·min) | Fitted | |
| Activation rate of IKK by Aktp | 0.0030 | [0.0019, 0.0046] | (mol·min) | Fitted | |
| Constitutive deactivation rate of IKK* | 15 | [5.3394, 48.7211] | min | Fitted | |
| Phosphorylation rate of IκBα by IKK* | 0.4500 | [0.4109, 0.5512] | min | Fitted | |
| Proteasomal degradation rate of pIκBα | 0.1500 | [0.1093, 0.1978] | min | Fitted | |
| Deactivation rate of IκBα by NFκB | 0.1500 | [0.0936, 0.2380] | min | Fitted | |
| Activation rate of NFκB by IκBα deactivation | 0.1500 | [0.0994, 0.2121] | min | Fitted | |
| Constitutive deactivation rate of NFκB | 0.0150 | [0.0124, 0.0272] | min | Fitted | |
| Phosphorylation rate of Akt by PDK1* | [, ] | (mol·min) | Fitted | ||
| Phosphorylation rate of Akt by mTORC2* | [, ] | (mol·min) | Fitted | ||
| Phosphorylation rate of Akt by PDK1* and mTORC2* | [, ] | (molmin) | Fitted | ||
| Dephosphorylation rate of Aktp by phosphatases | 1 | [0.5422, 7.0427] | min | Derived | |
| Phosphorylation rate of the TSC2 subunit of the TSC1-TSC2 complex (complex immediately dissociates) | 10 | [6.8726, 30.3895] | min | Fitted | |
| Dephosphorylation rate of TSC2p and its association with TSC1 | 1 | [0.3783, 8.2627] | min | Derived | |
| TSC2 phosphorylation Hill coefficient | 5 | unitless | Derived | ||
| Enzyme quantity producing half-maximum TSC2 phosphorylation rate | 0.15·[Akt] | mol | Fitted | ||
| Rate of Rheb guanine nucleotide exchange catalyzed by TSC1-TSC2 complex | [, ] | (mol·min) | Derived | ||
| Rate of RhebGDP to RhebGTP exchange | 1 | [0.3020, 10.5309] | min | Derived | |
| Activation rate of mTORC1 by RhebGTP | [, 0.0047] | (mol·min) | Fitted | ||
| Constitutive deactivation rate of mTORC1* | 1 | [0.2653, 4.7164] | min | Derived | |
| Activation rate of mTORC2 by PI3K* | 0.0030 | [2.7360e-4, 0.0993] | (mol·min) | Fitted | |
| Deactivation rate of mTORC2* by mTORC1 | 0.0030 | [0.0023, 0.0102] | (mol·min) | Fitted | |
| Constitutive deactivation rate of mTORC2* | 3 | [1.3631, 3.8319] | min | Fitted | |
| Activation rate of PTEN by FOXP3* | [, 0.0010] | (mol·min) | Derived | ||
| Constitutive activation of PTEN | 1 | [0.1925, 18.9529] | min | Derived | |
| Deactivation of PTEN* by TCR complex | 20 | [11.4372, 106.1915] | min | Fitted | |
| PTEN* deactivation Hill coefficient | 10 | unitless | Derived | ||
| Ligand-bound TCR quantity producing half-maximum PTEN* deactivation | 0.75·[TCR] | mol | Derived | ||
| Activation rate of IL-2 activity | [574.7247, ] | mol·min | Fitted | ||
| Hill coefficient for AP1-induced IL-2 activity | 2 | unitless | Derived | ||
| Hill coefficient for NFAT-induced IL-2 activity | 2 | unitless | Derived | ||
| Hill coefficient for NFκB-induced IL-2 activity | 2 | unitless | Derived | ||
| AP1 quantity producing half-maximum IL-2 activity | 0.1·[AP1] | mol | Derived | ||
| NFAT quantity producing half-maximum IL-2 activity | 0.3·[NFAT] | mol | Derived | ||
| NFκB quantity producing half-maximum IL-2 activity | 0.1·[NFκB] | mol | Derived | ||
| Deactivation rate of IL-2 activity by FOXP3 | [, 0.0093] | (mol·min) | Fitted | ||
| Constitutive deactivation rate of IL-2 | 1 | [0.1832, 8.9847] | min | Derived | |
| Activation rate of FOXP3 activity | [748.5767, ] | mol·min | Fitted | ||
