
Effects of Denaturants on Early-Stage Prion Conversion: Insights from Molecular Dynamics Simulations


Abstract
1. Introduction

2. Materials and Methods
3. Results
3.1. Secondary Structure Evolution

3.2. Essential Collective Dynamics Analysis



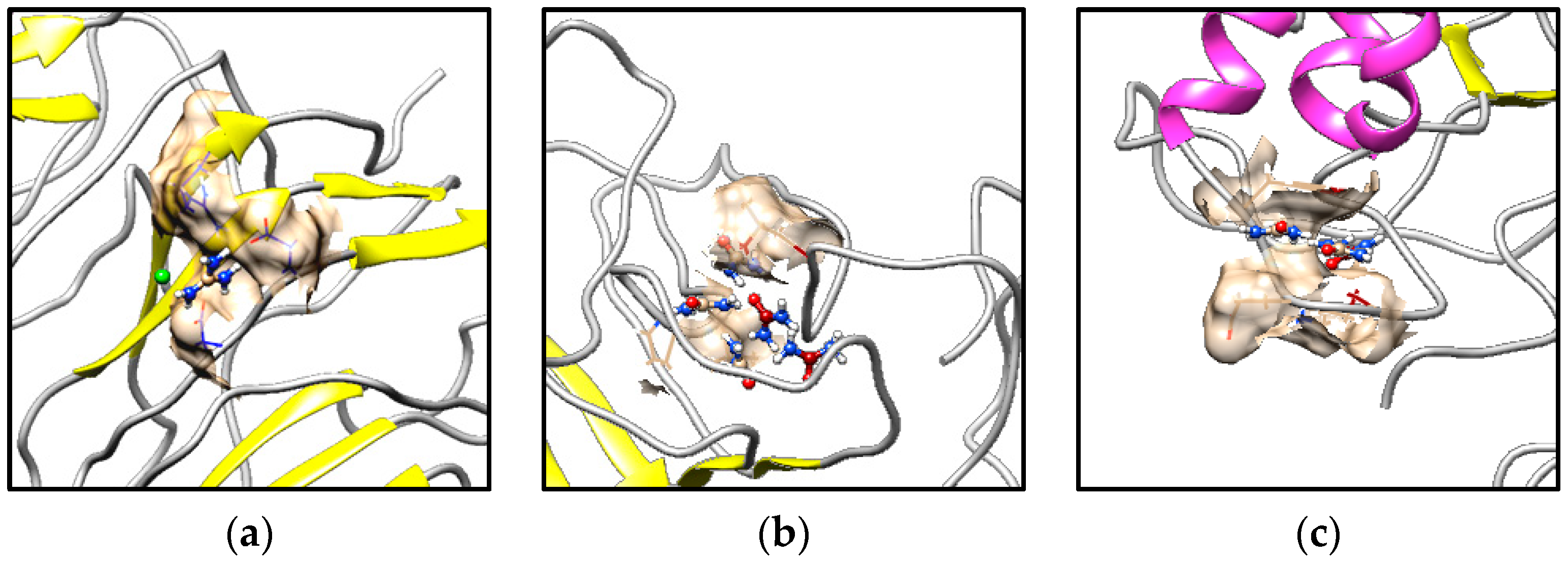
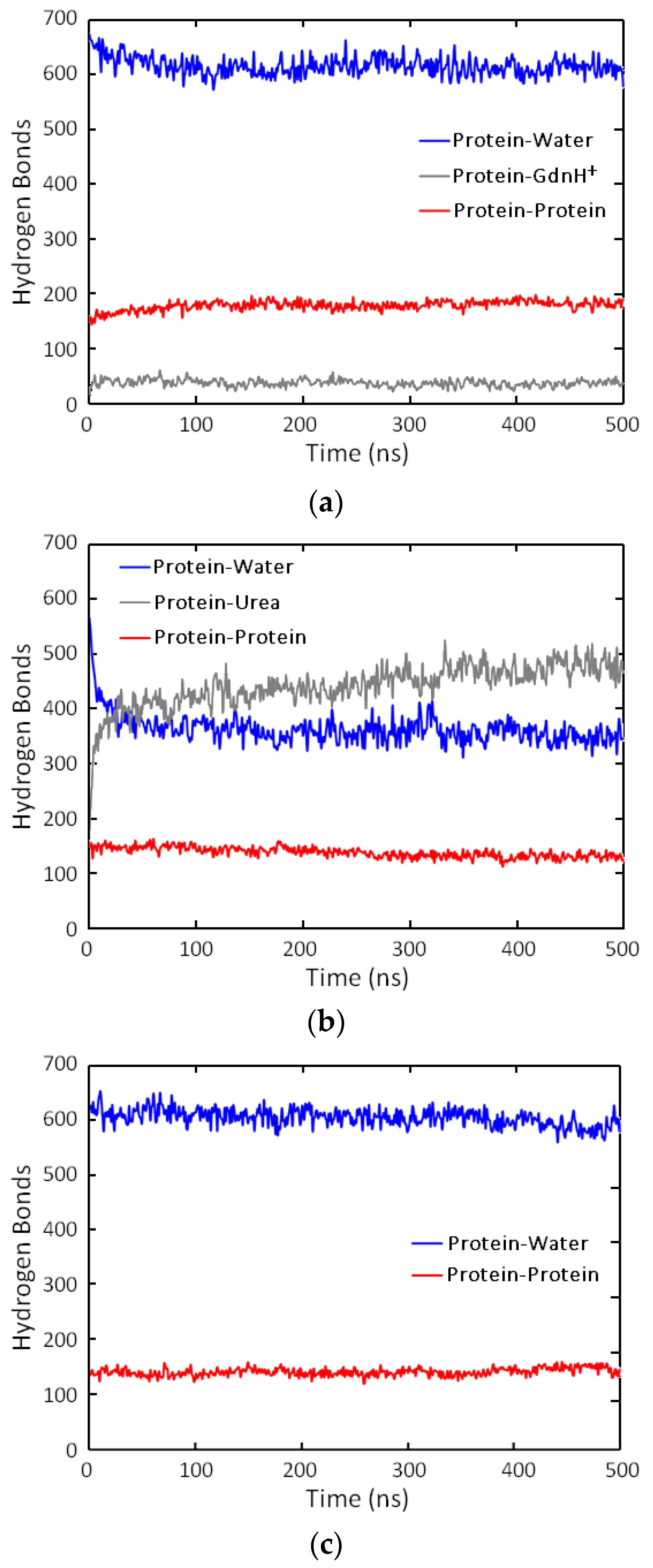
3.3. Hydrogen Bonding Under Denaturant Conditions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PrP | Prion protein |
| TSE | Transmissible spongiform encephalopathy |
| GdnHCl | Guanidine hydrochloride |
| GdnH+ | Guanidinium cation |
| MD | Molecular dynamics |
| WTD | White-tailed deer |
| CWD | Chronic wasting disease |
| 4RβS | Four-rung β-solenoid |
| EM | Electron microscopy |
| cryo-EM | Electron cryomicroscopy |
| HET-s | Heterokaryon incompatibility protein s |
| PIRIBS | Parallel-in-register β-sheets |
| NMR | Nuclear magnetic resonance |
| ECD | Essential collective dynamics |
| PCA | Principal component analysis |
| d | ECD pair correlation descriptor (Equation (2)) |
| ECD backbone flexibility descriptor (Equation (3)) | |
| RMSD | Root-mean-square deviation |
| SB | Salt bridge |
| HB | Hydrogen bond |
Appendix A
References
- Louros, N.; Schymkowitz, J.; Rousseau, F. Mechanisms and pathology of protein misfolding and aggregation. Nat. Rev. Mol. Cell Biol. 2023, 24, 912–933. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef]
- Goedert, M. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 2015, 349, 1255555. [Google Scholar] [CrossRef] [PubMed]
- Casey, C.; Sleator, R.D. Prions: Structure, function, evolution, and disease. Arch. Microbiol. 2025, 207, 1. [Google Scholar] [CrossRef]
- Orge, L.; Lima, C.; Machado, C.; Tavares, P.; Mendonça, P.; Carvalho, P.; Silva, J.; de Lurdes Pinto, M.; Bastos, E.; Pereira, J.C.; et al. Neuropathology of Animal Prion Diseases. Biomolecules 2021, 11, 466. [Google Scholar] [CrossRef]
- Silva, C.J. Chronic Wasting Disease (CWD) in Cervids and the Consequences of a Mutable Protein Conformation. ACS Omega 2022, 7, 12474–12492. [Google Scholar] [CrossRef]
- Zerr, I.; Ladogana, A.; Mead, S.; Hermann, P.; Forloni, G.; Appleby, B.S. Creutzfeldt-Jakob disease and other prion diseases. Nat. Rev. Dis. Primers 2024, 10, 14. [Google Scholar] [CrossRef]
- Shoup, D.; Priola, S.A. Cell biology of prion strains in vivo and in vitro. Cell Tissue Res. 2023, 392, 269–283. [Google Scholar] [CrossRef]
