
Comparative Analysis of Electrochemical and Thermochemical Hydrogenation of Biomass-Derived Phenolics for Sustainable Biofuel and Chemical Production


Abstract
1. Introduction
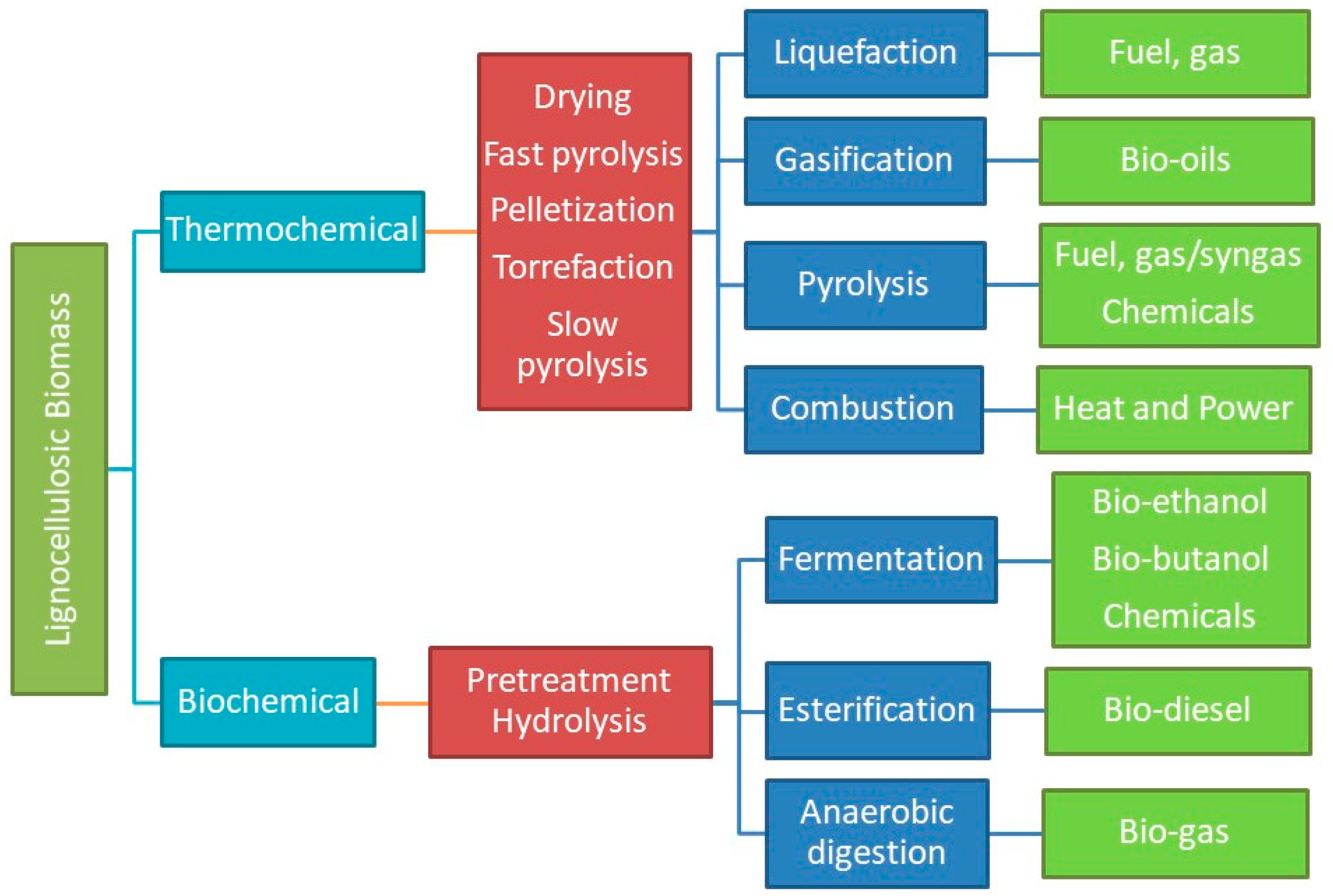
2. Thermochemical Conversion Processes for Biomass Valorization
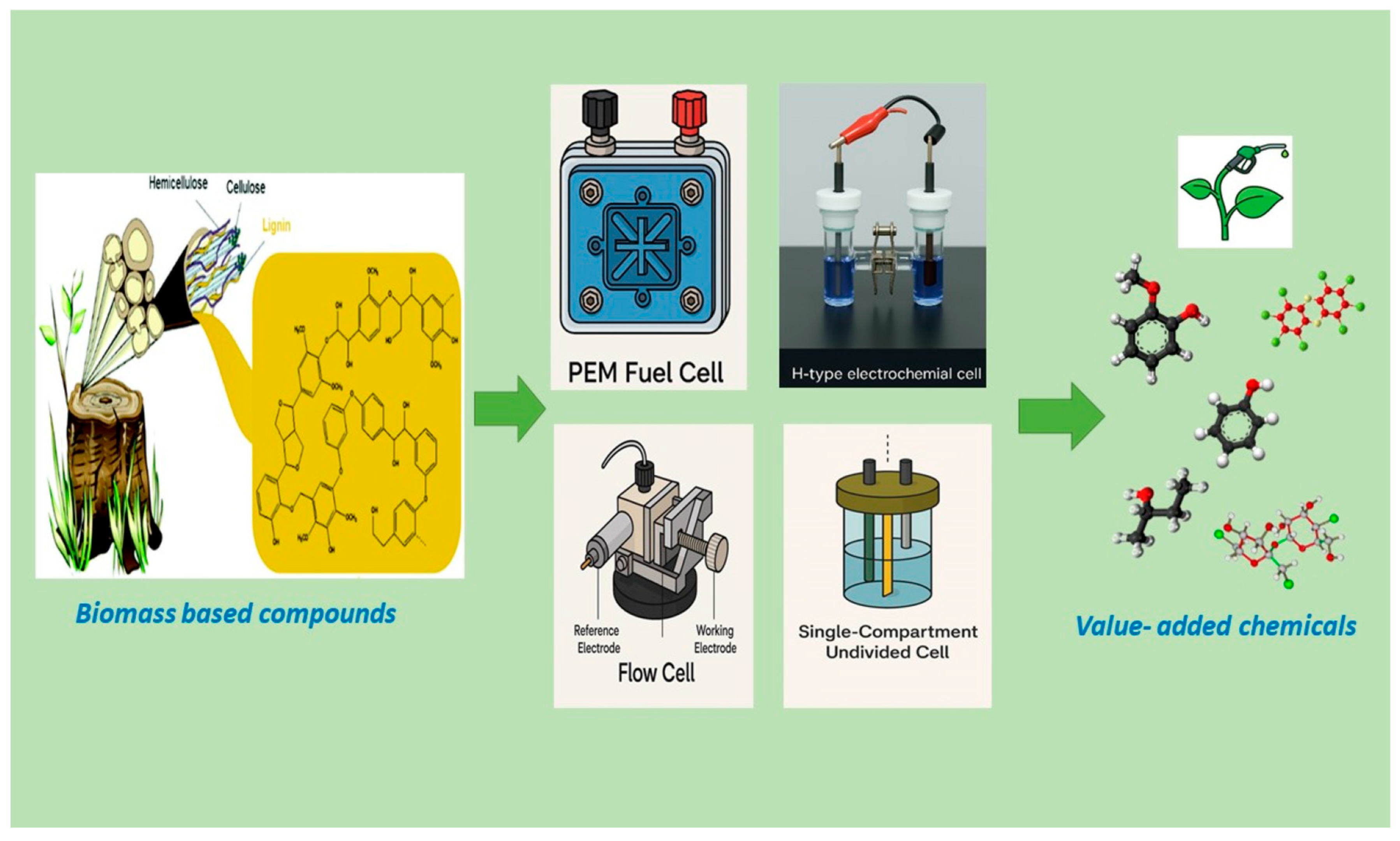
3. Electrochemical Hydrogenation (ECH)
3.1. The Role of Electrochemical Processes in Sustainable Chemical Conversion and Energy Systems
3.2. Controlled Selectivity and Low Carbon Emission in Electrochemical Conversion: Advancing Precision and Sustainability in Chemical Processes
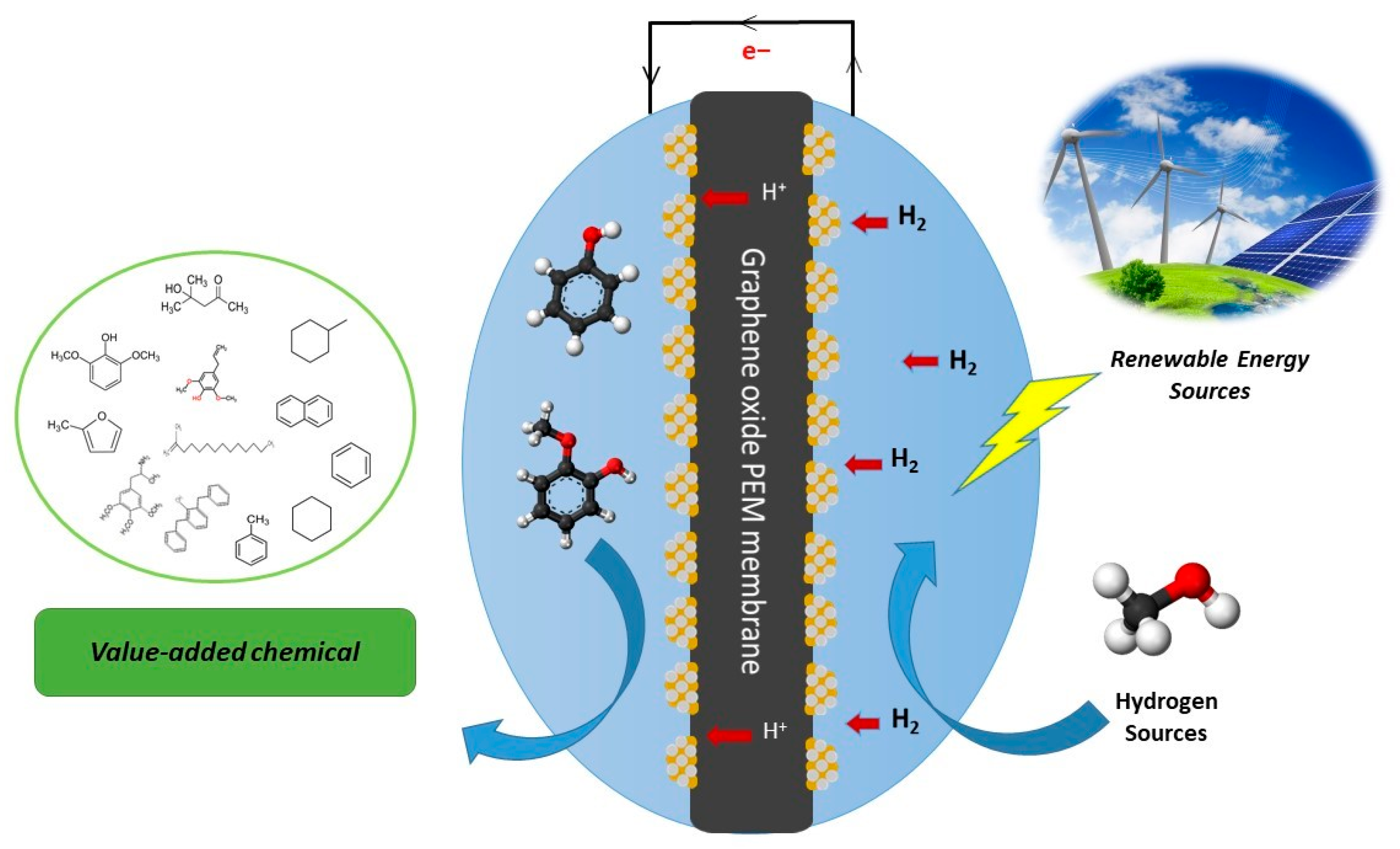
3.3. Electrochemical Conversion in PEM Fuel Cells: Distinct Physical and Chemical Advantages
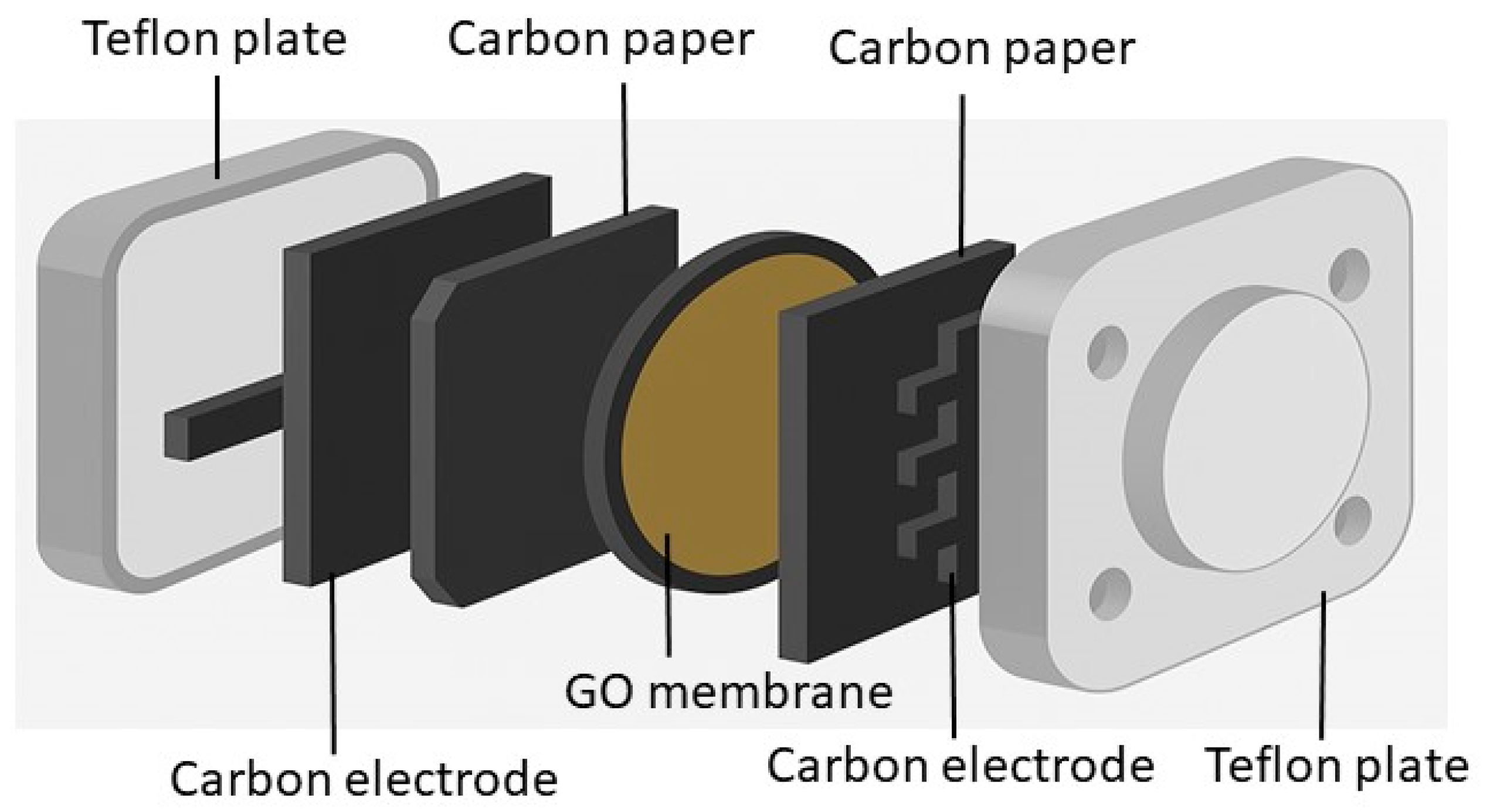
3.4. Proton-Exchange Membranes (PEMs) and Their Functional Role in Electrochemical Devices
3.5. Ion-Exchange Membranes in Electrochemical Systems: Functionality, Types, and Applications
3.6. Metal–Organic Frameworks (MOFs): Structural Advantages and Applications in Electrochemical Systems
4. Electrochemical Hydrogenation of Biomass-Derived Compounds Using Various Electrocatalysts, Membranes, and Electrode Systems
5. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ubando, A.T.; Felix, C.B.; Chen, W.-H. Biorefineries in Circular Bioeconomy: A Comprehensive Review. Bioresour. Technol. 2020, 299, 122585. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S. Catalytic Transformation of Biomass into Sustainable Carbocycles: Recent Advances, Prospects, and Challenges. ChemPlusChem 2025, 90, e202400568. [Google Scholar] [CrossRef]
- Velvizhi, G.; Jacqueline, P.J.; Shetti, N.P.; Latha, K.; Mohanakrishna, G.; Aminabhavi, T.M. Emerging Trends and Advances in Valorization of Lignocellulosic Biomass to Biofuels. J. Environ. Manag. 2023, 345, 118527. [Google Scholar] [CrossRef]
- Deng, W.; Feng, Y.; Fu, J.; Guo, H.; Guo, Y.; Han, B.; Jiang, Z.; Kong, L.; Li, C.; Liu, H.; et al. Biomass Refining and Valorization Toward Carbon Neutrality with High Efficiency, Low Cost, and Low Carbon Emission: A Review. Green Energy Environ. 2023, 8, 10–114. [Google Scholar] [CrossRef]
- Williams, C.L.; Westover, T.L.; Emerson, R.M.; Tumuluru, J.S.; Li, C. Sources of Biomass Feedstock Variability and the Potential Impact on Biofuels Production. BioEnergy Res. 2016, 9, 1–14. [Google Scholar] [CrossRef]
- Lee, R.A.; Lavoie, J.-M. From First- to Third-Generation Biofuels: Challenges of Producing a Commodity from a Biomass of Increasing Complexity. Animal Front. 2013, 3, 6–11. [Google Scholar] [CrossRef]
- Calvo-Flores, F.G.; Martin-Martinez, F.J. Lignin as Renewable Raw Material. Front. Chem. 2022, 10, 973417. [Google Scholar] [CrossRef]
- Adeleke, A.A.; Ikubanni, P.P.; Orhadahwe, T.A.; Christopher, C.T.; Akano, J.M.; Agboola, O.O.; Adegoke, S.O.; Balogun, A.O.; Ibikunle, R.A. A Review of Biomass Gasification as a Renewable Energy Source. Heliyon 2021, 7, e05973. [Google Scholar] [CrossRef]
- Cardona-Alzate, C.A.; Serna-Loaiza, S.; Ortiz-Sanchez, M. Trends in the Design and Use of Biorefineries in Colombia: A Review. J. Sustain. Dev. Energy Water Environ. Syst. 2020, 8, 88–117. [Google Scholar] [CrossRef]
- Modak, A.; Maiti, D. Recent Advances in Transition Metal-Catalyzed Direct C–H Arylation. Org. Biomol. Chem. 2016, 14, 21–35. [Google Scholar] [CrossRef]
- Grover, H.K.; Emmett, M.R.; Kerr, M.A. Recent Advances in the Synthesis of Cyclopropanes. Org. Biomol. Chem. 2015, 13, 655–671. [Google Scholar] [CrossRef] [PubMed]
- Colquhoun, H.M.; Holton, J.; Thompson, D.J.; Twigg, M.V. New Pathways for Organic Synthesis; Springer US: Boston, MA, USA, 1984; pp. 67–140. [Google Scholar]
- Vassilev, S.V.; Baxter, D.; Andersen, L.K.; Vassileva, C.G. An Overview of the Chemical Composition of Biomass. Fuel 2010, 89, 913–933. [Google Scholar] [CrossRef]
- Wang, H.; Pu, Y.; Ragauskas, A.; Yang, B. From Lignin to Valuable Products—Strategies, Challenges, and Prospects. Bioresour. Technol. 2019, 271, 449–461. [Google Scholar] [CrossRef]
- He, J.; Liu, M.; Huang, K.; Walker, T.W.; Maravelias, C.T.; Dumesic, J.A.; Huber, G.W. Integrated Catalytic Conversion of γ-Valerolactone to Liquid Alkenes for Transportation Fuels. Green Chem. 2017, 19, 3642–3653. [Google Scholar] [CrossRef]
- Kobayashi, H.; Yabushita, M.; Komanoya, T.; Hara, K.; Fujita, I.; Fukuoka, A. Production of Aromatics from Lignin via Catalytic Fast Pyrolysis Using Hybrid Mesoporous Silica. ACS Catal. 2013, 3, 581–587. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y.; Qian, C.; An, L.; Wang, W.; Li, X.; Shao, X.; Li, Z. Catalytic Conversion of Biomass-Derived Compounds into Biofuels: Recent Advances. RSC Adv. 2023, 13, 9466–9478. [Google Scholar] [CrossRef]
- Ravasco, J.M.J.M.; Gomes, R.F.A. Recent Developments in Catalytic Lignin Depolymerization Strategies. ChemSusChem 2021, 14, 3047–3053. [Google Scholar] [CrossRef]
- Tews, I.J.; Garcia-Perez, M. Advanced Oxidative Techniques for the Treatment of Aqueous Liquid Effluents from Biomass Thermochemical Conversion Processes: A Review. Energy Fuels 2022, 36, 60–79. [Google Scholar] [CrossRef]
- Mathanker, A.; Das, S.; Pudasainee, D.; Khan, M.; Kumar, A.; Gupta, R. A Review of Hydrothermal Liquefaction of Biomass for Biofuels Production with a Special Focus on the Effect of Process Parameters, Co-Solvents, and Extraction Solvents. Energies 2021, 14, 4916. [Google Scholar] [CrossRef]
- Durak, H. Comprehensive Assessment of Thermochemical Processes for Sustainable Waste Management and Resource Recovery. Processes 2023, 11, 2092. [Google Scholar] [CrossRef]