| Hill coefficient for AP1-induced FOXP3 activity | 2 | unitless | Derived | ||
| Hill coefficient for NFAT-induced FOXP3 activity | 2 | unitless | Derived | ||
| AP1 quantity producing half-maximum FOXP3 activity | 0.1·[AP1] | mol | Derived | ||
| NFAT quantity producing half-maximum FOXP3 activity | 0.1·[NFAT] | mol | Derived | ||
| Deactivation rate of FOXP3 activity by mTOR | [, ] | (molmin) | Fitted | ||
| Constitutive deactivation rate of FOXP3 activity | 1 | [0.3685, 4.7328] | min | Derived |
Appendix 2: Parameter Sensitivity Analysis
Figure A1 shows the results of the parameter sensitivity analysis used to determine which parameters should be considered for parameter identification. The sensitivity indices are standardized by rows, enabling the parameters to be ranked for each model state independently. For a given model state, a value shown in red denotes that the state is relatively sensitive to perturbations in the corresponding parameter. On the other hand, a value shown in green implies that the particular state is insensitive to the parameter in question. As shown in Figure A1, it can be common for certain states to be only sensitive to a fraction of the parameters. This blocking structure is leveraged during the parameter identification process to reduce the dimension of the uncertain parameter space when calibrating individual modules.
Appendix 3: Calcium Flux Measurements
Jurkat T leukemia cells (Jurkat clone E6.1; ATCC) were harvested in log phase growth (4–8 × 10 cell/mL), washed once with Hank’s Balanced Salt Solution (HBSS) without phenol red. Phenol red-free, calcium containing HBSS (Lonza) was used for the remainder of the experiment. Cells were resuspended at 2 × 10 cells/mL in HBSS supplemented with 2.5 mM probenecid (unloaded control) or in HBSS with 1 μM Fluo-4 AM, 2.5 mM probenecid and 10 μL/mL PowerLoad (Invitrogen, Carlsbad, USA). Cells were incubated for 30 min at room temperature in the dark. Following incubation, cells were washed twice with 1 mL HBSS with 2.5 mM probenecid and then resuspended in 100 μL HBSS plus 2.5 mM probenecid. Cells were loaded into a black-walled, clear bottom 96-well plate. Wells with cells were surrounded two wells deep with 100 μL water each as a temperature buffer. The plate was covered in foil, and cells were allowed to settle for 5 min at 37 C.
Figure A1.
Parameter sensitivity indices. Values are standardized by rows. Red = most sensitive; Green = least sensitive.
Figure A1.
Parameter sensitivity indices. Values are standardized by rows. Red = most sensitive; Green = least sensitive.
Fluorescence was measured on a BioTek plate reader (BioTek Instruments, Winooski, VT, USA) using the following parameters: excitation wavelength, 495 nm; emission, 520 nm; temperature, 37 C. Prior to stimulation, a background reading was taken. The plate was ejected from the plate reader and 10 μg/mL αCD3ϵ or 1 μg/mL ionomycin (positive control) were added to the wells using a repeat dispense pipette. Following the addition of stimulants, a kinetic measurement run was done over 5–10 min, measuring fluorescence every 10 s. From the obtained data, the background reading was subtracted.