- Alves Conceição, C.; Assis de Lemos, G.; Barros, C.A.; Vieira, T.C. What is the role of lipids in prion conversion and disease? Front. Mol. Neurosci. 2023, 15, 1032541. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Calvo, P.; Callender, J.A.; Sigurdson, C.J. Short and sweet: How glycans impact prion conversion, cofactor interactions, and cross-species transmission. PLoS Pathog. 2021, 17, e1009123. [Google Scholar] [CrossRef] [PubMed]
- Shafiq, M.; Da Vela, S.; Amin, L.; Younas, N.; Harris, D.A.; Zerr, I.; Altmeppen, H.C.; Svergun, D.; Glatzel, M. The prion protein and its ligands: Insights into structure-function relationships. Biochim. Biophys. Acta 2022, 1869, 119240. [Google Scholar] [CrossRef]
- Surewicz, W.K.; Jones, E.M.; Apetri, A.C. The emerging principles of mammalian prion propagation and transmissibility barriers: Insight from studies in vitro. Acc. Chem. Res. 2006, 39, 654–662. [Google Scholar] [CrossRef]
- Spagnolli, G.; Requena, J.R.; Biasini, E. Understanding prion structure and conversion. Prog. Mol. Biol. Transl. Sci. 2020, 175, 19–30. [Google Scholar] [PubMed]
- Bocharova, O.V.; Breydo, L.; Parfenov, A.S.; Salnikov, V.V.; Baskakov, I.V. In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrPSc. J. Mol. Biol. 2005, 346, 645–659. [Google Scholar] [CrossRef]
- Fridmanis, J.; Toleikis, Z.; Sneideris, T.; Ziaunys, M.; Bobrovs, R.; Smirnovas, V.; Jaudzems, K. Aggregation Condition–Structure Relationship of Mouse Prion Protein Fibrils. Int. J. Mol. Sci. 2021, 22, 9635. [Google Scholar] [CrossRef]
- Hosszu, L.L.P.; Wells, M.A.; Jackson, G.S.; Jones, S.; Batchelor, M.; Clarke, A.R.; Craven, C.J.; Waltho, J.P.; Collinge, J. Definable equilibrium states in the folding of human prion protein. Biochemistry 2005, 44, 16649–16657. [Google Scholar] [CrossRef] [PubMed]
- Julien, O.; Chatterjee, S.; Thiessen, A.; Graether, S.P.; Sykes, B.D. Differential stability of the bovine prion protein upon urea unfolding. Prot. Sci. 2009, 18, 2172–2182. [Google Scholar] [CrossRef]
- Julien, O.; Chatterjee, S.; Bjorndahl, T.C.; Sweeting, B.; Acharya, S.; Semenchenko, V.; Chakrabartty, A.; Pai, E.F.; Wishart, D.S.; Sykes, B.D.; et al. Relative and regional stabilities of the hamster, mouse, rabbit, and bovine prion proteins toward urea unfolding assessed by nuclear magnetic resonance and circular dichroism spectroscopies. Biochemistry 2011, 50, 7536–7545. [Google Scholar] [CrossRef]