- Venkatachalam, C.D.; Ravichandran, S.R.; Sengottian, M. Lignocellulosic and Algal Biomass for Bio-Crude Production Using Hydrothermal Liquefaction: Conversion Techniques, Mechanism and Process Conditions: A Review. Environ. Eng. Res. 2021, 27, 200555. [Google Scholar] [CrossRef]
- Li, Q.; Faramarzi, A.; Zhang, S.; Wang, Y.; Hu, X.; Gholizadeh, M. Progress in Catalytic Pyrolysis of Municipal Solid Waste. Energy Convers. Manag. 2020, 226, 113525. [Google Scholar] [CrossRef]
- Kan, T.; Strezov, V.; Evans, T.J. Lignocellulosic Biomass Pyrolysis: A Review of Product Properties and Effects of Pyrolysis Parameters. Renew. Sustain. Energy Rev. 2016, 57, 1126–1140. [Google Scholar] [CrossRef]
- Du, Y.; Ju, T.; Meng, Y.; Lan, T.; Han, S.; Jiang, J. A Review on Municipal Solid Waste Pyrolysis of Different Composition for Gas Production. Fuel Process. Technol. 2021, 224, 107026. [Google Scholar] [CrossRef]
- Zaccariello, L.; Montagnaro, F. Fluidised Bed Gasification of Biomasses and Wastes to Produce Hydrogen-Rich Syn-Gas—A Review. J. Chem. Technol. Biotechnol. 2023, 98, 1878–1887. [Google Scholar] [CrossRef]
- Sikarwar, V.S.; Zhao, M.; Clough, P.; Yao, J.; Zhong, X.; Memon, M.Z.; Shah, N.; Anthony, E.J.; Fennell, P.S. An Overview of Advances in Biomass Gasification. Energy Environ. Sci. 2016, 9, 2939–2977. [Google Scholar] [CrossRef]
- Seo, M.W.; Lee, S.H.; Nam, H.; Lee, D.; Tokmurzin, D.; Wang, S.; Park, Y.-K. Recent Advances of Thermochemical Conversion Processes for Biorefinery. Bioresour. Technol. 2022, 343, 126109. [Google Scholar] [CrossRef]
- Martins, M.M.; Carvalheiro, F.; Girio, F. An Overview of Lignin Pathways of Valorization: From Isolation to Refining and Conversion into Value-Added Products. Biomass Convers. Biorefin. 2024, 14, 3183–3207. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Phan, D.-P.; Sarwar, A.; Tran, M.H.; Lee, O.K.; Lee, E.Y. Valorization of Industrial Lignin to Value-Added Chemicals by Chemical Depolymerization and Biological Conversion. Ind. Crops Prod. 2021, 161, 113219. [Google Scholar] [CrossRef]
- Amen-Chen, C.; Pakdel, H.; Roy, C. Production of Monomeric Phenols by Thermochemical Conversion of Biomass: A Review. Bioresour. Technol. 2001, 79, 277–299. [Google Scholar] [CrossRef]
- Gosselink, R.J.A.; de Jong, E.; Guran, B.; Abächerli, A. Co-Ordination Network for Lignin—Standardisation, Production and Applications Adapted to Market Requirements (EUROLIGNIN). Ind. Crops Prod. 2004, 20, 121–129. [Google Scholar] [CrossRef]
- International Air Transport Association (IATA). Air Transport Association (IATA). A Global Approach to Reducing Aviation Emissions. In First Stop: Carbon-Neutral Growth from 2020; IATA: Geneva, Switzerland, 2009. [Google Scholar]
- Hileman, J.I.; Wong, H.M.; Ortiz, D.; Brown, N.; Maurice, L.; Rumizen, M. The Feasibility and Potential Environmental Benefits of Alternative Fuels for Commercial Aviation. In Proceedings of the 26th International Congress of the Aeronautical Sciences, Anchorage, AK, USA, 14–19 September 2008; pp. 5–8. [Google Scholar]
- Kim, B.Y.; Fleming, G.G.; Lee, J.J.; Waitz, I.A.; Clarke, J.-P.; Balasubramanian, S.; Malwitz, A.; Klima, K.; Locke, M.; Holsclaw, C.A.; et al. System for Assessing Aviation’s Global Emissions (SAGE), Part 1: Model Description and Inventory Results. Transp. Res. Part D Transp. Environ. 2007, 12, 325–346. [Google Scholar] [CrossRef]
- Speight, J.G. Handbook of Industrial Hydrocarbon Processes; Gulf Professional Publishing: Oxford, UK, 2019. [Google Scholar]
- Enright, C. Aviation Fuel Standard Takes Flight. ASTM Stand. News 2011, 39, 624. [Google Scholar]
- Hileman, J.I.; Stratton, R.W. Alternative Jet Fuel Feasibility. Transp. Policy 2014, 34, 52–62. [Google Scholar] [CrossRef]
- Shafer, L.; Striebich, R.; Gomach, J.; Edwards, T. Chemical Class Composition of Commercial Jet Fuels and Other Specialty Kerosene Fuels. In Proceedings of the 14th AIAA/AHI Space Planes and Hypersonic Systems and Technologies Conference, Canberra, Australia, 6–9 November 2006. [Google Scholar]
- Hu, P.; Xu, W.T.; Tian, L.; Zhu, H.C.; Li, F.B.; Qi, X.T.; Lu, Q.Q. Electrocatalytic Hydrogenation of Olefins. Angew. Chem. Int. Ed. 2025, 64, e202501215. [Google Scholar] [CrossRef]
- Hileman, J.I.; Ortiz, D.S.; Bartis, J.T.; Wong, H.M.; Donohoo, P.E.; Weiss, M.A.; Waitz, I.A. Near-Term Feasibility of Alternative Jet Fuels; Rand Corporation: Santa Monica, CA, USA, 2009. [Google Scholar]
- Green, S.K.; Lee, J.; Kim, H.J.; Tompsett, G.A.; Kim, W.B.; Huber, G.W. The Electrocatalytic Hydrogenation of Furanic Compounds in a Continuous Electrocatalytic Membrane Reactor. Green Chem. 2013, 15, 1869. [Google Scholar] [CrossRef]
- Zhang, K.D.; Sun, Y.W.; Chen, Z.Y.; Dong, M.Y.; Fu, H.Q.; Xu, Y.M.; Zou, Y.; Hu, M.Q.; Fu, B.; Wang, X.; et al. Catalyst Development for Electrochemical Hydrogenation of Biomass-Derived Platform Molecules. Chem. Phys. Rev. 2024, 5, 041301. [Google Scholar] [CrossRef]
- Xu, W.; Yu, C.; Chen, J.; Liu, Z. Electrochemical Hydrogenation of Biomass-Based Furfural in Aqueous Media by Cu Catalyst Supported on N-Doped Hierarchically Porous Carbon. Appl. Catal. B 2022, 305, 121062. [Google Scholar] [CrossRef]
- Li, K.; Sun, Y. Electrocatalytic Upgrading of Biomass-Derived Intermediate Compounds to Value-Added Products. Chem. Eur. J. 2018, 24, 18258–18270. [Google Scholar] [CrossRef]
- Carneiro, J.; Nikolla, E. Electrochemical Conversion of Biomass-Based Oxygenated Compounds. Annu. Rev. Chem. Biomol. Eng. 2019, 10, 85–104. [Google Scholar] [CrossRef]
- Zhou, P.; Zhang, J. Electrochemical Transformation of Biomass-Derived Oxygenates. Sci. China Chem. 2023, 66, 1011–1031. [Google Scholar] [CrossRef]
- Zhang, L.; Rao, T.U.; Wang, J.; Ren, D.; Sirisommboonchai, S.; Choi, C.; Machida, H.; Huo, Z.; Norinaga, K. A Review of Thermal Catalytic and Electrochemical Hydrogenation Approaches for Converting Biomass-Derived Compounds to High-Value Chemicals and Fuels. Fuel Process. Technol. 2022, 226, 107097. [Google Scholar] [CrossRef]