Appendix 4: Phospho-IκBα Measurements
Jurkat cells were grown in RPMI 1640 (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 7.5% heat-inactivated fetal bovine serum (Bio-West, Logan, UT, USA), 1 mM sodium pyruvate (Life Technologies, Carlsbad, CA, USA), 12.5 mM HEPES pH 7.4 (Sigma-Aldrich, St. Louis, MO, USA), 12 μM sodium bicarbonate (Sigma-Aldrich, St. Louis, MO, USA) 50 μM 2-mercaptoethanol (Sigma-Aldrich, St. Louis, MO, USA), 50 μg/mL streptomycin and 50 units/mL penicillin in an incubator at 37 °C in humidified air containing 5% carbon dioxide. Cells were harvested in log-phase growth at a density of 1 × 10 cells per treatment. The treatments were stimulated with either 2 μg/mL αCD3 or 2 μg/mL αCD3 plus αCD28 in a 37 °C water bath for up to 30 min. Samples of 1 × 10 cells were taken at the indicated time points and lysed in 1% NP40 lysis buffer (1% NP40, 25 mM Tris, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1 mM NaV, 10 mM NaF, 10 μg/mL each of aprotinin and leupeptin) for 15 min on ice. Lysates were centrifuged for 5 min at 18,000× g at 4 C. The supernatant was added to the same volume of 2X protein solubilizing mixture (PSM, 25% () sucrose, 2.5% () sodium dodecyl sulfate, 25 mM Tris, 2.5 mM EDTA, 0.05% bromophenol blue) and boiled for five min. Proteins were separated via SDS-PAGE, blotted for α-tubulin, phospho-IκB (S32/36) and IκB (Cell Signaling Technology, Beverly, MA, USA). IRDye 800 and 680 secondary anti-mouse and anti-rabbit antibodies (Li-Cor Biosciences, Lincoln, OR, USA) were used for signal detection using an Odyssey infrared scanner. Blot images were analyzed using ImageJ to produce quantitative data for model comparison.
References
- Yamane, H.; Paul, W.E. Cytokines of the gamma(c) family control CD4(+) T-cell differentiation and function. Nat. Immunol. 2012, 13, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.F.; Paul, W.E. Heterogeneity and plasticity of T helper cells. Cell Res. 2010, 20, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Doherty, P.C.; Zinkernagel, R.M. Biological role for major histocompatibility antigens. Lancet 1975, 1, 1406–1409. [Google Scholar] [CrossRef]
- Marrack, P.; Hannum, C.; Harris, M.; Haskins, K.; Kubo, R.; Pigeon, M.; Shimonkevitz, R.; White, J.; Kappler, J. Antigen-specific, major histocompatibility complex-restricted T-cell receptors. Immunol. Rev. 1983, 76, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Imboden, J.; Hardy, K.; Manger, B.; Terhorst, C.; Stobo, J. The role of the T3-antigen receptor complex in T-cell activation. Annu. Rev. Immunol. 1986, 4, 593–619. [Google Scholar] [CrossRef] [PubMed]
- Veillette, A.; Bookman, M.A.; Horak, E.M.; Bolen, J.B. The CD4 and CD8 T-cell surface-antigens are associated with the internal membrane tyrosine-protein kinase p56Lck. Cell 1988, 55, 301–308. [Google Scholar] [CrossRef]
- Palacios, E.H.; Weiss, A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 2004, 23, 7990–8000. [Google Scholar] [CrossRef] [PubMed]
- Hermiston, M.L.; Xu, Z.; Weiss, A. CD45: A critical regulator of signaling thresholds in immune cells. Annu. Rev. Immunol. 2003, 21, 107–137. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.E.; Johnson, P. Modulation of immune cell signalling by the leukocyte common tyrosine phosphatase, CD45. Cell. Signal. 2010, 22, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.C.; Iwashima, M.; Turck, C.W.; Weiss, A. ZAP-70—A 70kd protein-tyrosine kinase that associates with the TCR zeta-chain. Cell 1992, 71, 649–662. [Google Scholar] [CrossRef]