- Milto, K.; Michailova, K.; Smirnovas, V. Elongation of mouse prion protein amyloid-like fibrils: Effect of temperature and denaturant concentration. PLoS ONE 2014, 9, e94469. [Google Scholar] [CrossRef] [PubMed]
- Swietnicki, W.; Morillas, M.; Chen, S.G.; Gambetti, P.; Surewicz, W.K. Aggregation and fibrillization of the recombinant human prion protein huPrP90–231. Biochemistry 2000, 39, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Polano, M.; Bek, A.; Benetti, F.; Lazzarino, M.; Legname, G. Structural insights into alternate aggregated prion protein forms. J. Mol. Biol. 2009, 393, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Paladino, A.; Vitagliano, L.; Graziano, G. The action of chemical denaturants: From globular to intrinsically disordered proteins. Biology 2023, 12, 754. [Google Scholar] [CrossRef]
- Camilloni, C.; Tiana, G. Urea and guanidinium chloride denature protein L in different ways in molecular dynamics simulations. Biophys. J. 2008, 94, 4654–4661. [Google Scholar] [CrossRef]
- Meloni, R.; Camilloni CTiana, G. Sampling the Denatured State of Polypeptides in Water, Urea, and Guanidine Chloride to Strict Equilibrium Conditions with the Help of Massively Parallel Computers. J. Chem. Theory Comput. 2014, 10, 846–854. [Google Scholar] [CrossRef]
- Parui, S.; Jana, B. Relative Solvent Exposure of the Alpha-Helix and Beta-Sheet in Water Determines the Initial Stages of Urea and Guanidinium Chloride-Induced Denaturation of Alpha/Beta Proteins. J. Phys. Chem. B 2019, 123, 8889–8900. [Google Scholar] [CrossRef]
- Heyda, J.; Kožíšek, M.; Bednárová, L.; Thompson, G.; Konvalinka, J.; Vondrášek, J.; Jungwirth, P. Urea and Guanidinium Induced Denaturation of a Trp-Cage Miniprotein. J. Phys. Chem. B 2011, 115, 8910–8924. [Google Scholar] [CrossRef]
- Meloni, R.; Tiana, G. Thermodynamic and structural effect of urea and guanidine chloride on the helical and on a hairpin fragment of GB1 from molecular simulations. Proteins 2017, 85, 753–763. [Google Scholar] [CrossRef]
- Mandal, M.; Mukhopadhyay, C. Microsecond molecular dynamics simulation of guanidinium chloride induced unfolding of ubiquitin. Phys. Chem. Chem. Phys. 2014, 16, 21706–21716. [Google Scholar] [CrossRef]