- Tian, S.; Wang, Z.; Gong, W.; Chen, W.; Feng, Q.; Xu, Q.; Chen, C.; Chen, C.; Peng, Q.; Gu, L.; et al. Temperature-Controlled Selectivity of Hydrogenation and Hydrodeoxygenation in the Conversion of Biomass Molecule by the Ru1/mpg-C3N4 Catalyst. J. Am. Chem. Soc. 2018, 140, 11161–11164. [Google Scholar] [CrossRef]
- Zhang, Y.; Shen, Y. Electrochemical Hydrogenation of Levulinic Acid, Furfural and 5-Hydroxymethylfurfural. Appl. Catal., B 2024, 343, 123576. [Google Scholar] [CrossRef]
- Francke, R.; Little, R.D. Redox Catalysis in Organic Electrosynthesis: Basic Principles and Recent Developments. Chem. Soc. Rev. 2014, 43, 2492–2521. [Google Scholar] [CrossRef]
- Akhade, S.A.; Singh, N.; Gutierrez, O.Y.; Lopez-Ruiz, J.; Wang, H.; Holladay, J.D.; Liu, Y.; Karkamkar, A.; Weber, R.S.; Padmaperuma, A.B.; et al. Electrocatalytic Hydrogenation of Biomass-Derived Organics: A Review. Chem. Rev. 2020, 120, 11370–11419. [Google Scholar] [CrossRef]
- Du, L.; Shao, Y.; Sun, J.; Yin, G.; Du, C.; Wang, Y. Electrocatalytic Valorisation of Biomass Derived Chemicals. Catal. Sci. Technol. 2018, 8, 3216–3232. [Google Scholar] [CrossRef]
- Qu, R.; Junge, K.; Beller, M. Hydrogenation of Carboxylic Acids, Esters, and Related Compounds over Heterogeneous Catalysts: A Step Toward Sustainable and Carbon-Neutral Processes. Chem. Rev. 2023, 123, 1103–1165. [Google Scholar] [CrossRef]
- De Luna, P.; Hahn, C.; Higgins, D.; Jaffer, S.A.; Jaramillo, T.F.; Sargent, E.H. What Would It Take for Renewably Powered Electrosynthesis to Displace Petrochemical Processes? Science 2019, 364, eaav3506. [Google Scholar] [CrossRef]
- Begildayeva, T.; Theerthagiri, J.; Min, A.; Moon, C.J.; Choi, M.Y. Density-Controlled Metalloporphyrin with Mutated Surface via Pulsed Laser for Oxidative Refining of Alcohols to Benzoic Acid and H2 Production Using Linear Tandem Electrolysis. Appl. Catal. B 2024, 350, 123907. [Google Scholar] [CrossRef]
- Naik Shreyanka, S.; Theerthagiri, J.; Lee, S.J.; Yu, Y.; Choi, M.Y. Multiscale Design of 3D Metal–Organic Frameworks (M−BTC, M: Cu, Co, Ni) via PLAL Enabling Bifunctional Electrocatalysts for Robust Overall Water Splitting. Chem. Eng. J. 2022, 446, 137045. [Google Scholar] [CrossRef]
- Aggarwala, P.; Sarkar, D.; Awasthi, K.; Menezes, W.P. Functional Role of Single-Atom Catalysts in Electrocatalytic Hydrogen Evolution: Current Developments and Future Challenges. Coord. Chem. Rev. 2022, 452, 214289. [Google Scholar] [CrossRef]
- Liu, J. Catalysis by Supported Single Metal Atoms. ACS Catal. 2017, 7, 34–59. [Google Scholar] [CrossRef]
- Wang, A.; Li, J.; Zhang, T. Heterogeneous Single-Atom Catalysis. Nat. Rev. Chem. 2018, 2, 65–81. [Google Scholar] [CrossRef]
- Li, J.; Stephanopoulos, M.F.; Xia, Y. Introduction: Heterogeneous Single-Atom Catalysis. Chem. Rev. 2020, 120, 11699–11702. [Google Scholar] [CrossRef]
- Yin, X.P.; Wang, H.J.; Tang, S.F.; Lu, X.L.; Shu, M.; Si, R.; Lu, T.B. Engineering the Coordination Environment of Single-Atom Platinum Anchored on Graphdiyne for Optimizing Electrocatalytic Hydrogen Evolution. Angew. Chem. Int. Ed. 2018, 57, 9382–9386. [Google Scholar] [CrossRef]
- Chen, W.; Pei, J.; He, C.T.; Wan, J.; Ren, H.; Wang, Y.; Dong, J.; Wu, K.; Cheong, W.C.; Mao, J.; et al. Single Tungsten Atoms Supported on MOF-Derived N-Doped Carbon for Robust Electrochemical Hydrogen Evolution. Adv. Mater. 2018, 30, 1800396. [Google Scholar] [CrossRef]
- Wan, J.; Zhao, Z.; Shang, H.; Peng, B.; Chen, W.; Pei, J.; Zheng, L.; Dong, J.; Cao, R.; Sarangi, R.; et al. In Situ Phosphatizing of Triphenylphosphine Encapsulated within Metal-Organic Frameworks to Design Atomic Co1-P1N3 Interfacial Structure for Promoting Catalytic Performance. J. Am. Chem. Soc. 2020, 142, 8431–8439. [Google Scholar] [CrossRef]
- Cui, X.; Li, W.; Ryabchuk, P.; Junge, K.; Beller, M. Bridging Homogeneous and Heterogeneous Catalysis by Heterogeneous Single-Metal-Site Catalysts. Nat. Catal. 2018, 1, 385–397. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, W.; Lu, X.F.; Chen, T.; Lou, X.W. (David). Implanting Isolated Ru Atoms into Edge-Rich Carbon Matrix for Efficient Electrocatalytic Hydrogen Evolution. Adv. Energy Mater. 2020, 10, 2000882. [Google Scholar] [CrossRef]
- Jia, Y.; Xiong, X.; Wang, D.; Duan, X.; Sun, K.; Li, Y.; Zheng, L.; Lin, W.; Dong, M.; Zhang, G.; et al. Atomically Dispersed Fe–N4 Modified with Precisely Located S for Highly Efficient Oxygen Reduction. Nano-Micro Lett. 2020, 12, 116. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chen, W.; Xiao, H.; Gong, Y.; Li, Z.; Zheng, L.; Zheng, X.; Yan, W.; Cheong, W.-C.; Shen, R.; et al. Fe Isolated Single Atoms on S, N Codoped Carbon by Copolymer Pyrolysis Strategy for Highly Efficient Oxygen Reduction Reaction. Adv. Mater. 2018, 30, 1800588. [Google Scholar] [CrossRef]
- Wang, J.; Fang, W.; Hu, Y.; Zhang, Y.; Dang, J.; Wu, Y.; Chen, B.; Zhao, H.; Li, Z. Single Atom Ru Doping 2H-MoS2 as Highly Efficient Hydrogen Evolution Reaction Electrocatalyst in a Wide pH Range. Appl. Catal. B Environ. 2021, 298, 120490. [Google Scholar] [CrossRef]
- Zhang, H.; Yu, L.; Chen, T.; Zhou, W.; Lou, X.W. (David). Surface Modulation of Hierarchical MoS2 Nanosheets by Ni Single Atoms for Enhanced Electrocatalytic Hydrogen Evolution. Adv. Funct. Mater. 2018, 28, 1807086. [Google Scholar] [CrossRef]
- Jiang, K.; Liu, B.; Luo, M.; Ning, S.; Peng, M.; Zhao, Y.; Lu, Y.R.; Chan, T.S.; de Groot, F.M.F.; Tan, Y. Single Platinum Atoms Embedded in Nanoporous Cobalt Selenide as Electrocatalyst for Accelerating Hydrogen Evolution Reaction. Nat. Commun. 2019, 10, 1743. [Google Scholar] [CrossRef]