- Iwashima, M.; Irving, B.A.; Vanoers, N.S.C.; Chan, A.C.; Weiss, A. Sequential interactions of the TCR with 2 distinct cytoplasmic tyrosine kinases. Science 1994, 263, 1136–1139. [Google Scholar] [CrossRef] [PubMed]
- Szabo, M.; Czompoly, T.; Kvell, K.; Talaber, G.; Bartis, D.; Nemeth, P.; Berki, T.; Boldizsar, F. Fine-tuning of proximal TCR signaling by ZAP-70 tyrosine residues in JurkaT-cells. Int. Immunol. 2012, 24, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Irvine, R.F. Inositol trisphosphate, a novel 2nd messenger in cellular signal transduction. Nature 1984, 312, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Nishibe, S.; Wahl, M.I.; Hernandez-Sotomayor, S.M.T.; Tonks, N.K.; Rhee, S.G.; Carpenter, G. Increase of the catalytic activity of phospholipase C-gamma-1 by tyrosine phosphorylation. Science 1990, 250, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.G.; Sloan-Lancaster, J.; Kitchen, J.; Trible, R.P.; Samelson, L.E. LAT: The ZAP-70 tyrosine kinase substrate that links T-cell receptor to cellular activation. Cell 1998, 92, 83–92. [Google Scholar] [CrossRef]
- Imboden, J.B.; Stobo, J.D. Transmembrane signaling by the T-cell antigen receptor - Perturbation of the T3-antigen receptor complex generates inositol phosphates and releases calcium-ions from intracellular stores. J. Exp. Med. 1985, 161, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Imboden, J.B.; Weiss, A.; Stobo, J.D. The antigen receptor on a human T-cell line initiates activation by increasing cytoplasmic free calcium. J. Immunol. 1985, 134, 663–665. [Google Scholar] [PubMed]
- Macian, F. NFAT proteins: Key regulators of T-cell development and function. Nat. Rev. Immunol. 2005, 5, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Roose, J.P.; Mollenauer, M.; Gupta, V.A.; Stone, J.; Weiss, A. A diacylglycerol-protein kinase C-RasGRP1 pathway directs Ras activation upon antigen receptor stimulation of T-cells. Mol. Cell. Biol. 2005, 25, 4426–4441. [Google Scholar] [CrossRef] [PubMed]
- Genot, E.; Cantrell, D.A. Ras regulation and function in lymphocytes. Curr. Opin. Immunol. 2000, 12, 289–294. [Google Scholar] [CrossRef]
- Isakov, N.; Altman, A. Protein kinase C theta in T-cell activation. Annu. Rev. Immunol. 2002, 20, 761–794. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Baltimore, D. I-kappa-B—A specific inhibitor of the NF-kappa-B transcription factor. Science 1988, 242, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-kappa B and rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef] [PubMed]
- Hacker, H.; Karin, M. Regulation and function of IKK and IKK-related kinases. Science’s STKE 2006, 2006, re13. [Google Scholar] [CrossRef] [PubMed]
- Jain, J.N.; McCaffrey, P.G.; Miner, Z.; Kerppola, T.K.; Lambert, J.N.; Verdine, G.L.; Curran, T.; Rao, A. The T-cell transcription factor NFAT(p) is a substrate for calcineurin and interacts with Fos and Jun. Nature 1993, 365, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Jain, J.; Loh, C.; Rao, A. Transcriptional regulation of the IL-2 gene. Curr. Opin. Immunol. 1995, 7, 333–342. [Google Scholar] [CrossRef]
- Smeets, R.L.; Fleuren, W.W.M.; He, X.; Vink, P.M.; Wijnands, F.; Gorecka, M.; Klop, H.; Bauerschmidt, S.; Garritsen, A.; Koenen, H.J.P.M.; et al. Molecular pathway profiling of T lymphocyte signal transduction pathways; Th1 and Th2 genomic fingerprints are defined by TCR and CD28-mediated signaling. BMC Immunol. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.D.; Ragheb, J.A.; Kitagawa-Sakakida, S.; Schwartz, R.H. Molecular regulation of interleukin-2 expression by CD28 co-stimulation and anergy. Immunol. Rev. 1998, 165, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Kane, L.P.; Lin, J.; Weiss, A. It’s all Rel-ative: NF-kappa B and CD28 costimulation of T-cell activation. Trends Immunol. 2002, 23, 413–420. [Google Scholar] [CrossRef]