- Ganguly, P.; Shea, J.-E. Distinct and Nonadditive Effects of Urea and Guanidinium Chloride on Peptide Solvation. J. Phys. Chem. Lett. 2019, 10, 7406–7413. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Mukhopadhyay, C. Atomistic mechanism of protein denaturation by urea. J. Phys. Chem. B 2008, 112, 7903–7908. [Google Scholar] [CrossRef]
- Eberini, I.; Emerson, A.; Sensi, C.; Ragona, L.; Ricchiuto, P.; Pedretti, A.; Gianazza, E.; Tramontano, A. Simulation of urea-induced protein unfolding: A lesson from bovine β-lactoglobulin. J. Mol. Graph. Model. 2011, 30, 24–30. [Google Scholar] [CrossRef]
- Rg, C.; Tallon, A.; Latch, E.K. Chronic Wasting Disease Research in North America: A systematic review highlighting species-wise and interdisciplinary research trends. Prion 2025, 19, 1–16. [Google Scholar] [CrossRef]
- Spagnolli, G.; Rigolli, M.; Orioli, S.; Sevillano, A.M.; Faccioli, P.; Wille, H.; Biasini, E.; Requena, J.R. Full atomistic model of prion structure and conversion. PLoS Pathog. 2019, 15, e1007864. [Google Scholar] [CrossRef]
- Wille, H.; Bian, W.; McDonald, M.; Kendall, A.; Colby, D.W.; Bloch, L.; Ollesch, J.; Borovinskiy, A.L.; Cohen, F.E.; Prusiner, S.B.; et al. Natural and synthetic prion structure from X-ray fiber diffraction. Proc. Natl. Acad. Sci. USA 2009, 106, 16990–16995. [Google Scholar] [CrossRef] [PubMed]
- Kamali-Jamil, R.; Vázquez-Fernández, E.; Tancowny, B.; Rathod, V.; Amidian, S.; Wang, X.; Tang, X.; Fang, A.; Senatore, A.; Hornemann, S.; et al. The ultrastructure of infectious L-type bovine spongiform encephalopathy prions constrains molecular models. PLoS Pathog. 2021, 17, e1009628. [Google Scholar] [CrossRef] [PubMed]
- Wasmer, C.; Lange, A.; Van Melckebeke, H.; Siemer, A.B.; Riek, R.; Meier, B.H. Amyloid fibrils of the HET-s (218–289) prion form a beta solenoid with a triangular hydrophobic core. Science 2008, 319, 1523–1526. [Google Scholar] [CrossRef]
- Manka, S.W.; Wenborn, A.; Collinge, J.; Wadsworth, J.D.F. Prion strains viewed through the lens of cryo-EM. Cell Tissue Res. 2023, 392, 167–178. [Google Scholar] [CrossRef]
- Ribes, J.M.; Patel, M.P.; Halim, H.A.; Berretta, A.; Tooze, S.A.; Klöhn, P.-C. Prion protein conversion at two distinct cellular sites precedes fibrillisation. Nat. Commun. 2023, 14, 8354. [Google Scholar] [CrossRef]
- BIOVIA Discovery Studio Visualizer, v. 19.1.0.1828; Dassault Systèmes: San Diego, CA, USA. Available online: https://www.3ds.com/products/biovia/discovery-studio (accessed on 17 July 2019).