- Ahmed, S.; Hussain, M.S.; Khan, M.K.; Kim, J. Innovations in Catalysis towards Efficient Electrochemical Reduction of CO2 to C1 Chemicals. J. Energy Chem. 2025, 107, 622–649. [Google Scholar] [CrossRef]
- Ahmed, S.; Khan, M.K.; Kim, J. Revolutionary Advancements in Carbon Dioxide Valorization via Metal-Organic Framework-Based Strategies. Carbon Capture Sci. Technol. 2025, 15, 100405. [Google Scholar] [CrossRef]
- Yan, X.H.; Wu, R.; Xu, J.B.; Luo, Z.; Zhao, T.S. A Monolayer Graphene–Nafion Sandwich Membrane for Direct Methanol Fuel Cells. J. Power Sources 2016, 311, 188–194. [Google Scholar] [CrossRef]
- Gagliardi, G.G.; El-Kharouf, A.; Borello, D. Recent Advances in Sustainable Jet Fuels: A Review. Fuel 2023, 345, 128252. [Google Scholar] [CrossRef]
- Asghar, M.R.; Zhang, W.; Huaneng, S.; Zhang, J.; Liu, H.; Xing, L.; Yan, X.; Qian, X. A Review of Proton Exchange Membranes Modified with Inorganic Nanomaterials for Fuel Cells. Energy Adv. 2025, 4, 185–223. [Google Scholar] [CrossRef]
- Mo, S.; Du, L.; Huang, Z.; Chen, J.; Zhou, Y.; Wu, P.; Meng, L.; Wang, N.; Xing, L.; Zhao, M.; et al. Recent Progress in Electrocatalytic Biomass Conversion. Electrochem. Energy Rev. 2023, 6, 28. [Google Scholar] [CrossRef]
- Wong, C.Y.; Wong, W.Y.; Ramya, K.; Khalid, M.; Loh, K.S.; Daud, W.R.W.; Lim, K.L.; Walvekar, R.; Kadhum, A.A.H. Recent Advances in Polymer Electrolyte Membranes for Fuel Cell Applications. Int. J. Hydrogen Energy 2019, 44, 6116–6135. [Google Scholar] [CrossRef]
- Karimi, M.B.; Mohammadi, F.; Hooshyari, K. Recent Advances in Hybrid Electrolyte Membranes for Fuel Cells. Int. J. Hydrogen Energy 2019, 44, 28919–28938. [Google Scholar] [CrossRef]
- Yan, X.; Li, H.; Lin, C.; Chen, J.; Han, A.; Shen, S.; Zhang, J. Nanostructured Electrocatalysts for Sustainable Energy Conversion. Sustain. Energy Fuels 2020, 4, 772–778. [Google Scholar] [CrossRef]
- Kleinhaus, J.T.; Wolf, J.; Pellumbi, K.; Wickert, L.; Viswanathan, S.C.; Junge Puring, K.; Siegmund, D.; Apfel, U.-P. Developing Electrochemical Hydrogenation Towards Industrial Application. Chem. Soc. Rev. 2023, 52, 7305–7332. [Google Scholar] [CrossRef]
- Yang, J.; Qin, H.; Yan, K.; Cheng, X.; Wen, J. Recent Advances in the Selective Hydrogenation of Biomass-Derived Compounds. Adv. Synth. Catal. 2021, 363, 5407–5416. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y.; Hummel, C.; Huang, G.; Ji, X.; Demiröz, E.; Urakawa, A.; Huang, S.; Zhao, R.; Lercher, J.; et al. Electrocatalytic Production of a Liquid Organic Hydrogen Carrier with Anodic Valorization of the Process: Review and Outlook. Energy Fuels 2025, 39, 132–165. [Google Scholar] [CrossRef]
- Moonnee, I.; Ahmad, M.S.; Inomata, Y.; Kiatkittipong, W.; Kida, T. Hybrid Nanomaterials for Bioenergy Conversion. Nanoscale 2024, 16, 20791–20810. [Google Scholar] [CrossRef]
- Luo, T.; Abdu, S.; Wessling, M. Selectivity of Ion Exchange Membranes: A Review. J. Membr. Sci. 2018, 555, 429–454. [Google Scholar] [CrossRef]
- Espinoza, C.; Díaz, J.C.; Kitto, D.; Kim, H.K.; Kamcev, J. Bound Water Enhances the Ion Selectivity of Highly Charged Polymer Membranes. ACS Appl. Mater. Interfaces 2024, 16, 45433–45446. [Google Scholar] [CrossRef]
- Wang, R.; Kapteijn, F.; Gascon, J. Engineering Metal–Organic Frameworks for the Electrochemical Reduction of CO2: A Minireview. Chem. Asian J. 2019, 14, 3452–3461. [Google Scholar] [CrossRef] [PubMed]
- Gulati, S.; Vijayan, S.; Kumar, S.; Harikumar, B.; Trivedi, M.; Varma, R.S. Recent Advances in the Application of MOF-Based Nanocatalysts for Direct CO2 Conversion to Value-Added Chemicals. Coord. Chem. Rev. 2023, 474, 214853. [Google Scholar] [CrossRef]
- Madagalam, M.; Bartoli, M.; Tagliaferro, A. A Short Overview on Graphene and Graphene-Related Materials for Electrochemical Gas Sensing. Materials 2024, 17, 2. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ren, Y.W.; Fan, F.; Wu, T.Y.; Wang, Z.; Zhang, Y.J.; Zhao, J.W.; Cui, G.L. Artificial Frameworks Towards Ion-Channel Construction in Proton Exchange Membranes. J. Power Sources 2023, 574, 233081. [Google Scholar] [CrossRef]
- Tellez-Cruz, M.M.; Escorihuela, J.; Solorza-Feria, O.; Compañ, V. Proton Exchange Membrane Fuel Cells (PEMFCs): Advances and Challenges. Polymers 2021, 13, 18. [Google Scholar] [CrossRef]
- Ahmad, S.; Nawaz, T.; Ali, A.; Orhan, M.F.; Samreen, A.; Kannan, A.M. An Overview of Proton Exchange Membranes for Fuel Cells: Materials and Manufacturing. Int. J. Hydrogen Energy 2022, 47, 22917–22942. [Google Scholar] [CrossRef]
- Komma, M.; Marth, A.; Maier, M.; Hutzler, A.; Böhm, T.; Thiele, S. Applicability of Graphene Oxide Interlayers in PEMs for Reducing Crossover in Electrochemical Acetone Hydrogenation Reactors. J. Electrochem. Soc. 2024, 171, 104502. [Google Scholar] [CrossRef]
- Hauenstein, P.; Seeberger, D.; Wasserscheid, P.; Thiele, S. Electrochemical Hydrogenation of Acetone in a PEM Reactor. Electrochem. Commun. 2020, 118, 106786. [Google Scholar] [CrossRef]
- Ding, Y.; Zheng, H.; Cheng, J.; Xu, H.; Sun, M.; Yan, G. Platinum Supported on Reduced Graphene Oxide as a Catalyst for the Electrochemical Hydrogenation of Soybean Oils. Solid State Sci. 2019, 92, 46–52. [Google Scholar] [CrossRef]
- García-Cruz, L.; Casado-Coterillo, C.; Irabien, Á.; Montiel, V.; Iniesta, J. High Performance of Alkaline Anion-Exchange Membranes Based on Chitosan/Poly(Vinyl Alcohol) Doped with Graphene Oxide for the Electrooxidation of Primary Alcohols. C 2016, 2, 10. [Google Scholar] [CrossRef]