- Powell, J.D.; Pollizzi, K.N.; Heikamp, E.B.; Horton, M.R. Regulation of Immune Responses by mTOR. Annu. Rev. Immunol. 2012, 30, 39–68. [Google Scholar] [CrossRef] [PubMed]
- Huehn, J.; Polansky, J.K.; Hamann, A. Epigenetic control of FOXP3 expression: The key to a stable regulatory T-cell lineage? Nat. Rev. Immunol. 2009, 9, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Gudapati, P.; Dragovic, S.; Spencer, C.; Joyce, S.; Killeen, N.; Magnuson, M.A.; Boothby, M. Mammalian Target of Rapamycin Protein Complex 2 Regulates Differentiation of Th1 and Th2 Cell Subsets via Distinct Signaling Pathways. Immunity 2010, 32, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Saez-Rodriguez, J.; Simeoni, L.; Lindquist, J.A.; Hemenway, R.; Bommhardt, U.; Arndt, B.; Haus, U.U.; Weismantel, R.; Gilles, E.D.; Klamt, S.; et al. A logical model provides insights into T-cell receptor signaling. PLoS Comput. Biol. 2007, 3, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Miskov-Zivanov, N.; Turner, M.S.; Kane, L.P.; Morel, P.A.; Faeder, J.R. The Duration of T-cell Stimulation Is a Critical Determinant of Cell Fate and Plasticity. Sci. Signal. 2013, 6, ra97. [Google Scholar] [CrossRef] [PubMed]
- Levchenko, A.; Bruck, J.; Sternberg, P.W. Scaffold proteins may biphasically affect the levels of mitogen-activated protein kinase signaling and reduce its threshold properties. Proc. Natl. Acad. Sci. USA 2000, 97, 5818–5823. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Levchenko, A.; Scott, M.L.; Baltimore, D. The I kappa B-NF-kappa B signaling module: Temporal control and selective gene activation. Science 2002, 298, 1241–1245. [Google Scholar] [CrossRef] [PubMed]
- O’Dea, E.L.; Barken, D.; Peralta, R.Q.; Tran, K.T.; Werner, S.L.; Kearns, J.D.; Levchenko, A.; Hoffmann, A. A homeostatic model of I kappa B metabolism to control constitutive NF-kappa B activity. Mol. Syst. Biol. 2007, 3. [Google Scholar] [CrossRef]
- Coombs, D.; Kalergis, A.M.; Nathenson, S.G.; Wofsy, C.; Goldstein, B. Activated TCRs remain marked for internalization after dissociation from pMHC. Nat. Immunol. 2002, 3, 926–931. [Google Scholar] [CrossRef] [PubMed]
- Wylie, D.C.; Hori, Y.; Dinner, A.R.; Chakraborty, A.K. A hybrid deterministic-stochastic algorithm for modeling cell signaling dynamics in spatially inhomogeneous environments and under the influence of external fields. J. Phys. Chem. B 2006, 110, 12749–12765. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y. A Systems Biology Study to Delineate the T-cell Receptor-Activated Erk-MAPK Signaling Pathway. Ph.D. Thesis, Purdue University, West Lafayette, IN, USA, 2005. [Google Scholar]
- Perley, J.P.; Mikolajczak, J.; Harrison, M.L.; Buzzard, G.T.; Rundell, A.E. Multiple Model-Informed Open-Loop Control of Uncertain Intracellular Signaling Dynamics. PLoS Comput. Biol. 2014, 10, e1003546. [Google Scholar] [CrossRef] [PubMed]
- Buzzard, G.T.; Xiu, D.B. Variance-Based Global Sensitivity Analysis via Sparse-Grid Interpolation and Cubature. Commun. Comput. Phys. 2011, 9, 542–567. [Google Scholar] [CrossRef]
- Buzzard, G.T. Global sensitivity analysis using sparse grid interpolation and polynomial chaos. Reliab. Eng. Syst. Saf. 2012, 107, 82–89. [Google Scholar] [CrossRef]