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Baral, P.K.; Swayampakula, M.; Aguzzi, A.; James, M.N. X-ray structural and molecular dynamical studies of the globular domains of cow, deer, elk and Syrian hamster prion proteins. J. Struct. Biol. 2015, 192, 37–47. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Stepanova, M. Dynamics of essential collective motions in proteins: Theory. Phys. Rev. E. 2007, 76, 051918. [Google Scholar] [CrossRef] [PubMed]
- Potapov, A.; Stepanova, M. Conformational modes in biomolecules: Dynamics and approximate invariance. Phys. Rev. E. 2012, 85 Pt 1, R02090. [Google Scholar] [CrossRef] [PubMed]
- Dorosh, L.; Stepanova, M. Probing oligomerization of amyloid beta peptide in silico. Mol. Biosyst. 2017, 13, 165–182. [Google Scholar] [CrossRef]
- Issack, B.B.; Berjanskii, M.; Wishart, D.S.; Stepanova, M. Exploring the essential collective dynamics of interacting proteins: Application to prion protein dimers. Proteins Struct. Funct. Bioinforma. 2012, 80, 1847–1865. [Google Scholar] [CrossRef]
- Wu, M.; Wille, H.; Stepanova, M. Essential collective dynamics analysis reveals nonlocal interactions of alpha-synuclein38-95 monomers with fibrillar seeds. J. Chem. Phys. 2022, 157, 235101. [Google Scholar] [CrossRef]
- Blinov, N.; Berjanskii, M.; Wishart, D.S.; Stepanova, M. Structural domains and main-chain flexibility in prion proteins. Biochemistry 2009, 48, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Santo, K.P.; Berjanskii, M.; Wishart, D.S.; Stepanova, M. Comparative analysis of essential collective dynamics and NMR-derived flexibility profiles in evolutionarily diverse prion proteins. Prion 2011, 5, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Mercer, R.C.C.; Daude, N.; Dorosh, L.; Fu, Z.-L.; Mays, C.E.; Gapeshina, H.; Wohlgemuth, S.L.; Acevedo-Morantes, C.Y.; Yang, J.; Cashman, N.R.; et al. A novel Gerstmann-Sträussler-Scheinker disease mutation defines a precursor for amyloidogenic 8 kDa PrP fragments and reveals N-terminal structural changes shared by other GSS alleles. PLoS Pathog. 2018, 14, e1006826. [Google Scholar] [CrossRef] [PubMed]
- Morillas, M.; Vanik, D.L.; Surewicz, W.K. On the mechanism of α-helix to β-sheet transition in the recombinant prion protein. Biochemistry 2001, 40, 6982–6987. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Denaturant | α-Helical Content% | β-Sheet Content% |
|---|---|---|
| GdnHCl | 20.4 ± 0.1 | 19.5 ± 0.1 |
| Urea | 16.7 ± 0.1 | 10.0 ± 0.1 |
| None | 16.6 ± 0.1 | 13.4 ± 0.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dorosh, L.; Wu, M.; Stepanova, M. Effects of Denaturants on Early-Stage Prion Conversion: Insights from Molecular Dynamics Simulations. Processes 2025, 13, 2151. https://doi.org/10.3390/pr13072151
Dorosh L, Wu M, Stepanova M. Effects of Denaturants on Early-Stage Prion Conversion: Insights from Molecular Dynamics Simulations. Processes. 2025; 13(7):2151. https://doi.org/10.3390/pr13072151
Chicago/Turabian StyleDorosh, Lyudmyla, Min Wu, and Maria Stepanova. 2025. "Effects of Denaturants on Early-Stage Prion Conversion: Insights from Molecular Dynamics Simulations" Processes 13, no. 7: 2151. https://doi.org/10.3390/pr13072151
APA StyleDorosh, L., Wu, M., & Stepanova, M. (2025). Effects of Denaturants on Early-Stage Prion Conversion: Insights from Molecular Dynamics Simulations. Processes, 13(7), 2151. https://doi.org/10.3390/pr13072151