- Gahlot, S.; Sharma, P.P.; Kulshrestha, V.; Jha, P.K. SGO/SPES-Based Highly Conducting Polymer Electrolyte Membranes for Fuel Cell Application. ACS Appl. Mater. Interfaces 2014, 6, 5595–5601. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Choi, B.G.; Choi, D.; Park, H.S. Nanoindentation of Annealed Nafion/Sulfonated Graphene Oxide Nanocomposite Membranes for the Measurement of Mechanical Properties. J. Membr. Sci. 2014, 451, 40–45. [Google Scholar] [CrossRef]
- Sharma, P.P.; Gahlot, S.; Bhil, B.M.; Gupta, H.; Kulshrestha, V. An Environmentally Friendly Process for the Synthesis of an fGO Modified Anion Exchange Membrane for Electro-Membrane Applications. RSC Adv. 2015, 5, 38712–38721. [Google Scholar] [CrossRef]
- Sahroni, I.; Kodama, T.; Ahmad, M.S.; Nakahara, T.; Inomata, Y.; Kida, T. Platinum Nanoparticles Supported on Functionalized Graphene Oxide for Electrocatalytic Applications. Nano Lett. 2024, 24, 3590–3597. [Google Scholar] [CrossRef]
- Hong, J.G.; Park, T.W. Ion-Exchange Membranes for Blue Energy Generation: A Short Overview Focused on Nanocomposites. J. Electrochem. Sci. Eng. 2023, 13, 12. [Google Scholar] [CrossRef]
- Strathmann, H.; Grabowski, A.; Eigenberger, G. Ion-Exchange Membranes in the Chemical Process Industry. Ind. Eng. Chem. Res. 2013, 52, 10364–10379. [Google Scholar] [CrossRef]
- Li, J.S.; Liu, C.P.; Ge, J.J.; Xing, W.; Zhu, J.B. Challenges and Strategies of Anion Exchange Membranes in Hydrogen-Electricity Energy Conversion Devices. Chem. Eur. J. 2023, 29, 26. [Google Scholar] [CrossRef]
- Liu, W.; Yin, X.B. Metal–Organic Frameworks for Electrochemical Applications. TrAC Trends Anal. Chem. 2016, 75, 86–96. [Google Scholar] [CrossRef]
- Chen, K.F.; Wang, X.L.; Hu, W.H.; Kong, Q.Q.; Pang, H.; Xu, Q. Modified Metal-Organic Frameworks for Electrochemical Applications. Small Struct. 2022, 3, 2100200. [Google Scholar] [CrossRef]
- Luo, L.Q.; Qian, X.D.; Wang, X.J. Bimetallic Metal-Organic Frameworks and Their Derivatives for Electrochemical Energy Conversion and Storage: Recent Progress, Challenges and Perspective. J. Energy Storage 2024, 98, 113052. [Google Scholar] [CrossRef]
- Amouzegar, K.; Savadogo, O. Electrocatalytic Hydrogenation of Phenol on Highly Dispersed Pt Electrodes. Electrochim. Acta 1994, 39, 557–559. [Google Scholar] [CrossRef]
- Martel, A.; Mahdavi, B.; Lessard, J.; Ménard, H.; Brossard, L.; Tountian, D.; Brisach-Wittmeyer, A.; Nkeng, P.; Poillerat, G.; Chagnes, A.; et al. Electrocatalytic Hydrogenation of Phenol on Various Electrode Materials. Can. J. Chem. 1997, 75, 1862–1867. [Google Scholar] [CrossRef]
- Amouzegar, K.; Savadogo, O. Electrocatalytic Hydrogenation of Phenol on Dispersed Pt: Effect of Metal Electrochemically Active Surface Area and Electrode Material. J. Appl. Electrochem. 1997, 27, 539–542. [Google Scholar] [CrossRef]
- Amouzegar, K.; Savadogo, O. Electrocatalytic Hydrogenation of Phenol on Dispersed Pt: Reaction Mechanism and Support Effect. Electrochim. Acta 1998, 43, 503–508. [Google Scholar] [CrossRef]
- Singh, N.; Sanyal, U.; Ruehl, G.; Stoerzinger, K.A.; Gutiérrez, O.Y.; Camaioni, D.M.; Fulton, J.L.; Lercher, J.A.; Campbell, C.T. Aqueous Phase Catalytic and Electrocatalytic Hydrogenation of Phenol and Benzaldehyde over Platinum Group Metals. J. Catal. 2020, 382, 372–384. [Google Scholar] [CrossRef]
- Cirtiu, C.M.; Hassani, H.O.; Bouchard, N.; Rowntree, P.A.; Me, H. Modification of the Surface Adsorption Properties of Alumina-Supported Pd Catalysts for the Electrocatalytic Hydrogenation of Phenol. Langmuir 2006, 22, 6414–6421. [Google Scholar] [CrossRef]
- Zhao, B.; Guo, Q.; Fu, Y. Electrocatalytic Hydrogenation of Lignin-Derived Phenol into Alkanes by Using Platinum Supported on Graphite. Electrochemistry 2014, 82, 954–959. [Google Scholar] [CrossRef]
- Song, Y.; Gutiérrez, O.Y.; Herranz, J.; Lercher, J.A. Aqueous Phase Electrocatalysis and Thermal Catalysis for the Hydrogenation of Phenol at Mild Conditions. Appl. Catal. B 2016, 182, 236–246. [Google Scholar] [CrossRef]
- Singh, N.; Song, Y.; Gutiérrez, O.Y.; Camaioni, D.M.; Campbell, C.T.; Lercher, J.A. Electrocatalytic Hydrogenation of Phenol over Platinum and Rhodium: Unexpected Temperature Effects Resolved. ACS Catal. 2016, 6, 7466–7470. [Google Scholar] [CrossRef]
- Lu, X.; Wang, J.; Peng, W.; Li, N.; Liang, L.; Cheng, Z.; Yan, B.; Yang, G.; Chen, G. Electrocatalytic Hydrogenation of Phenol by Active Sites on Pt-Decorated Shrimp Shell Biochar Catalysts: Performance and Internal Mechanism. Fuel 2023, 331, 125845. [Google Scholar] [CrossRef]
- Zhou, L.; Zhu, X.; Su, H.; Lin, H.; Lyu, Y.; Zhao, X.; Chen, C.; Zhang, N.; Xie, C.; Li, Y.; et al. Identification of the Hydrogen Utilization Pathway for the Electrocatalytic Hydrogenation of Phenol. Sci. China Chem. 2021, 64, 1586–1595. [Google Scholar] [CrossRef]
- Du, Y.; Chen, X.; Liang, C. Selective Electrocatalytic Hydrogenation of Phenols over Ternary Pt3RuSn Alloy. Mol. Catal. 2023, 535, 112831. [Google Scholar] [CrossRef]
- Gu, Z.; Zhang, Z.; Ni, N.; Hu, C.; Qu, J. Simultaneous Phenol Removal and Resource Recovery from Phenolic Wastewater by Electrocatalytic Hydrogenation. Environ. Sci. Technol. 2022, 56, 4356–4366. [Google Scholar] [CrossRef]
- Kassim, A.B.; Rice, C.L.; Kuhn, A.T. Formation of Sorbitol by Cathodic Reduction of Glucose. J. Appl. Electrochem. 1981, 11, 261–267. [Google Scholar] [CrossRef]
- Pintauro, P.N.; Johnson, D.K.; Park, K.; Baizer, M.M.; Nobe, K. The Paired Electrochemical Synthesis of Sorbitol and Gluconic Acid in Undivided Flow Cells. I. J. Appl. Electrochem. 1984, 14, 209–220. [Google Scholar] [CrossRef]
- Lessard, J.; Belot, G.; Couture, Y.; Desjardins, S.; Roy, C. The Use of Hydrogen Generated at the Electrode Surface for Electrohydrogenation of Organic Compounds. Int. J. Hydrogen Energy 1993, 18, 681–684. [Google Scholar] [CrossRef]