- Donahue, M.M.; Buzzard, G.T.; Rundell, A.E. Robust Parameter Identification with Adaptive Sparse Grid-based Optimization for Nonlinear Systems Biology Models. In Proceedings of the IEEE 2009 American Control Conference, St. Louis, MO, USA, 10–12 June 2009; IEEE: New York, NY, USA, 2009; Volumes 1–9, pp. 5055–5060. [Google Scholar]
- Von Essen, M.; Bonefeld, C.M.; Siersma, V.; Rasmussen, A.B.; Lauritsen, J.P.H.; Nielsen, B.L.; Geisler, C. Constitutive and ligand-induced TCR degradation. J. Immunol. 2004, 173, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Geisler, C. TCR trafficking in resting and stimulated T-cells. Crit. Rev. Immunol. 2004, 24, 67–85. [Google Scholar] [CrossRef] [PubMed]
- Menne, C.; Sorensen, T.M.; Siersma, V.; von Essen, M.; Odum, N.; Geisler, C. Endo- and exocytic rate constants for spontaneous and protein kinase C-activated T-cell receptor cycling. Eur. J. Immunol. 2002, 32, 616–626. [Google Scholar] [CrossRef]
- Okada, M.; Nada, S.; Yamanashi, Y.; Yamamoto, T.; Nakagawa, H. Csk - A protein-tyrosine kinase involved in regulation of src family kinases. J. Biol. Chem. 1991, 266, 24249–24252. [Google Scholar] [PubMed]
- Plas, D.R.; Johnson, R.; Pingel, J.T.; Matthews, R.J.; Dalton, M.; Roy, G.; Chan, A.C.; Thomas, M.L. Direct regulation of ZAP-70 by SHP-1 in T-cell antigen receptor signaling. Science 1996, 272, 1173–1176. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, I.; Hemmer, B.; Vergelli, M.; Martin, R.; Biddison, W.E.; Germain, R.N. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat. Immunol. 2003, 4, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Lipniacki, T.; Hat, B.; Faeder, J.R.; Hlavacek, W.S. Stochastic effects and bistability in T-cell receptor signaling. J. Theor. Biol. 2008, 254, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Buday, L.; Egan, S.E.; Viciana, P.R.; Cantrell, D.A.; Downward, J. A complex of Grb2 adapter protein, SOS exchange factor, and a 36-kDa membrane-bound tyrosine phosphoprotein is implicated in Ras activation in T-cells. J. Biol. Chem. 1994, 269, 9019–9023. [Google Scholar] [PubMed]
- Clipstone, N.A.; Crabtree, G.R. Identification of calcineurin as a key signaling enzyme in lyphocyte-T activation. Nature 1992, 357, 695–697. [Google Scholar] [CrossRef] [PubMed]
- Rudd, C.E.; Taylor, A.; Schneider, H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009, 229, 12–26. [Google Scholar] [CrossRef] [PubMed]
- McNeill, L.; Salmond, R.J.; Cooper, J.C.; Carret, C.K.; Cassady-Cain, R.L.; Roche-Molina, M.; Tandon, P.; Holmes, N.; Alexander, D.R. The differential regulation of lck kinase phosphorylation sites by CD45 is critical for T-cell receptor signaling responses. Immunity 2007, 27, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.D.; Conroy, L.A.; Byth, K.F.; Hederer, R.A.; Howlett, S.; Takemoto, Y.; Holmes, N.; Alexander, D.R. Aberrant TCR-Mediated signaling in CD45-null thymocytes involves dysfunctional regulation of Lck, Fyn, TCR-zeta and ZAP-70. J. Immunol. 1997, 158, 5773–5782. [Google Scholar] [PubMed]
- Kretschmer, K.; Apostolou, I.; Hawiger, D.; Khazaie, K.; Nussenzweig, M.C.; von Boehmer, H. Inducing and expanding regulatory T-cell populations by foreign antigen. Nat. Immunol. 2005, 6, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.S. Calcium signaling mechanisms in T lymphocytes. Annu. Rev. Immunol. 2001, 19, 497–521. [Google Scholar] [CrossRef] [PubMed]
- Dogan, G. Confidence Interval Estimation in System Dynamics Models: Bootstrapping vs. Likelihood Ratio Method. In Proceedings of the 22nd International Conference of the System Dynamics Society, Oxford, UK, 2004.
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).