- Nilges, P.; Schröder, U. Electrochemistry for Biofuel Generation: Production of Furans by Electrocatalytic Hydrogenation of Furfurals. Energy Environ. Sci. 2013, 6, 2925–2931. [Google Scholar] [CrossRef]
- Li, Z.; Garedew, M.; Lam, C.H.; Jackson, J.E.; Miller, D.J.; Saffron, C.M. Mild Electrocatalytic Hydrogenation and Hydrodeoxygenation of Bio-Oil Derived Phenolic Compounds Using Ruthenium Supported on Activated Carbon Cloth. Green Chem. 2012, 14, 2540–2549. [Google Scholar] [CrossRef]
- Wijaya, Y.P.; Grossmann-Neuhaeusler, T.; Dhewangga Putra, R.D.; Smith, K.J.; Kim, C.S.; Gyenge, E.L. Electrocatalytic Hydrogenation of Guaiacol in Diverse Electrolytes Using a Stirred Slurry Reactor. ChemSusChem 2020, 13, 629–639. [Google Scholar] [CrossRef]
- Wijaya, Y.P.; Smith, K.J.; Kim, C.S.; Gyenge, E.L. Synergistic Effects between Electrocatalyst and Electrolyte in the Electrocatalytic Reduction of Lignin Model Compounds in a Stirred Slurry Reactor. J. Appl. Electrochem. 2021, 51, 51–63. [Google Scholar] [CrossRef]
- Peng, T.; Zhuang, T.; Yan, Y.; Qian, J.; Dick, G.R.; de Bueren, J.B.; Hung, S.-F.; Zhang, Y.; Wang, Z.; Wicks, J.; et al. Ternary Alloys Enable Efficient Production of Methoxylated Chemicals via Selective Electrocatalytic Hydrogenation of Lignin Monomers. J. Am. Chem. Soc. 2021, 143, 17226–17235. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.H.; Lowe, C.B.; Li, Z.; Longe, K.N.; Rayburn, J.T.; Caldwell, M.A.; Houdek, C.E.; Maguire, J.B.; Saffron, C.M.; Miller, D.J.; et al. Electrocatalytic Upgrading of Model Lignin Monomers with Earth Abundant Metal Electrodes. Green Chem. 2015, 17, 601–609. [Google Scholar] [CrossRef]
- Garedew, M.; Young-Farhat, D.; Jackson, J.E.; Saffron, C.M. Electrocatalytic Upgrading of Phenolic Compounds Observed after Lignin Pyrolysis. ACS Sustain. Chem. Eng. 2019, 7, 8375–8386. [Google Scholar] [CrossRef]
- Wang, M.; Peng, T.; Yang, C.; Liang, B.; Chen, H.; Kumar, M.; Zhang, Y.; Zhao, W. Electrocatalytic Hydrogenation of Lignin Monomer to Methoxy-Cyclohexanes with High Faradaic Efficiency. Green Chem. 2022, 24, 142–146. [Google Scholar] [CrossRef]
- Xin, L.; Zhang, Z.; Qi, J.; Chadderdon, D.J.; Qiu, Y.; Warsko, K.M.; Li, W. Electricity Storage in Biofuels: Selective Electrocatalytic Reduction of Levulinic Acid to Valeric Acid or γ-Valerolactone. ChemSusChem 2013, 6, 674–686. [Google Scholar] [CrossRef]
- Nilges, P.; Dos Santos, T.R.; Harnisch, F.; Schröder, U. Electrochemistry for Biofuel Generation: Electrochemical Conversion of Levulinic Acid to Octane. Energy Environ. Sci. 2012, 5, 5231–5235. [Google Scholar] [CrossRef]
- Dos Santos, T.R.; Nilges, P.; Sauter, W.; Harnisch, F.; Schröder, U. Electrochemistry for the Generation of Renewable Chemicals: Electrochemical Conversion of Levulinic Acid. RSC Adv. 2015, 5, 26634–26643. [Google Scholar] [CrossRef]
- Zhao, B.; Chen, M.; Guo, Q.; Fu, Y. Electrocatalytic Hydrogenation of Furfural to Furfuryl Alcohol Using Platinum Supported on Activated Carbon Fibers. Electrochim. Acta 2014, 135, 139–146. [Google Scholar] [CrossRef]
- Jung, S.; Biddinger, E.J. Electrocatalytic Hydrogenation and Hydrogenolysis of Furfural and the Impact of Homogeneous Side Reactions of Furanic Compounds in Acidic Electrolytes. ACS Sustain. Chem. Eng. 2016, 4, 6500–6508. [Google Scholar] [CrossRef]
- Wang, F.; Xu, M.; Wei, L.; Wei, Y.; Hu, Y.; Fang, W.; Zhu, C.G. Fabrication of La-Doped TiO2 Film Electrode and Investigation of Its Electrocatalytic Activity for Furfural Reduction. Electrochim. Acta 2015, 153, 170–174. [Google Scholar] [CrossRef]
- Lopez-Ruiz, J.A.; Andrews, E.; Akhade, S.A.; Lee, M.S.; Koh, K.; Sanyal, U.; Yuk, S.F.; Karkamkar, A.J.; Derewinski, M.A.; Holladay, J.; et al. Understanding the Role of Metal and Molecular Structure on the Electrocatalytic Hydrogenation of Oxygenated Organic Compounds. ACS Catal. 2019, 9, 9964–9972. [Google Scholar] [CrossRef]
- Liu, W.; You, W.; Gong, Y.; Deng, Y. High-Efficiency Electrochemical Hydrodeoxygenation of Bio-Phenols to Hydrocarbon Fuels by a Superacid-Noble Metal Particle Dual-Catalyst System. Energy Environ. Sci. 2020, 13, 917–927. [Google Scholar] [CrossRef]
- Zhou, Y.; Gao, Y.; Zhong, X.; Jiang, W.; Liang, Y.; Niu, P.; Li, M.; Zhuang, G.; Li, X.; Wang, J. Electrocatalytic Upgrading of Lignin-Derived Bio-Oil Based on Surface-Engineered PtNiB Nanostructure. Adv. Funct. Mater. 2019, 29, 1807651. [Google Scholar] [CrossRef]
- Yu, Y.; Lee, S.J.; Theerthagiri, J.; Fonseca, S.; Pinto, L.M.C.; Maia, G.; Choi, M.Y. Reconciling of Experimental and Theoretical Insights on the Electroactive Behavior of C/Ni Nanoparticles with AuPt Alloys for Hydrogen Evolution Efficiency and Non-Enzymatic Sensor. Chem. Eng. J. 2022, 435, 134790. [Google Scholar] [CrossRef]
- Kwon, Y.; Birdja, Y.Y.; Raoufmoghaddam, S.; Koper, M.T. Electrocatalytic Hydrogenation of 5-Hydroxymethylfurfural in Acidic Solution. ChemSusChem 2015, 8, 1745–1751. [Google Scholar] [CrossRef]
- Ciotti, A.; Rahaman, M.; Yeung, C.W.S.; Li, T.; Reisner, E.; García-Melchor, M. Driving Electrochemical Organic Hydrogenations on Metal Catalysts by Tailoring Hydrogen Surface Coverages. J. Am. Chem. Soc. 2025, 147, 13158–13168. [Google Scholar] [CrossRef]
- Ledezma-Yanez, I.; Wallace, W.D.Z.; Sebastián-Pascual, P.; Climent, V.; Feliu, J.M.; Koper, M.T.M. Interfacial Water Reorganization as a pH-Dependent Descriptor of the Hydrogen Evolution Rate on Platinum Electrodes. Nat. Energy 2017, 2, 17031. [Google Scholar] [CrossRef]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Nørskov, J.K.; Jaramillo, T.F. Combining Theory and Experiment in Electrocatalysis: Insights into Materials Design. Science 2017, 355, eaad4998. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jonsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Bondue, C.J.; Koper, M.T.M. Electrochemical Reduction of the Carbonyl Functional Group: The Importance of Adsorption Geometry, Molecular Structure, and Electrode Surface Structure. J. Am. Chem. Soc. 2019, 141, 12071–12078. [Google Scholar] [CrossRef]
- Bondue, C.J.; Calle-Vallejo, F.; Figueiredo, M.C.; Koper, M.T.M. Structural Principles to Steer the Selectivity of the Electrocatalytic Reduction of Aliphatic Ketones on Platinum. Nat. Catal. 2019, 2, 243–250. [Google Scholar] [CrossRef]
- Skúlason, E.; Karlberg, G.S.; Rossmeisl, J.; Bligaard, T.; Greeley, J.; Jónsson, H.; Nørskov, J.K. Density Functional Theory Calculations for the Hydrogen Evolution Reaction in an Electrochemical Double Layer on the Pt(111) Electrode. Phys. Chem. Chem. Phys. 2007, 9, 3241–3251. [Google Scholar] [CrossRef]
- Mahmood, N.; Yao, Y.; Zhang, J.-W.; Pan, L.; Zhang, X.; Zou, J.-J. Electrocatalysts for Hydrogen Evolution in Alkaline Electrolytes: Mechanisms, Challenges, and Prospective Solutions. Adv. Sci. 2018, 5, 1700464. [Google Scholar] [CrossRef]
- Strmcnik, D.; Lopes, P.P.; Genorio, B.; Stamenkovic, V.R.; Markovic, N.M. Design Principles for Hydrogen Evolution Reaction Catalyst Materials. Nano Energy 2016, 29, 29–36. [Google Scholar] [CrossRef]
- Zhao, B.-H.; Chen, F.; Wang, M.; Cheng, C.; Wu, Y.; Liu, C.; Yu, Y.; Zhang, B. Economically Viable Electrocatalytic Ethylene Production with High Yield and Selectivity. Nat. Sustain. 2023, 6, 827–837. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Catalyst/Electrocatalyst/Electrode | Membrane | Products | Ref. |
|---|---|---|---|---|
| Phenol | Pt/Pt, Pt/C, Pd/C,Ru/C,Rh/C Rh/Ni, Ru/Ni, RaNi, Pt alloys: Cr, V, Co, Ir | Nafion-324 | Cyclohexanol, Cyclohexanone | [107,108,109,110,111] |
| Pt/G,Rh/G,Pd/G,Rh/C,Ru/TiO2, Pt/SSB,RVC-Pt mesh, RVC-graphite, Pt3RuSn/CC, Pt1Rh1/MCN | Nafion-117 | Cyclohexanol, Cyclohexanone, Cyclohexane | [112,113,114,115,116,117,118,119] | |
| Glucose | Pb(Hg), Zn, | Nafion, type XR 475 | Sorbitol | [120,121] |
| RaNi, | Nafion-324 | Sorbitol | [122] | |
| 5-Hydroxymethyl Furfural (5-HMF) | Cu | Nafion-117 | 2,5-dimethylfuran (DMF), 2,5-di (hydroxymethyl)furan, 5-methylfur furyl alcohol, and 5-methylfuran-2-carbaldehyde. | [123] |
| Guaiacol | Ru/ACC, Pt gauze, Ti gauze, Ni gauze, PtRhAu | Nafion-117 | Phenol, 2-Methoxycyclohexanol, 2-Methoxycyclohexanone | [124,125,126,127] |
| Ra-Ni | Nafion-117 | Phenol, 3-Methoxycyclohexanol, 4-Methoxycyclohexanol | [128] | |
| Rh/ACC | Nafion | Cyclohexanol | [129] | |
| RhPtRu | Bipolar membrane | Methoxy-cyclohexanes | [130] | |
| Levulinic acid (LVA) | Pb, Cu, Fe, Ni, C | Nafion-117 Fumasep® FKE (FuMA-Tech GmbH, Ludwigsburg, Germany) | Valeric acid, γ-valerolactone | [131,132,133] |
| Furfural | Pt, Pb, Ni, Cu, Pt/ACF, C, Fe, Ti/La nano-TiO2, Ti/nano-TiO2, Rh/C, Pt/C, Pd/C, | Nafion Nafion-117 Nafion-115 Fumasep® FKE | Furfuryl alcohol, methyl furan, 2-methylfuran, Pinacol, furoic acid, tetrahydrofurfuryl alcohol, 2-methyltetrahydrofuran | [42,134,135,136,137] |
| Phenolic compounds | PtNiB/CMK-3, Graphite rod | Nafion-117 | Cyclohexanol, cyclohexanone, Cyclohexane | [138,139] |
| Electrochemical Hydrogenation (ECH) | Thermochemical Hydrogenation (TCH) | |
|---|---|---|
| Energy Efficiency | It efficiently employs electrical energy and requires only a small amount of thermal energy. This system can work with renewable energy sources, and energy recovery is possible through built-in half-reactions. For example, producing two outputs in a single cell has shown to result in energy savings of up to 50%. | The process demands the production of external hydrogen (H2) and heating, with the generation and compression of hydrogen leading to extra energy expenses. There is a restricted use of waste heat, and co-reactions are typically not taken advantage of, potentially causing a reduction in overall energy efficiency. |
| Selectivity | Low temperatures and adjustable potentials facilitate achieving high selectivity for the target product, simultaneously reducing side reactions. For instance, under optimized alloy composition and reaction conditions, a single-product selectivity exceeding 95% can be attained. Additionally, by adjusting the potential and the reaction environment, it is possible to select intermediate products by stopping the reaction at a predetermined stage. | When there is an adequate supply of hydrogen (H2), thermochemical hydrogenation typically facilitates a targeted transformation. Nonetheless, higher temperatures can heighten the likelihood of side reactions. In cases where the catalyst possesses acidic sites, unwanted processes like deoxygenation might take place. It is challenging to accumulate intermediate products unless the procedure is deliberately halted, given that the reaction naturally progresses towards the end product from a thermodynamic perspective. |
| Reaction Conditions | Functions effectively under gentle conditions, specifically at atmospheric pressure and within the temperature range of 20–60 °C. This method can be readily utilized in aqueous environments or in slightly organic-modified settings, as hydrogen is generated on-site from water, eliminating the need for an external gas supply. The procedure is fundamentally safe and does not necessitate intricate pressurized apparatus. | The process may necessitate the use of high-pressure hydrogen gas, usually ranging from several bar to twenty bar, depending on the specific reaction conditions. Elevated temperatures, typically exceeding 100 °C, are frequently utilized. In certain cases, organic solvents or biphasic systems might be required, with the solubility of hydrogen being a potential constraint. The complexity and expense of the operation are increased due to the need for specialized pressurized equipment and heating systems. |
| Catalyst Activity and Reusability | The low operational temperatures significantly reduce the risks of catalyst sintering and coke accumulation, leading to prolonged catalyst activity. Catalysts are typically very stable; for example, the Pt/SSB catalyst showcased a conversion rate of 94% after undergoing three cycles. Regeneration of the catalyst surface can be facilitated by re-hydrogenating the poisoning agents, which is accomplished by modifying the electrode potential. When the catalyst is applied to the electrode, it is straightforward to keep it contained within the reactor; alternatively, if the catalyst is in suspension, it can be easily filtered and reused. | Metal particles can sinter under thermal conditions, reducing the active surface area for reactions. Elevated temperatures can create byproducts like coke and polymers, contaminating the catalyst surface. Periodic regeneration methods, such as hydrogen treatment or oxidative cleaning, may be needed. Extracting solid catalysts from pressurized reactors is challenging since they are typically in fixed-bed setups and replaced upon deactivation. Nonetheless, many commercial catalysts can work effectively for hundreds of hours, with their stability influenced by feedstock purity and operating temperature. |
| Surface Poisoning and Adsorption | The buildup of specific intermediate species on a catalyst’s surface can result in catalyst poisoning, primarily due to insufficient hydrogen (H) coverage. This issue can be alleviated by either modifying the applied potential or increasing the proton flux, as indicated in the literature. For example, during electrochemical hydrogenation (ECH) processes, raising the current or potential enhances the rehydrogenation of phenolic species that are adsorbed, thus helping to restore the electrode surface. Additionally, organic surface modifiers like carboxylates can be intentionally adsorbed onto the electrode to influence the reaction pathway. Furthermore, the adsorption strength of hydrogen on the metal surface can be adjusted through alloying, which enables the optimization of the relationship between the hydrogen evolution reaction (HER) and electrochemical hydrogenation (ECH). | At high temperatures, hydrocarbon derivatives may polymerize on catalysts, leading to deactivation. To address this, elevated hydrogen (H2) partial pressures are maintained, which helps to prevent poisoning and allows regeneration by hydrogenating accumulated species. In thermal catalytic hydrogenation (TCH), the active surface’s potential is not externally controlled; instead, the adsorption characteristics depend on the catalyst formulation. Metals like palladium (Pd) can form hydride phases under TCH, affecting reaction kinetics, though this is less significant than in electrochemical catalytic hydrogenation (ECH). Catalyst poisoning regeneration often involves increasing H2 pressure or replacing the deactivated catalyst. |
| Industrial Applicability and Scalability | Modular and Scalable Design: Electrochemical cells can be arranged in series or parallel configurations to meet specific capacity requirements. They utilize similar infrastructure to current technologies, including water electrolysis and electroplating. Safety Benefits: Since high-pressure hydrogen is not stored, the likelihood of explosion is significantly reduced. When powered by renewable energy sources, this process is completely sustainable. The operation occurs at low temperature and pressure, leading to decreased equipment costs. Moreover, it is well suited for decentralized applications, such as on-site wastewater treatment. An example of this is the effective electrochemical hydrogenation (ECH) of phenol and guaiacol in continuous flow and membrane reactor systems, which has established a framework for larger-scale implementation. | Traditional and Commonly Employed in Industry: Fixed-bed reactors, batch autoclaves, and continuous reactors are available for large-scale applications. These systems necessitate hydrogen supply and storage, making them more appropriate for centralized, large facilities. Processing feedstocks with low hydrogen concentrations is not efficient; for instance, hydrogenating phenol in wastewater under high pressure is not feasible. Most current processes rely on hydrogen derived from fossil fuels, resulting in a substantial carbon footprint. Although the implementation of green hydrogen is an option, it tends to be expensive. At present, economies of scale favor large-volume production, but this situation may not be beneficial for small-scale or decentralized operations. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Durak, H. Comparative Analysis of Electrochemical and Thermochemical Hydrogenation of Biomass-Derived Phenolics for Sustainable Biofuel and Chemical Production. Processes 2025, 13, 1581. https://doi.org/10.3390/pr13051581
Durak H. Comparative Analysis of Electrochemical and Thermochemical Hydrogenation of Biomass-Derived Phenolics for Sustainable Biofuel and Chemical Production. Processes. 2025; 13(5):1581. https://doi.org/10.3390/pr13051581
Chicago/Turabian StyleDurak, Halil. 2025. "Comparative Analysis of Electrochemical and Thermochemical Hydrogenation of Biomass-Derived Phenolics for Sustainable Biofuel and Chemical Production" Processes 13, no. 5: 1581. https://doi.org/10.3390/pr13051581
APA StyleDurak, H. (2025). Comparative Analysis of Electrochemical and Thermochemical Hydrogenation of Biomass-Derived Phenolics for Sustainable Biofuel and Chemical Production. Processes, 13(5), 1581. https://doi.org/10.3390/pr13051581